

Abstract
Objective:
To determine the short-term and long-term effects of dichlorphenamide (DCP) on attack frequency and quality of life in hyperkalemic (HYP) and hypokalemic (HOP) periodic paralysis.
Methods:
Two multicenter randomized, double-blind, placebo-controlled trials lasted 9 weeks (Class I evidence), followed by a 1-year extension phase in which all participants received DCP. Forty-four HOP and 21 HYP participants participated. The primary outcome variable was the average number of attacks per week over the final 8 weeks of the double-blind phase.
Results:
The median attack rate was lower in HOP participants on DCP than in participants on placebo (0.3 vs 2.4, p = 0.02). The 9-week mean change in the Physical Component Summary score of the Short Form–36 was also better in HOP participants receiving DCP (treatment effect = 7.29 points, 95% confidence interval 2.26 to 12.32, p = 0.006). The median attack rate was also lower in HYP participants on DCP (0.9 vs 4.8) than in participants on placebo, but the difference in median attack rate was not significant (p = 0.10). There were no significant effects of DCP on muscle strength or muscle mass in either trial. The most common adverse events in both trials were paresthesia (47% DCP vs 14% placebo, both trials combined) and confusion (19% DCP vs 7% placebo, both trials combined).
Conclusions:
DCP is effective in reducing the attack frequency, is safe, and improves quality of life in HOP periodic paralysis.
Classification of evidence:
These studies provide Class I evidence that DCP significantly reduces attack frequency in HOP but lacked the precision to support either efficacy or lack of efficacy of DCP in HYP.
The periodic paralyses (PPs) are inherited skeletal muscle channelopathies characterized by attacks of weakness associated with serum potassium change, and typically triggered by exercise followed by rest.1–3 Hypokalemic PP (HOP) is caused by mutations in CACNA1S in the majority4,5; a small proportion (<10%) is due to SCN4A mutations.6–10 Hyperkalemic PP (HYP) is caused by mutations in SCN4A.11,12 Although the PP are seldom fatal, they cause major disability and persistent interattack weakness may develop.13,14 There is no established standardized treatment.15–18
It has been suggested that dichlorphenamide (DCP) may be more effective than acetazolamide15,19,20 in both preventing attacks and improving interictal strength in PP.21 In 2000, 2 randomized, double-blind, placebo-controlled crossover trials22 showed that DCP was effective in the prevention of episodic weakness in both HOP and HYP. These initial crossover trials had a short follow-up duration and did not assess quality of life.
We report the results of 2 multicenter randomized, double-blind, placebo-controlled parallel-group trials of DCP in HOP and in HYP. A 9-week double-blind phase was followed by a 1-year uncontrolled extension phase during which all participants received DCP to determine the short- and long-term effects of DCP.
The study is listed on clinicaltrials.gov (NCT00494507).
METHODS
Primary objective.
To determine the short-term (with Class I evidence) and long-term (with Class II evidence) effects of DCP on attack frequency and quality of life in HYP and HOP.
Study design.
Two trials (HOP and HYP) were conducted with the same protocol. Each trial was divided into 2 phases: a randomized, double-blind, placebo-controlled 9-week phase followed by a 1-year uncontrolled extension phase during which all participants received treatment with DCP. The original design included an acetazolamide group in each trial. There is a widespread view among PP patients that acetazolamide is inferior to DCP (based on personal experience or that of family members) so the acetazolamide arm had to be abandoned to facilitate recruitment. Randomization began in April 2007 and follow-up ended in November 2013.
Standard protocol approvals, registrations, and patient consents.
The protocol was approved by the institutional review board at each of the 12 participating centers: 10 in the United States, 1 in the United Kingdom, and 1 in Italy. All participants gave written informed consent. The trial sponsor (National Institute of Neurological Disorders and Stroke) established a Data and Safety Monitoring Board that monitored the study (but did not review efficacy outcomes).
Participants.
Men and women aged 18 years and older with genetically definite, clinically definite, or clinically probable HOP or HYP defined according to accepted diagnostic criteria17,23 (table e-1, appendix e-1, and appendix e-2 on the Neurology® Web site at Neurology.org) were eligible. Participants were required to have distinct regular episodes of weakness with an average frequency of at least 1 per week and fewer than 3 per day either on or off treatment. Exclusion criteria are set out in table e-2.
Randomization and blinding.
Participants were randomized to DCP or matching placebo (manufactured and supplied by Taro Pharmaceutical Industries, Haifa Bay, Israel) for the first 9 weeks. Further details of randomization are provided in appendix e-1. An independent blinded physician at each site monitored the laboratory test results for safety.
Intervention.
DCP is 30 times more potent than acetazolamide in vitro.24 If a participant was taking either acetazolamide or DCP at enrollment, that medication was immediately stopped and study drug was substituted; there were no washout periods. Participants already taking acetazolamide at enrollment were assigned an equivalent dosage of study medication, calculated as 20% of the acetazolamide dosage. Those taking DCP at enrollment received the same dosage of study medication, and those on no carbonic anhydrase inhibitor received 50 mg bid. HOP patients continued using potassium supplements as required for acute attacks. Study drug was started during a 3-day inpatient visit. If a participant experienced an intolerable adverse event, dosage reduction was permitted. Participants continued the double-blind dosage at entry into the 1-year extension phase, after which dosage adjustment to mimic clinical practice was permitted.
Outcome variables.
Double-blind phase.
Details regarding participant evaluation are provided in appendix e-1. The primary measure of efficacy was the average number of attacks per week over the final 8 weeks of the 9-week double-blind phase as captured by daily telephone report to an electronic diary (interactive voice response).25 The first week of diary data was excluded to avoid contamination of the efficacy assessment by the participant's previous treatment, if any. Secondary outcome variables included severity-weighted attack rate (sum of attack severity scores from 1 to 10 over the final 8 weeks of the 9-week double-blind phase divided by the number of weeks), total attack duration per week (sum of attack durations across all distinct attacks over the final 8 weeks of the double-blind phase divided by the number of weeks), and, for the HOP trial only, the endpoint of acute worsening during the double-blind phase. Acute worsening was defined as an intolerable increase in frequency or severity of attacks, necessitating withdrawal from the 9-week double-blind phase and immediate initiation of the 1-year extension phase. Additional secondary outcome variables included 9-week changes in muscle strength measured clinically (manual muscle testing [MMT]) and quantitatively (maximal voluntary isometric contraction testing),26,27 lean body mass (measured by DEXA),28 and quality of life (Short Form-36 [SF-36] v2).29,30
Safety outcome variables included occurrences of individual adverse events and changes from baseline to week 9 in vital signs, laboratory test results, and neuropsychological test results (Symbol Digit Modalities Test,31 Profile of Mood States,32 and Trail-Making Test Parts A and B).33
One-year uncontrolled extension phase.
Outcomes assessed during the uncontrolled extension phase included attack rate, severity-weighted attack rate, total attack duration per week from weeks 54 to 61, and changes from weeks 9 to 61 in measures of strength and lean body mass. Adverse events and other safety outcomes were also assessed.
Statistics.
Power calculations based on results from a previous trial22 using a bootstrap method (see supplementary material for details) yielded a target sample size of 60 HOP participants (30 per group) and 80 HYP participants (40 per group). Due to delays in recruitment, the trial was stopped before the target sample sizes were achieved.
Attack rates were compared between the DCP and placebo groups using Wilcoxon rank sum tests. Participants who reached the endpoint of acute worsening and those who did not report any postbaseline data were included by assigning them an attack rate that was higher than any other observed during the trial. Treatment effects and associated 95% confidence intervals were estimated from the bootstrap distribution of the difference in medians between the DCP and placebo groups (median and 2.5th and 97.5th percentiles).34 Missing diary entries prior to week 9 were treated as indicating no attack on that day. For participants who prematurely withdrew from the trial, attack rates were computed using all available diary data up until the time of withdrawal. Similar analyses were performed for severity-weighted attack rate and total attack duration per week.
The proportions of HOP participants on DCP or placebo reaching the endpoint of acute worsening were compared with Fisher exact test.
Analysis of covariance, adjusting for the baseline value of the outcome variable, was used to compare the DCP and placebo groups with respect to the mean change from baseline to week 9 in strength, lean body mass, and quality of life outcomes. Missing data at week 9 were accommodated in the analyses using regression-based multiple imputation with the baseline value of the outcome variable and treatment group included in the imputation model.35 Changes from baseline to week 9 in vital signs and neuropsychological test results were analyzed similarly. One-sample t tests were used to evaluate the significance of mean changes in strength and lean body mass outcomes from week 9 to week 61. Diary outcomes and adverse events and other safety data from the uncontrolled extension phase were analyzed descriptively.
The analyses of data from the 9-week double-blind phase included all randomized participants in accordance with the intention-to-treat principle, with one exception noted in the Results. All p values reported are 2-tailed and a significance level of 5% was used for hypothesis testing.
RESULTS
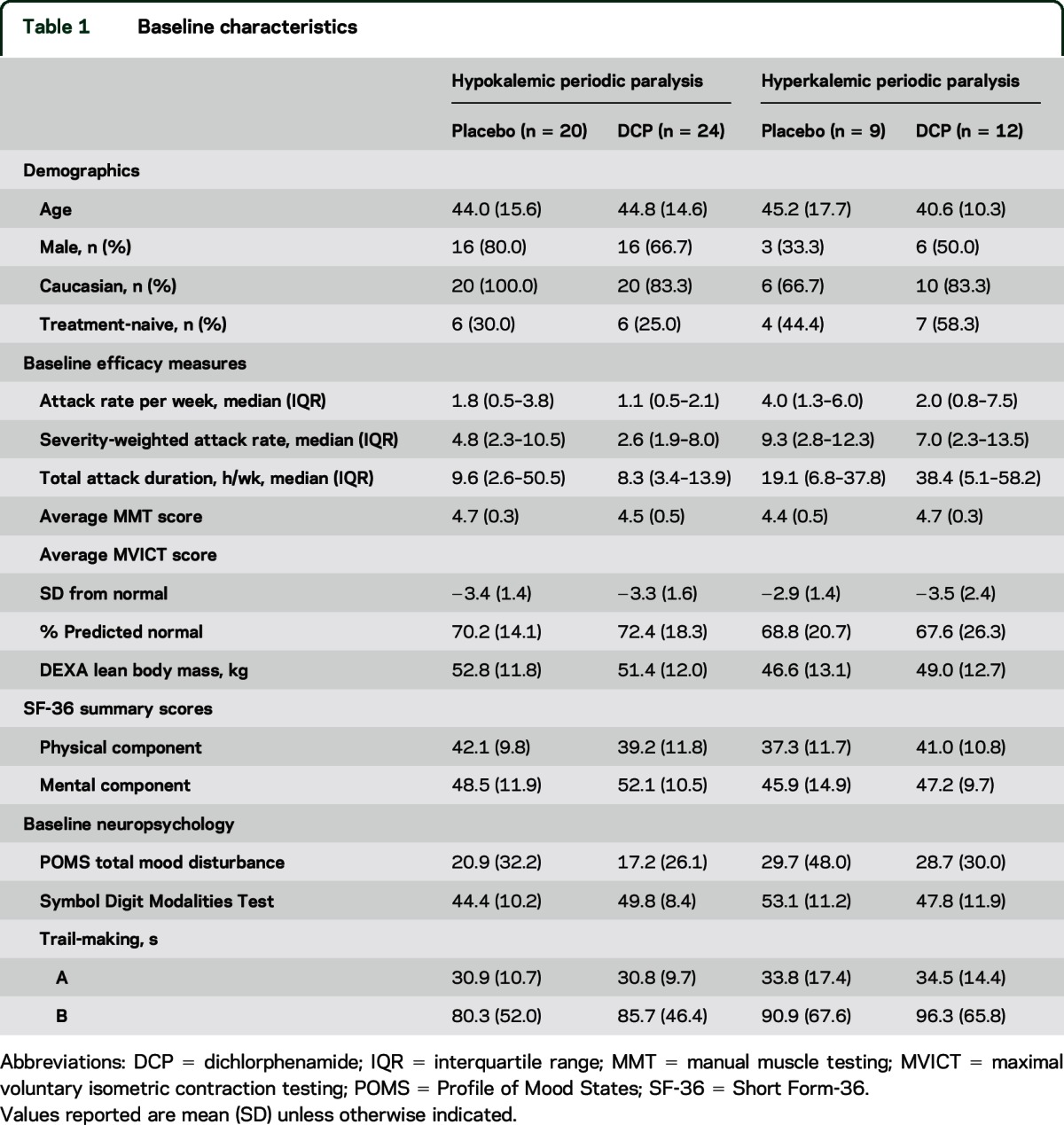
Participant numbers and flow are shown in the figure. Baseline characteristics are presented in table 1 and discussed in appendix e-1. The participant with negative genetics who was withdrawn from the HOP placebo group represents a misdiagnosis. This individual is from a family with 5 clinically and genetically affected members so the negative genetic test suggests a phenocopy rather than true HOP. Information regarding compliance with study medication, dosage, and assessment of the double-blind is presented in appendix e-3.
Figure. Flow diagram.

*Participants who reached the endpoint of acute worsening during the double-blind phase had an early week 9 visit and moved directly into the 52-week uncontrolled extension phase. **Data from one participant in the hyperkalemic periodic paralysis (PP) trial were included in the 9-week phase, but not in the 52-week uncontrolled extension phase as the participant had been randomized to acetazolamide in this phase (the trial was initially designed to include a comparison against acetazolamide; however, the acetazolamide arm had to be abandoned owing to poor recruitment). Forty-five participants were randomized to the hypokalemic PP (HOP) trial, but data from one participant assigned to receive dichlorphenamide (DCP) were not used because the participant did not meet diagnostic criteria for HOP (this participant is not shown in this figure). The decision to not include the participant was made prior to unblinding. One participant in the HOP trial assigned to receive placebo in the double-blind phase was mistakenly treated with placebo during the extension phase; data from this participant were not included in the analyses of data from the extension phase.
Table 1.
Baseline characteristics
Double-blind phase.
Efficacy.
HOP trial.
In the double-blind phase, median attack rate, severity-weighted attack rate, and attack duration were significantly lower with DCP than with placebo (table 2). Five participants from the placebo group but none in the DCP group reached the endpoint of acute worsening (p = 0.01, Fisher exact test).
Table 2.
Treatment effects on efficacy outcomes in the double-blind phase
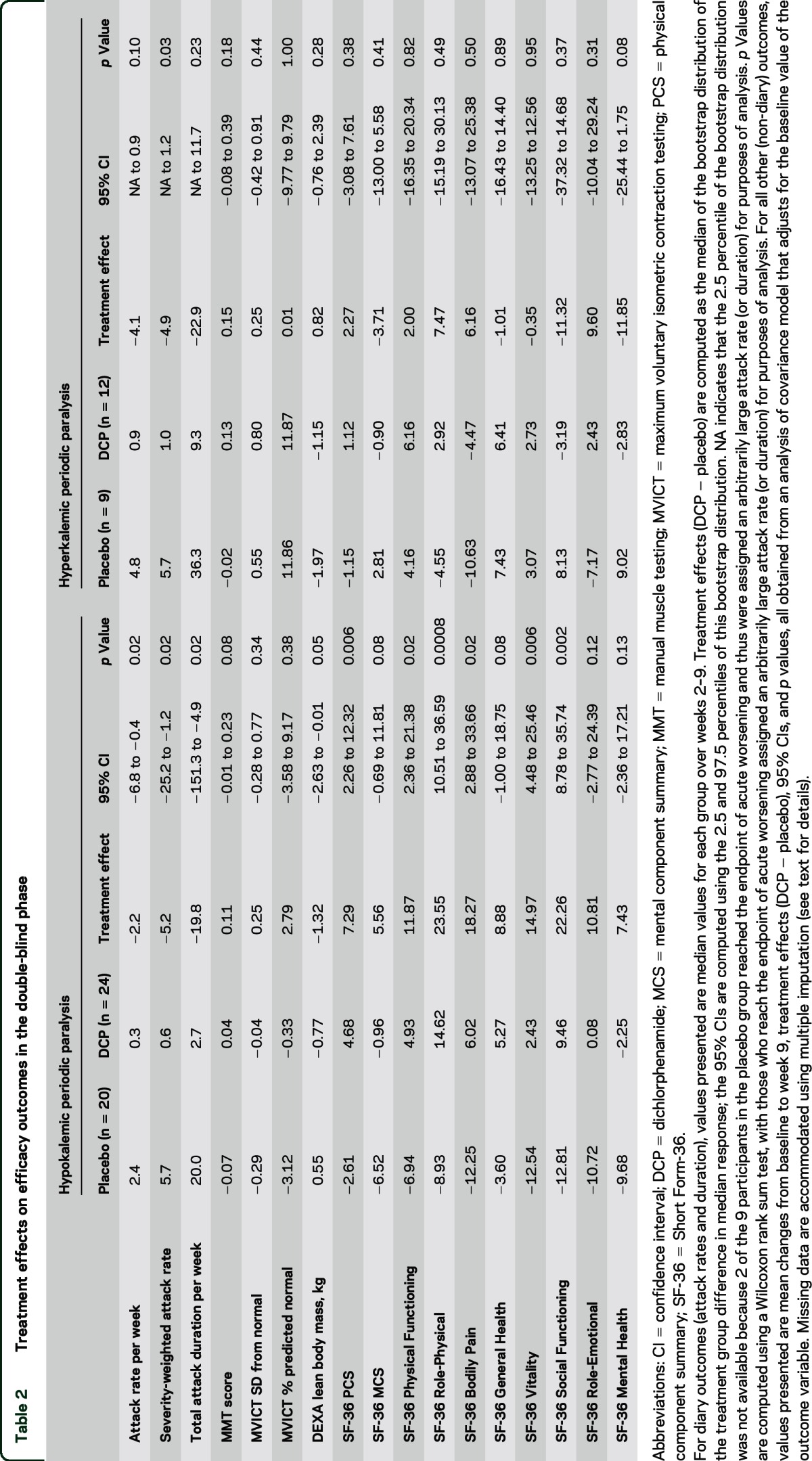
No treatment effects were apparent on muscle strength, but the DCP group lost lean body mass on average compared to a slight increase in the placebo group (table 2). The mean change in SF-36 v2 Physical Component Summary score indicated quality of life improvement with DCP compared to placebo. Beneficial effects of DCP were also apparent on SF-36 v2 subscale scores including Physical Function, Role-Physical, Bodily Pain, Vitality, and Social Functioning (table 2).
HYP trial.
The median severity-weighted attack rate was significantly lower in the DCP group than in the placebo group (table 2). Group differences in the median weekly attack rate and duration did not reach significance. No treatment effects were apparent on measures of muscle strength, lean body mass, and quality of life.
Safety.
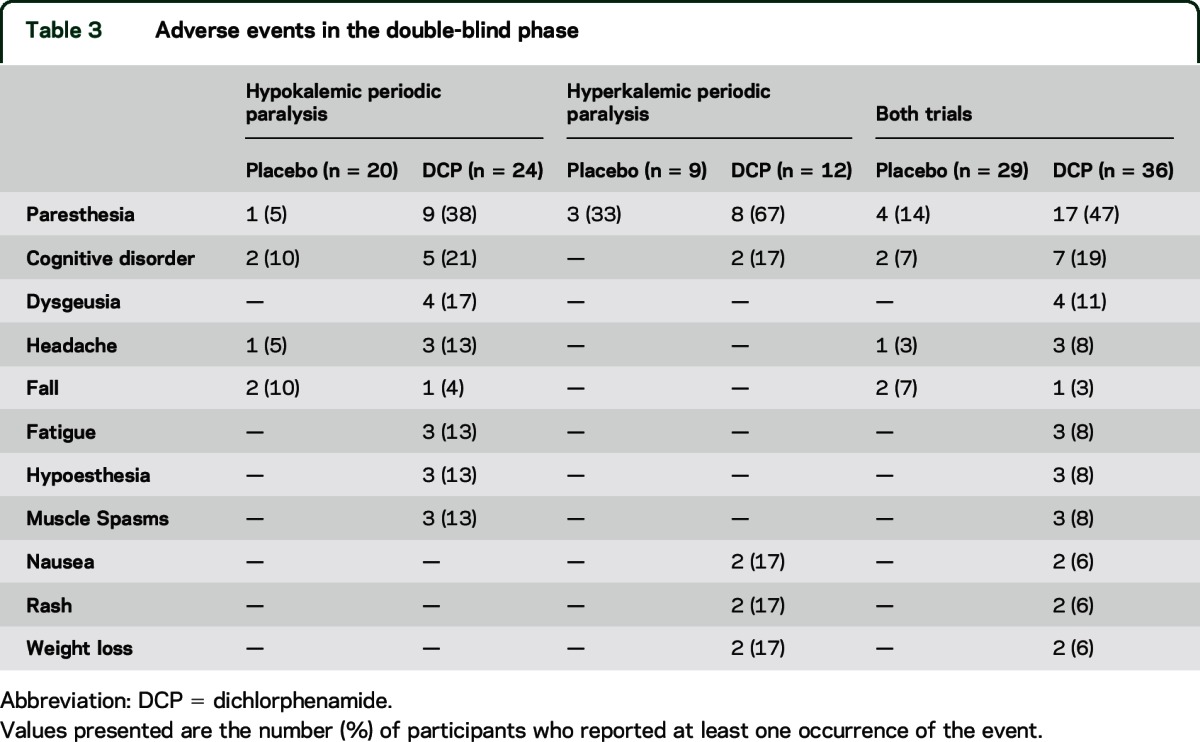
Table 3 shows the adverse events most commonly observed in the double-blind phase.
Table 3.
Adverse events in the double-blind phase
HOP trial.
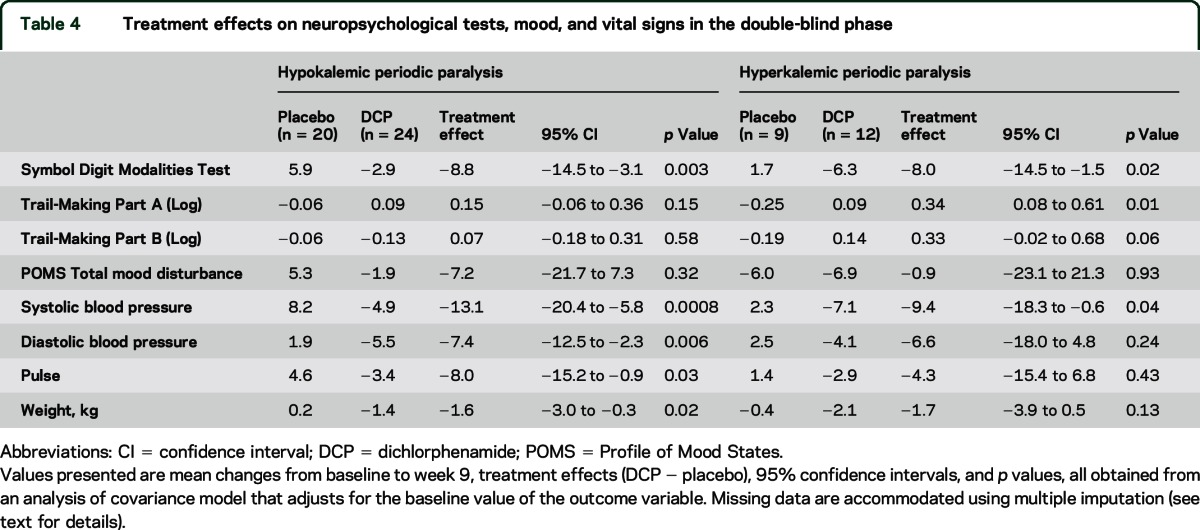
Paresthesia was more common with DCP than with placebo (38% vs 5%, p = 0.01). Cognitive disorder (21% vs 10%) and dysgeusia (17% vs 0%) were also more common with DCP, but these group differences were not statistically significant. One participant withdrew from the double-blind phase due to adverse events, primarily “mental fog.” One participant developed a rash on DCP. One participant had a serious adverse event (fractured humerus). DCP was associated with decreases in blood pressure, pulse, and weight, as well as worsening in performance on the Symbol Digit Modalities Test (table 4).
Table 4.
Treatment effects on neuropsychological tests, mood, and vital signs in the double-blind phase
HYP trial.
Eight of the 12 participants (67%) on DCP experienced paresthesias compared to 3 out of 9 (33%) on placebo (p = 0.20). Cognitive disorder occurred in 2 DCP-treated participants but in none on placebo. Two participants developed a rash on DCP. One of these was a serious adverse event and required emergency disclosure of treatment assignment; the rash resolved after discontinuation of DCP and 5 days of hospitalization. DCP was associated with a decrease in systolic blood pressure and with worse performance on the Symbol Digit Modalities Test and Trail-Making test (part A) compared with placebo (table 4).
Open-label uncontrolled extension phase.
Efficacy.
HOP trial.
At the end of the uncontrolled extension phase (weeks 54–61), the median (interquartile range [IQR]) attack rate was reduced to 0.2 (0.0–1.0) in participants who had been on placebo (n = 14) and 0.0 (0.0–0.1) in participants who had been on DCP (n = 17) in the double-blind phase. The median (IQR) severity-weighted attack rate was 0.4 (0.0–2.6) in participants who had been on placebo and 0.0 (0.0–0.3) in participants who had been on DCP. The weekly total attack duration was 2.1 (0.0–21.4) in participants who had been on placebo and 0.0 (0.0–0.3) in participants who had been on DCP. From weeks 9–61, there was a small mean increase in composite MMT score (n = 28; 0.07 points, 95% confidence interval [CI] 0.004–0.14, p = 0.04) that was not corroborated by quantitative muscle strength testing. There was a reduction in mean lean body mass at week 61 compared to week 9 (n = 24; −2.00 kg, 95% CI −3.31 to −0.69 kg, p = 0.004).
HYP trial.
At the end of the uncontrolled extension phase (weeks 54–61), the median (IQR) attack rate was reduced to 0.9 (0.0–1.4) in participants who had been on placebo (n = 7) and 0.3 (0.1–1.3) in participants who had been on DCP (n = 9) in the double-blind phase. The median (IQR) severity-weighted attack rate was 2.4 (0.0–3.9) in participants who had been on placebo and 0.9 (0.3–2.5) in participants who had been on DCP. The weekly total attack duration was 8.0 (0.0–107.6) in participants who had been on placebo and 9.9 (0.2–21.0) in participants who had been on DCP. No significant changes in muscle strength scores or muscle mass were observed during the extension phase.
Safety.
HOP trial.
Of the adverse events during the extension phase that were classified as possibly, probably, or definitely related to treatment (table e-3), the most common was paresthesia, which was assigned in 10 of 40 participants (25%) as probably related and in 2 of 40 (5%) as possibly related. The second most common was cognitive disorder. Of the 39 participants who underwent the final assessment for renal tract calculi, 7 developed new calculi during the trial and 2 showed an increase in size or number of preexisting calculi.
Ten HOP participants withdrew from the extension phase of the trial. One of these was discovered to have pancreatic cancer. Five experienced tiredness, fatigue, or mental slowness sufficient to impair performance at work. One, who had never tried carbonic anhydrase inhibitors, experienced worsening attacks and was discovered to have a sodium channel mutation (R222W mutation). The drug was stopped and the participant withdrawn from the trial. One participant withdrew due to painful kidney stones, and 2 others withdrew for intolerable but unspecified adverse events.
HYP trial.
Of the adverse events during the extension phase that were classified as possibly, probably, or definitely related to treatment (table e-3), the most common was paresthesia, which was assigned in 2 of 17 participants (11.8%) as possibly related, in 6 of 17 (35.3%) as probably related, and in 1 of 17 (5.9%) as definitely related. The only other adverse event that occurred in more than one participant was memory impairment, which was assigned in 1 of 17 (6%) as possibly related to treatment and in 1 of 17 (6%) as probably related to treatment. Of the 14 participants who underwent the final assessment for renal tract calculi, only 1 developed new calculi.
DISCUSSION
This parallel-group trial strengthens the evidence22 for the beneficial effect of DCP in HOP. In addition, it shows that DCP improves quality of life in HOP, and that its efficacy is maintained in the long term.
The majority of HOP participants on DCP in this trial experienced less than 1 attack per week compared to more than 2 attacks per week on placebo. A quarter of the HOP participants on placebo but none of those on DCP had intolerable attacks and had to be moved into the extension phase before completing the 9-week double-blind phase. Upon entering the uncontrolled phase, HOP participants previously treated with placebo appeared to benefit substantially from the introduction of DCP, and there was continued control of attacks in participants already taking DCP. This is an important observation suggesting persisting efficacy of DCP well beyond the time window of previous trials of this agent.
For the majority of participants, DCP was well-tolerated, and the benefits on quality of life appear to outweigh the side effects. Twelve of the 65 randomized participants (18%; 9 HOP and 3 HYP) withdrew from the trials due to adverse events while being treated with DCP, most during the long-term extension phase (8 HOP and 1 HYP). In both trials, the most common adverse event was paresthesia, but this was transitory and never the cause of withdrawal. Kidney stones, another potential problem with carbonic anhydrase inhibitors,36 were a cause of drop-out in only one participant. The second most common adverse event was slowed cognition; patients, particularly those in cognitively demanding professions, should be counseled about this side effect. There was an apparent decrease in muscle mass associated with DCP in the HOP trial; however, this may be due to an artifact (reduced muscle water content) of DCP-induced diuresis.
The main shortcomings of these trials were the lack of comparison against acetazolamide and the limited number of participants. The PP are exceedingly rare diseases (HOP prevalence 1/100,000; HYP prevalence 1/250,000).1–3 In the United States, inclusion of acetazolamide in the original design discouraged many potential participants. In the European Union, local ethics and research and development bureaucracy was a major barrier, with Italy and the United Kingdom entering the trial late, and France not at all.
Insufficient numbers of participants precluded analysis by genetic subgroup. Mutations were identified in 75% of HOP participants and in 67% of HYP participants either prior to or following enrollment. The most common were T704M in the HYP trial and R528H and R1239H in the HOP trial. Qualitative assessment of the effect of DCP treatment in subgroups defined by mutation type did not reveal any major subgroup differences in the effect of DCP on attacks other than worsening of attacks in the HOP patient with NaV1.4 pR222W. Genetic confirmation of included patients was lacking in more than 25%. This is in agreement with published data3 and supports the idea that the study reflects patients with PP in the general population.
DCP was available for off-label prescription for PP until 2002, when the drug stopped being marketed for glaucoma. As a consequence, DCP became unavailable for patients with PP in many countries until Food and Drug Administration approval in August 2015. Our findings support the idea that DCP would benefit many patients with PP.
Supplementary Material
ACKNOWLEDGMENT
The authors acknowledge the contributions of the Clinical Trials Coordination Center (CTCC) and Statistical Center at the University of Rochester, NY. Data management services provided by the University of Rochester Clinical Trials Coordination Center: Tori Ross, Cathy Covert, Alicia Brocht, Emily Flagg, Cornelia Kamp, Cindy Casaceli. Assistance with statistical analysis was provided by Arthur Watts. Data and Safety Monitoring Board Members (DSMB): Eric Hoffman, PhD, Robert Miller, MD, Jeffery Rosenfeld, PhD, MD, Donald Sanders, MD, David Schoenfeld, PhD, Scott Janis, PhD, NINDS DSMB liaison; Independent Medical Monitor: Seward Rutkove, MD, Beth Israel Deaconess Medical Center. Taro Pharmaceutical Industries supplied dichlorphenamide and matching placebo. The authors thank the study participants and their families for their commitment and participation in this international collaboration.
GLOSSARY
- CI
confidence interval
- DCP
dichlorphenamide
- HOP
hypokalemic periodic paralysis
- HYP
hyperkalemic periodic paralysis
- IQR
interquartile range
- MMT
manual muscle testing
- PP
periodic paralysis
- SF-36
Short Form-36
Footnotes
Editorial, page 1366
Supplemental data at Neurology.org
AUTHOR CONTRIBUTIONS
V.A. Sansone and J. Burge, coinvestigators, had full access to all of the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. Study concept and design: R.C. Griggs (coordinator). Acquisition of data: V.A. Sansone, J. Burge, R. Tawil (coinvestigators), J. Kissel (investigator), E. Ciafaloni (coinvestigator), P. Shieh (investigator), J.W. Ralph (investigator), A. Amato (investigator), S.C. Cannon (investigator), J. Trivedi (coinvestigator), R. Barohn (investigator), B. Crum (investigator), H. Mitsumoto (investigator). Analysis and interpretation of data: V.A. Sansone, J. Burge, M.P. McDermott (biostatistician), R. Barohn, R.C. Griggs, M.G. Hanna (investigator). Statistical analysis: M.P. McDermott. Clinical coordination and site monitoring: P.C. Smith. Administrative, technical, and material support: B. Herr. Critical revision of the manuscript for important intellectual content: R. Tawil, J. Kissel, E. Ciafaloni, P. Shieh, J.W. Ralph, A. Amato, S.C. Cannon, J. Trivedi, R. Barohn, B. Crum, H. Mitsumoto, A. Pestronk (investigator), G. Meola (investigator), R. Conwit. Clinical evaluator monitor: S. Pandya. Drafting of the manuscript: V.A. Sansone, J. Burge, M.P. McDermott, R.C. Griggs, M.G. Hanna. Study supervision: P.C. Smith, B. Herr, V.A. Sansone, R. Barohn, R. Tawil, J. Kissel, E. Ciafaloni, P. Shieh, J.W. Ralph, A. Amato, S.C. Cannon, J. Trivedi, M.G. Hanna, and R.C. Griggs.
STUDY FUNDING
This study was supported by NIH National Institute of Neurological Disorders and Stroke award R01 NS045686 (Robert C. Griggs, principal investigator); the Muscular Dystrophy Association award 93615; NIH NCATS awards UL1 TR000042, UL1 TR000040, UL1TR001070, UL1TR000001, UL1TR000124, UL1 TR000004, UL1 TR001105, UL1 TR000135, and NIH NCRR; NCATS awards UL1 RR024160 and UL1 RR024131; NIH NCRR awards C06 RR20092, M01-02635, and 1 UL1 RR025758; NIH NINDS awards U10NS077384, R37-AR42703, and RO1-AR063182 (S. Cannon, principal investigator); and MRC Centre Grant and UCL Biomedical Research Centre Grant (M. Hanna, principal investigator, Institute of Neurology, Queen Square, London). NINDS and MDA funding was provided for participant travel support.
DISCLOSURE
V. Sansone and J. Burge report no disclosures relevant to the manuscript. M. McDermott is a consultant for Cerebral Assessment Systems, Inc. and the New York State Department of Health and was a consultant for AstraZeneca and Asubio Pharmaceuticals. He serves or has served on the Data and Safety Monitoring Boards for studies sponsored by Novartis Pharmaceuticals Corporation, AstraZeneca, ATyr Pharma, Catabasis Pharmaceuticals, Inc., the ALS Association, and the Muscular Dystrophy Association. He receives research support from NIH, FDA, NYSTEM, the Spinal Muscular Atrophy Foundation, and Rhythm Pharmaceuticals, Inc. He serves on the Editorial Board of Movement Disorders and as an Editor for Chance. P. Smith reports no disclosures relevant to the manuscript. B. Herr is partially funded by NIH for clinical trial research. R. Tawil, S. Pandya, J. Kissel, E. Ciafaloni, P. Shieh, and J. Ralph report no disclosures relevant to the manuscript. A. Amato is Associate Editor of Neurology®. S. Cannon and J. Trivedi report no disclosures relevant to the manuscript. R. Barohn receives remuneration from Complete Publication Solutions, Indiana University, Columbia University, American Academy of Neurology, Texas Neurological Society, Clinical Connexion LLC, University of Texas at San Antonio, University of Colorado, NuFactor, Grifols Therapeutics, Walgreens Infusion Services, Impractice Resources LLC, S.A. Boney & Associates Ltd., Plan 365 Inc., and University of Washington. B. Crum and H. Mitsumoto report no disclosures relevant to the manuscript. A. Pestronk has received travel expenses & honoraria paid by the Myositis Association, receives revenue related to antibody patent licenses and speaker honoraria from Athena, owns stock in Johnson & Johnson, directs the Washington University Neuromuscular Clinical Laboratory, which performs antibody testing, and receives research support from the NIH, Cytokinetics, Biogen Idec, ISIS, Genzyme, GSK, Sanofi, and Ultragenyx. G. Meola, R. Conwit, and M. Hanna report no disclosures relevant to the manuscript. R. Griggs receives consulting fees from Marathon, PTC, VM, and Taro Pharmaceuticals. He is PI of the NINDS trial that funded this study. Drug and placebo for the study were provided by Taro Pharmaceuticals. Dr. Griggs receives an editorial stipend from the American Academy of Neurology and royalties from Elsevier and Oxford Publications for medical textbooks. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Venance SL, Cannon SC, Fialho D, et al. The primary periodic paralyses: diagnosis, pathogenesis and treatment. Brain 2006;129:8–17. [DOI] [PubMed] [Google Scholar]
- 2.Miller TM, Dias da Silva MR, Miller HA, et al. Correlating phenotype and genotype in the periodic paralyses. Neurology 2004;63:1647–1655. [DOI] [PubMed] [Google Scholar]
- 3.Horga A, Raja Rayan DL, Matthews E, et al. Prevalence study of genetically defined skeletal muscle channelopathies in England. Neurology 2013;80:1472–1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ptacek LJ, George AL, Jr, Griggs RC, et al. Identification of a mutation in the gene causing hyperkalemic periodic paralysis. Cell 1991;67:1021–1027. [DOI] [PubMed] [Google Scholar]
- 5.Fouad G, Dalakas M, Servidei S, et al. Genotype-phenotype correlations of DHP receptor alpha 1-subunit gene mutations causing hypokalemic periodic paralysis. Neuromuscul Disord 1997;7:33–38. [DOI] [PubMed] [Google Scholar]
- 6.Bulman DE, Scoggan KA, van Oene MD, et al. A novel sodium channel mutation in a family with hypokalemic periodic paralysis. Neurology 1999;53:1932–1936. [DOI] [PubMed] [Google Scholar]
- 7.Davies NP, Eunson LH, Samuel M, Hanna MG. Sodium channel gene mutations in hypokalemic periodic paralysis: an uncommon cause in the UK. Neurology 2001;57:1323–1325. [DOI] [PubMed] [Google Scholar]
- 8.Bendahhou S, Cummins TR, Griggs RC, Fu YH, Ptacek LJ. Sodium channel inactivation defects are associated with acetazolamide-exacerbated hypokalemic periodic paralysis. Ann Neurol 2001;50:417–420. [DOI] [PubMed] [Google Scholar]
- 9.Jurkat-Rott K, Mitrovic N, Hang C, et al. Voltage-sensor sodium channel mutations cause hypokalemic periodic paralysis type 2 by enhanced inactivation and reduced current. Proc Natl Acad Sci USA 2000;97:9549–9554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sternberg D, Maisonobe T, Jurkat-Rott K, et al. Hypokalaemic periodic paralysis type 2 caused by mutations at codon 672 in the muscle sodium channel gene SCN4A. Brain 2001;124:1091–1099. [DOI] [PubMed] [Google Scholar]
- 11.Ptacek LJ, Tawil R, Griggs RC, et al. Sodium channel mutations in acetazolamide-responsive myotonia congenita, paramyotonia congenita, and hyperkalemic periodic paralysis. Neurology 1994;44:1500–1503. [DOI] [PubMed] [Google Scholar]
- 12.Rojas CV, Wang JZ, Schwartz LS, Hoffman EP, Powell BR, Brown RH., Jr A Met-to-Val mutation in the skeletal muscle Na+ channel alpha-subunit in hyperkalemic periodic paralysis. Nature 1991;354:387–389. [DOI] [PubMed] [Google Scholar]
- 13.Sansone VA, Ricci C, Montanari M, Apolone G, Rose M, Meola G. Measuring quality of life impairment in skeletal muscle channelopathies. Eur J Neurol 2012;19:1470–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cavel-Greant D, Lehmann-Horn F, Jurkat-Rott K. The impact of permanent muscle weakness on quality of life in periodic paralysis: a survey of 66 patients. Acta Myol 2012;31:126–133. [PMC free article] [PubMed] [Google Scholar]
- 15.Griggs RC, Engel WK, Resnick JS. Acetazolamide treatment of hypokalemic periodic paralysis: prevention of attacks and improvement of persistent weakness. Ann Intern Med 1970;73:39–48. [DOI] [PubMed] [Google Scholar]
- 16.Resnick JS, Engel WK, Griggs RC, Stam AC. Acetazolamide prophylaxis in hypokalemic periodic paralysis. N Engl J Med 1968;278:582–586. [DOI] [PubMed] [Google Scholar]
- 17.Sansone V, Meola G, Links TP, Panzeri M, Rose MR. Treatment for periodic paralysis. Cochrane Database Syst Rev 2008:CD005045. [DOI] [PubMed] [Google Scholar]
- 18.Dejthevaporn C, Papsing C, Phakdeekitcharoen B, et al. Long-term effectiveness of acetazolamide on permanent weakness in hyperkalemic periodic paralysis. Neuromuscul Disord 2013;23:445–449. [DOI] [PubMed] [Google Scholar]
- 19.Viskoper RJ, Licht A, Fidel J, Chaco J. Acetazolamide treatment in hypokalemic periodic paralysis: a metabolic and electromyographic study. Am J Med Sci 1973;266:118–123. [PubMed] [Google Scholar]
- 20.Vroom FW, Jarrell MA, Maren TH. Acetazolamide treatment of hypokalemic periodic paralysis: probable mechanism of action. Arch Neurol 1975;32:385–392. [DOI] [PubMed] [Google Scholar]
- 21.Dalakas MC, Engel WK. Treatment of “permanent” muscle weakness in familial hypokalemic periodic paralysis. Muscle Nerve 1983;6:182–186. [DOI] [PubMed] [Google Scholar]
- 22.Tawil R, McDermott MP, Brown R, Jr, et al. Randomized trials of dichlorphenamide in the periodic paralyses: Working Group on Periodic Paralysis. Ann Neurol 2000;47:46–53. [PubMed] [Google Scholar]
- 23.Lehmann-Horn F, Rudel R, Ricker K. Non-dystrophic myotonias and periodic paralyses: a European Neuromuscular Center Workshop held 4–6 October 1992, Ulm, Germany. Neuromuscul Disord 1993;3:161–168. [DOI] [PubMed] [Google Scholar]
- 24.Gonzales-Jimenez E, Leopold IH. Effect of dichlorphenamide on the intraocular pressure of humans. AMA Arch Ophthalmol 1958;60:427–436. [DOI] [PubMed] [Google Scholar]
- 25.Piette JD. Interactive voice response systems in the diagnosis and management of chronic disease. Am J Manag Care 2000;6:817–827. [PubMed] [Google Scholar]
- 26.The FSH-DY Group. A prospective, quantitative study of the natural history of facioscapulohumeral muscular dystrophy (FSHD): implications for therapeutic trials. The FSH-DY Group. Neurology 1997;48:38–46. [DOI] [PubMed] [Google Scholar]
- 27.Kissel JT, McDermott MP, Mendell JR, et al. Randomized, double-blind, placebo-controlled trial of albuterol in facioscapulohumeral dystrophy. Neurology 2001;57:1434–1440. [DOI] [PubMed] [Google Scholar]
- 28.Skalsky AJ, Han JJ, Abresch RT, McDonald CM. Regional and whole-body dual-energy X-ray absorptiometry to guide treatment and monitor disease progression in neuromuscular disease. Phys Med Rehabil Clin N Am 2012;23:67–73. [DOI] [PubMed] [Google Scholar]
- 29.McHorney CA, Ware JE, Jr, Raczek AE. The MOS 36-Item Short-Form Health Survey (SF-36): II: psychometric and clinical tests of validity in measuring physical and mental health constructs. Med Care 1993;31:247–263. [DOI] [PubMed] [Google Scholar]
- 30.Ware JE, Jr, Sherbourne CD. The MOS 36-item short-form health survey (SF-36): I: conceptual framework and item selection. Med Care 1992;30:473–483. [PubMed] [Google Scholar]
- 31.Smith A. Symbol Digit Modalities Test: Manual. Los Angeles: Western Psychological Services; 1982. [Google Scholar]
- 32.Martin DT, Andersen MB, Gates W. Using Profile of Mood States (POMS) to monitor high-intensity training in cyclists: group versus case studies. Sport Psychologist 2000;14:138–156. [Google Scholar]
- 33.Reitan RM. Validity of the trail making test as an indicator of organic brain damage. Percept Mot Skills 1958;8:271–276. [Google Scholar]
- 34.Efron B, Tibshirani RJ. An Introduction to the Bootstrap. New York: Chapman and Hall; 1993. [Google Scholar]
- 35.Little R, Yau L. Intent-to-treat analysis for longitudinal studies with drop-outs. Biometrics 1996;52:1324–1333. [PubMed] [Google Scholar]
- 36.Tawil R, Moxley RT, III, Griggs RC. Acetazolamide-induced nephrolithiasis: implications for treatment of neuromuscular disorders. Neurology 1993;43:1105–1106. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.