

Abstract
Background
Glaucoma is a leading cause of irreversible blindness. Pathogenic variants in the Myocilin gene (MYOC) cause juvenile open angle glaucoma (JOAG) in 8–36 % of cases, and display an autosomal dominant inheritance with high penetrance. Molecular diagnosis is important for early identification as therapies are effective in minimizing vision loss and MYOC variants can be associated to severe glaucoma. MYOC variants are usually inherited, however a fifth of carriers do not report a family history. The occurrence of de novo MYOC variants is currently unknown.
Case presentation
In this study we investigated a 14 year old male Caucasian patient diagnosed with JOAG, and no family history of glaucoma. A novel probably deleterious MYOC:p.(Pro254Leu) variant was identified in the index case. This variant was not present in the parents or the siblings.
Conclusion
This is the second report of a de novo MYOC variant in a sporadic case of JOAG and it is currently unknown if this mechanism occurs more frequently. This finding emphasizes the importance of screening individuals with JOAG for MYOC mutations irrespective of a negative family history.
Keywords: De novo variant, Juvenile open angle glaucoma, Genetic testing, Glaucoma, Myocilin
Background
Glaucoma is one of the leading causes of irreversible blindness affecting over 60 million individuals worldwide [1]. Primary open angle glaucoma (POAG, MIM 137760) is the most common type and is characterized by changes in the optic nerve head with corresponding visual field loss in the presence of an open anterior chamber angle [2]. Juvenile open angle glaucoma (JOAG) refers to a younger age at diagnosis usually defined by an onset before 30–40 years old and associated with a more severe phenotype [3, 4]. Therapies for POAG aim at controlling intraocular pressure (IOP) and are usually effective in minimizing disease progression [5–7]. However, the early stages are often asymptomatic and half of the cases remain undiagnosed, making it challenging to implement treatment before irreversible vision loss occurs.
Pathogenic sequence variants in the MYOC gene (MIM 601652) have been first described in association with JOAG in 1997 [8]. Since then, they have been consistently identified in 2–4 % of adult-onset POAG [9, 10] and in 8–36 % of JOAG [9, 11, 12] among different ethnicities. MYOC comprises three exons which encode a protein consisting of two major domains, an N-terminal myosin-like domain and a C-terminal olfactomedin-like domain [13]. Most disease causing variants are clustered within exon 3 in the olfactomedin domain [14]. The pathophysiology is not fully understood but it has been postulated that the accumulation of misfolded proteins lead to endoplasmic reticulum stress, which compromises the trabecular meshwork cells regulating the IOP [15]. MYOC pathogenic variants are inherited in an autosomal dominant fashion and are often associated with high IOP, younger age at diagnosis and strong family history and can result in severe glaucoma and blindness if left untreated [9, 10, 16].
The majority of MYOC carriers report a family history of glaucoma, however sporadic cases still account for 20 % of mutation carriers [9]. It is currently unknown whether sporadic cases could be explained by de novo variants. In this study, we report a JOAG sporadic case with a novel de novo MYOC variant, and discuss the occurrence of de novo variants in MYOC associated glaucoma and the implications for the patient and his family.
Case presentation
Clinical presentation
The pedigree of the family is shown in Fig. 1a. The index case and his family were referred to the Australian and New Zealand Registry of Advanced Glaucoma (ANZRAG) through his treating ophthalmologist [17]. The proband was a 14 year old Caucasian male patient (II-1). He was referred to an ophthalmologist following a routine optometrist review for his glasses prescription which revealed high IOP. Following examination, he was diagnosed with JOAG. His IOP at presentation were 31 mmHg in the right eye and 32 mmHg in the left. His vertical cup-to-disc ratio was 0.85 right and 0.8 left, and he had central field loss involving fixation in the right eye (Humphrey Field Analyzer, Zeiss) (Fig. 2a). His visual acuity was 20/20 in both eyes. His IOP was initially under control with latanoprost and brimonidine/timolol. However he underwent bilateral trabeculectomies following his most recent IOP which were 40 mmHg. Optic nerve appearances and retinal nerve fiber layer loss (Spectralis®, Heidelberg Engineering) are depicted in Fig. 2b and c. His parents and two siblings had normal eye examinations.
Fig. 1.
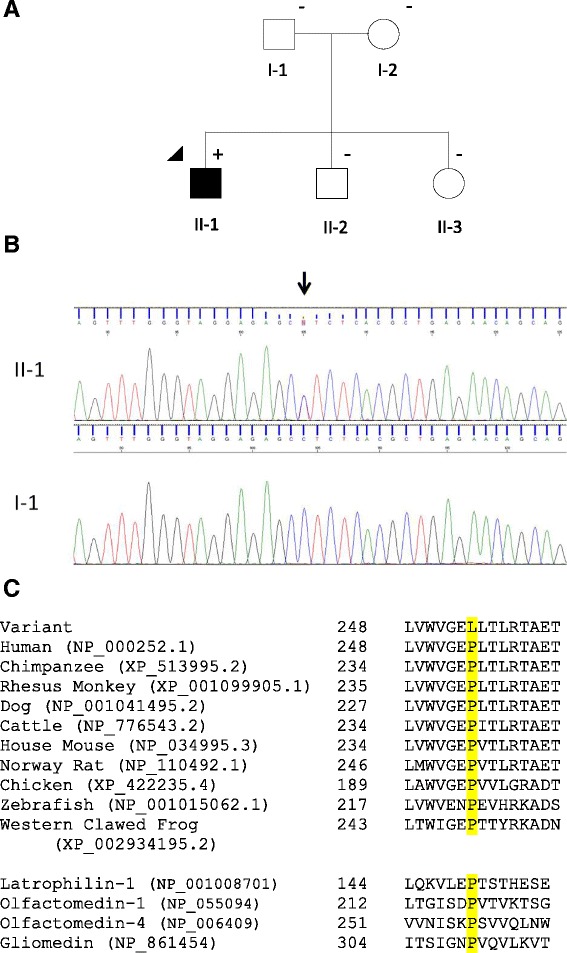
Pedigree and genetic analysis. a Pedigree of the family. Round symbols indicate female; square symbols, male; fully filled symbols, open angle glaucoma; unfilled symbols, unaffected; arrow, proband; plus/minus, presence/absence of the MYOC:p.(Pro254Leu) variant. b Chromatogram showing the presence of MYOC:c.761C > T, p.(Pro254Leu) sequence variant in individual II-1 at the top (affected) and its absence in individual I-1 at the bottom (unaffected). The black arrow marks the heterozygous variant. c. Alignment of MYOC protein sequences corresponding to residues 248 through 262 (NP_000252.1), against different species, and of different human olfactomedin proteins. The residue of interest, p.(Pro254Leu), is highlighted in yellow. Reference sequences IDs of the genes/species aligned are shown in brackets
Fig. 2.
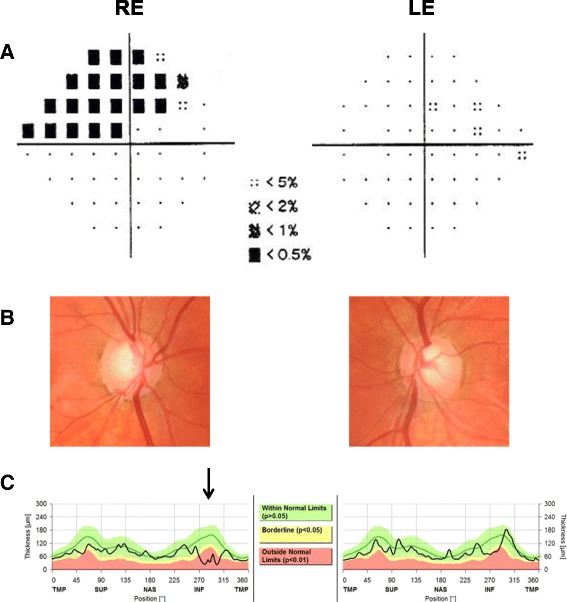
Clinical presentation of the index case. Glaucomatous defects in index case. a Visual field pattern deviation showing a superior arcuate defect involving fixation in the right eye (Humphrey Field analyser, Zeiss). b Optic discs photos showing a right infratemporal notch and disc haemorrhage. c Optical coherence tomography showing inferior retinal nerve fibre layer loss more prominent in the right eye than the left as shown by the black arrow (Spectralis®, Heidelberg Engineering). RE: right eye, LE: left eye, TMP: temporal, SUP: superior, NAS: nasal, INF: inferior
Genetic testing
Genetic testing was performed through the National Association of Testing Authorities (NATA) accredited laboratories of SA Pathology at the Flinders Medical Centre in Adelaide, Australia. The proband was sequenced for the 3 coding exons of the MYOC gene as previously described [9]. A heterozygous substitution of Thymine for Cytosine at nucleotide 761 of the MYOC exon 3 coding sequence was identified (MYOC:c.761C > T), encoding a missense substitution of Proline to Leucine at position 254 (p.(Pro254Leu)) (Fig. 1b). No other variants were identified in the MYOC gene of the proband. JOAG can also be associated with CYP1B1 variants [18]. The coding region of the CYP1B1 gene was sequenced to exclude other causative genes. No disease-causing variants were identified in CYP1B1.
The p.(Pro254Leu) variant is novel since it was absent from the MYOC Database (www.myocilin.com), NCBI dbSNP (www.ncbi.nlm.nih.gov/SNP/), and the Exome Aggregation Consortium (http://exac.broadinstitute.org/) which comprises exome sequence data spanning 60 706 unrelated individuals. A search of the scientific literature also failed to identify any reference to this variant. However, a recent study reported a MYOC variant at the same residue p.(Pro254Arg) in a patient with JOAG and his affected mother [19]. SIFT and Polyphen-2 both predicted this variant to be deleterious, with sequence alignment demonstrating this position to be highly conserved among vertebrates and other olfactomedin domain-containing proteins (Fig. 1c). MYOC is a well characterized gene and codon position 254 resides in the core hydrophobic β-sheet belt of the olfactomedin domain, which is important in protein-protein interactions and is sensitive to aggregation in the presence of substitutions [20]. The p.(Pro254Leu) variant is likely pathogenic based on bioinformatics prediction, invariant conservation of this residue, and characterization of the protein structure. MYOC disease-causing variants can be associated with severe glaucoma and blindness [9]. In the view of the genetic result and the patient’s most recent IOP, bilateral trabeculectomies were performed to better control his IOP and minimize damage on his optic nerves.
This variant was not detected in either parent of the index case (Fig. 1b). The marker profile comparison using the AmpFLSTR® Identifiler® PCR Amplification Kit confirmed a profile consistent with the proband being the biological child of the stated parents, indicating p.(Pro254Leu) has arisen de novo in the proband. A de novo MYOC pathogenic variant, p.(Val251Ala), has been previously reported once in a JOAG case [21]. Interestingly, this variant was located three amino acids from p.(Pro254Leu) which was identified in this study.
While the occurrence of de novo pathogenic variants in the genome vary considerably based on genomic location, they are estimated to be common and have been linked to many sporadic diseases [22]. Conditions with dominant inheritance and modest fitness effect are more commonly inherited than caused by de novo variants, and this is the situation for MYOC associated glaucoma which is usually inherited. For example, a founder effect with an origin prior to the European settlement of Australia has been suggested for the most common MYOC disease-causing variant, p.Gln368Ter, in some families [23]. However, we previously reported that 20 % of MYOC carriers do not report a family history of the disease [9]. Although this may be explained by individuals not being aware of a diagnosis in their families, or relatives being undiagnosed, it is possible that variants occur de novo in some families. MYOC variants are often identified in older individuals with parents usually unavailable for testing, making it difficult to evaluate whether variants are inherited or sporadic. This case is the second report of a de novo MYOC variant, emphasizing that a sporadic variant should be considered when evaluating the likelihood of MYOC variants in cases with no family history of JOAG or POAG.
De novo variants arise either in the germline or during embryogenesis. If present in the germline cells of one parent, they can represent a recurrence risk in siblings of the variant carrier. We have previously shown that MYOC genetic testing is important for early identification of at-risk individuals and appropriate interventions to minimize irreversible vision loss [9, 24]. To exclude a recurrence risk resulting from germline mosaicism, both siblings of the proband were subsequently tested for the MYOC variant. Our testing revealed that neither sibling carried the MYOC p.(Pro254Leu) variant, eliminating an inherited risk of developing MYOC associated glaucoma.
Conclusion
In conclusion, we report a novel de novo MYOC variant considered pathogenic in a patient with sporadic JOAG. This is the second report of a MYOC de novo variant, and it is currently unknown if this mechanism occurs more frequently. This case also highlights that MYOC testing should not be restricted to individuals with a positive family history of glaucoma.
Consent
Ethics approval was obtained from the Southern Adelaide and Flinders University Clinical Research Ethics Committee. The study conformed to the tenets of the Declaration of Helsinski and follows the National Health and Medical Research Council statement of ethical conduct in research involving humans. Written informed consents were obtained from each participating family member. A copy of the written consent is available for review by the Series Editor of this journal.
Acknowledgments
This project has been supported by The RANZCO Eye Foundation (www.eyefoundation.org.au, Sydney, Australia), Glaucoma Australia (www.glaucoma.org.au) and is currently funded by the Australian National Health and Medical Research Council (NHMRC) Centers of Research Excellence Grant 1023911 (2012–2016). Jamie E Craig is an NHMRC Practitioner Fellow and Kathryn P Burdon is supported by an NHMRC Research Fellowship.
Abbreviations
- ANZRAG
Australian and New Zealand Registry of Advanced Glaucoma
- IOP
intraocular pressure
- JOAG
juvenile open angle glaucoma
- MYOC
Myocilin
- NATA
National Association of Testing Authorities
- POAG
primary open angle glaucoma
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
ES participated in the design of the study, interpreted the data and drafted the manuscript. KPB participated in the design of the study and critically revised the manuscript. BR recruited the family in the study and critically revised the manuscript. AD carried out the molecular genetic studies, interpreted the results and critically revised the manuscript. JBR performed ophthalmological examination of the patients and critically revised the manuscript. JEC participated in the design of the study and critically revised the manuscript. All authors read and approved the final manuscript.
Contributor Information
Emmanuelle Souzeau, Phone: +61 8 8404 2035, Email: emmanuelle.souzeau@flinders.edu.au.
Kathryn P. Burdon, Email: kathryn.burdon@utas.edu.au
Bronwyn Ridge, Email: Bronwyn.usher@flinders.edu.au.
Andrew Dubowsky, Email: Andrew.dubowsky@health.sa.gov.au.
Jonathan B. Ruddle, Email: jruddle@unimelb.edu.au
Jamie E. Craig, Email: Jamie.craig@flinders.edu.au
References
- 1.Quigley HA, Broman AT. The number of people with glaucoma worldwide in 2010 and 2020. Br J Ophthalmol. 2006;90(3):262–267. doi: 10.1136/bjo.2005.081224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wolfs RC, Klaver CC, Ramrattan RS, van Duijn CM, Hofman A, de Jong PT. Genetic risk of primary open-angle glaucoma. Population-based familial aggregation study. Arch Ophthalmol. 1998;116(12):1640–1645. doi: 10.1001/archopht.116.12.1640. [DOI] [PubMed] [Google Scholar]
- 3.Wiggs JL, Damji KF, Haines JL, Pericak-Vance MA, Allingham RR. The distinction between juvenile and adult-onset primary open-angle glaucoma. Am J Hum Genet. 1996;58(1):243–244. [PMC free article] [PubMed] [Google Scholar]
- 4.Turalba AV, Chen TC. Clinical and genetic characteristics of primary juvenile-onset open-angle glaucoma (JOAG) Semin Ophthalmol. 2008;23(1):19–25. doi: 10.1080/08820530701745199. [DOI] [PubMed] [Google Scholar]
- 5.Heijl A, Leske MC, Bengtsson B, Hyman L, Hussein M. Reduction of intraocular pressure and glaucoma progression: results from the Early Manifest Glaucoma Trial. Arch Ophthalmol. 2002;120(10):1268–1279. doi: 10.1001/archopht.120.10.1268. [DOI] [PubMed] [Google Scholar]
- 6.The Advanced Glaucoma Intervention Study (AGIS): 7. The relationship between control of intraocular pressure and visual field deterioration.The AGIS Investigators. Am J Ophthalmol. 2000;130(4):429-440. [DOI] [PubMed]
- 7.The effectiveness of intraocular pressure reduction in the treatment of normal-tension glaucoma. Collaborative Normal-Tension Glaucoma Study Group. Am J Ophthalmol. 1998;126(4):498-505. [DOI] [PubMed]
- 8.Stone EM, Fingert JH, Alward WL, et al. Identification of a gene that causes primary open angle glaucoma. Science. 1997;275(5300):668–670. doi: 10.1126/science.275.5300.668. [DOI] [PubMed] [Google Scholar]
- 9.Souzeau E, Burdon KP, Dubowsky A, et al. Higher prevalence of myocilin mutations in advanced glaucoma in comparison with less advanced disease in an Australasian disease registry. Ophthalmology. 2013;120(6):1135–1143. doi: 10.1016/j.ophtha.2012.11.029. [DOI] [PubMed] [Google Scholar]
- 10.Fingert JH, Heon E, Liebmann JM, et al. Analysis of myocilin mutations in 1703 glaucoma patients from five different populations. Hum Mol Genet. 1999;8(5):899–905. doi: 10.1093/hmg/8.5.899. [DOI] [PubMed] [Google Scholar]
- 11.Shimizu S, Lichter PR, Johnson AT, et al. Age-dependent prevalence of mutations at the GLC1A locus in primary open-angle glaucoma. Am J Ophthalmol. 2000;130(2):165–177. doi: 10.1016/S0002-9394(00)00536-5. [DOI] [PubMed] [Google Scholar]
- 12.Wiggs JL, Allingham RR, Vollrath D, et al. Prevalence of mutations in TIGR/Myocilin in patients with adult and juvenile primary open-angle glaucoma. Am J Hum Genet. 1998;63(5):1549–1552. doi: 10.1086/302098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Green CM, Kearns LS, Wu J, et al. How significant is a family history of glaucoma? Experience from the Glaucoma Inheritance Study in Tasmania. Clin Experiment Ophthalmol. 2007;35(9):793–799. doi: 10.1111/j.1442-9071.2007.01612.x. [DOI] [PubMed] [Google Scholar]
- 14.Hewitt AW, Craig JE, Mackey DA. Complex genetics of complex traits: the case of primary open-angle glaucoma. Clin Experiment Ophthalmol. 2006;34(5):472–484. doi: 10.1111/j.1442-9071.2006.01268.x. [DOI] [PubMed] [Google Scholar]
- 15.Anholt RR, Carbone MA. A molecular mechanism for glaucoma: endoplasmic reticulum stress and the unfolded protein response. Trends Mol Med. 2013;19(10):586–593. doi: 10.1016/j.molmed.2013.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hewitt AW, Mackey DA, Craig JE. Myocilin allele-specific glaucoma phenotype database. Hum Mutat. 2008;29(2):207–211. doi: 10.1002/humu.20634. [DOI] [PubMed] [Google Scholar]
- 17.Souzeau E, Goldberg I, Healey PR, et al. Australian and New Zealand Registry of Advanced Glaucoma: methodology and recruitment. Clin Experiment Ophthalmol. 2012;40(6):569–575. doi: 10.1111/j.1442-9071.2011.02742.x. [DOI] [PubMed] [Google Scholar]
- 18.Souzeau E, Hayes M, Zhou T, et al. Occurrence of CYP1B1 Mutations in Juvenile Open-Angle Glaucoma With Advanced Visual Field Loss. JAMA Ophthalmol. 2015;133(7):826–833. doi: 10.1001/jamaophthalmol.2015.0980. [DOI] [PubMed] [Google Scholar]
- 19.Yang Y, Shi Y, Huang X, et al. Identification of a novel MYOC mutation in a Chinese family with primary open-angle glaucoma. Gene. 2015;571(2):188–193. doi: 10.1016/j.gene.2015.06.042. [DOI] [PubMed] [Google Scholar]
- 20.Donegan RK, Hill SE, Freeman DM, et al. Structural basis for misfolding in myocilin-associated glaucoma. Hum Mol Genet. 2015;24(8):2111–2124. doi: 10.1093/hmg/ddu730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kuchtey J, Chowdhury UR, Uptegraft CC, Fautsch MP, Kuchtey RW. A de novo MYOC mutation detected in juvenile open angle glaucoma associated with reduced myocilin protein in aqueous humor. Eur J Med Genet. 2013;56(6):292–296. doi: 10.1016/j.ejmg.2013.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Veltman JA, Brunner HG. De novo mutations in human genetic disease. Nat Rev Genet. 2012;13(8):565–575. doi: 10.1038/nrg3241. [DOI] [PubMed] [Google Scholar]
- 23.Baird PN, Craig JE, Richardson AJ, et al. Analysis of 15 primary open-angle glaucoma families from Australia identifies a founder effect for the Q368STOP mutation of myocilin. Hum Genet. 2003;112(2):110–116. doi: 10.1007/s00439-002-0865-5. [DOI] [PubMed] [Google Scholar]
- 24.Souzeau E, Glading J, Keane M, et al. Predictive genetic testing experience for myocilin primary open-angle glaucoma using the Australian and New Zealand Registry of Advanced Glaucoma. Genet Med. 2014;16(7):558–563. doi: 10.1038/gim.2013.196. [DOI] [PubMed] [Google Scholar]