

Abstract
Background
Acropora cervicornis, a threatened, keystone reef-building coral has undergone severe declines (>90 %) throughout the Caribbean. These declines could reduce genetic variation and thus hamper the species’ ability to adapt. Active restoration strategies are a common conservation approach to mitigate species' declines and require genetic data on surviving populations to efficiently respond to declines while maintaining the genetic diversity needed to adapt to changing conditions. To evaluate active restoration strategies for the staghorn coral, the genetic diversity of A. cervicornis within and among populations was assessed in 77 individuals collected from 68 locations along the Florida Reef Tract (FRT) and in the Dominican Republic.
Results
Genotyping by Sequencing (GBS) identified 4,764 single nucleotide polymorphisms (SNPs). Pairwise nucleotide differences (π) within a population are large (~37 %) and similar to π across all individuals. This high level of genetic diversity along the FRT is similar to the diversity within a small, isolated reef. Much of the genetic diversity (>90 %) exists within a population, yet GBS analysis shows significant variation along the FRT, including 300 SNPs with significant FST values and significant divergence relative to distance. There are also significant differences in SNP allele frequencies over small spatial scales, exemplified by the large FST values among corals collected within Miami-Dade county.
Conclusions
Large standing diversity was found within each population even after recent declines in abundance, including significant, potentially adaptive divergence over short distances. The data here inform conservation and management actions by uncovering population structure and high levels of diversity maintained within coral collections among sites previously shown to have little genetic divergence. More broadly, this approach demonstrates the power of GBS to resolve differences among individuals and identify subtle genetic structure, informing conservation goals with evolutionary implications.
Keywords: Genotyping by sequencing, Coral reefs, Population genomics, Restoration genetics, Florida reef tract
Background
Caribbean coral reef communities have lost nearly 80 % of coral cover since the early 1980s [1] due to multiple interacting factors such as overfishing, eutrophication, climate change, storm damage, grazer die-off, and disease [2, 3]. Amongst Caribbean corals, the genus Acropora has experienced particularly large declines over the last 30 years, with losses exceeding 95 % in some areas [4] and up to 90 % region-wide [5], a decline unparalleled in the fossil record [6]. The staghorn coral Acropora cervicornis is the fastest growing Caribbean coral [7] and is thought to reproduce largely by fragmentation [8]. Thus, active restoration propagates coral fragments in nurseries prior to outplanting to depleted reefs and is an effective coral restoration technique [9]. Active restoration is especially important for reef-building corals that provide the bulk of the three-dimensional complexity on reefs and support critical ecological functions for many other reef-associated species. Restoration efforts must consider how corals will respond to changing environments in today’s oceans, where organisms may rely on a variety of responses, including physiological acclimatization or evolutionary adaptation [10–13]. With the increase in the number and scope of reef and coral restoration programs around the world, detailed knowledge is needed concerning the role that genetic diversity can play in the survivorship or remaining coral populations and the re-establishment of depleted populations based on nursery propagation.
Recent bottlenecks in the abundance of A. cervicornis can negatively impact this species’ genetic diversity. Reduced populations may lose uniquely adapted individuals and rare alleles, each important for adaptation and potential recovery. Reduced genetic diversity also can compromise successful sexual reproduction by decreasing the potential of cross-fertilization (acroporids have low self-fertilization success [14]). Since A. cervicornis appears to undergo limited sexual recruitment, in part due to spatial gaps between existing populations, enhancing densities using nursery-reared coral colonies has become a focal point for increasing the chances of a successful mass-spawning event [9]. To repopulate reefs and increase population densities, greater knowledge on the genetic structure of A. cervicornis is needed so management strategies can be tailored to the appropriate areas and spatial scales. Specifically since the potential for evolutionary adaptation is related to genetic diversity and is critical for the survivorship of any species in today’s changing environments [10], evaluation of genetic variation is needed to help recognize potential evolutionary outcomes and management repercussions.
Genetic variation in A. cervicornis shows significant regional structure (e.g., between Florida and the Bahamas) for populations separated by more than 500 km in both nuclear and mitochondrial genes, suggesting restricted gene flow over large distances and potentially isolated populations [15]. Yet, within a smaller region like the Florida Reef Tract (FRT), analysis of A. cervicornis using microsatellites showed little population differentiation and no significant population structure [16]; these results were confirmed with mitochondrial control region sequences that showed no significant population structure for staghorn corals within the FRT based on data from 52 individuals [17]. Although most genetic diversity is related to large distances among regions, population structure was detected over smaller spatial scales (as small as 2 km) in 3 of the 20 areas examined [15]. This rare, fine scale structure was attributed to one-way introgression of A. palmata into A. cervicornis [15]. The finding of moderate genetic structure among regions in the Caribbean separated by more than 500 km suggests that these distant areas require independent conservation and management practices. Approaches that provide higher differentiation at smaller scales would highlight the need for more local management and restoration strategies. These approaches require new techniques to resolve any meaningful genetic variation.
Recently, the ability to quantify genetic variation has greatly improved with the use of next-generation sequence technologies [18]. It is now possible to genotype large numbers of individuals at thousands of loci using Genotyping by Sequencing (GBS) [19]. Here we use GBS to investigate the genetic diversity within and among A. cervicornis populations using individuals collected throughout the FRT) with individuals from the Dominican Republic used as an outgroup (Fig. 1; map was drawn using ESRI ArcMap 10.2). All individuals (except “Wild”) are harbored in a network of in-situ nurseries, which represent critical repositories of genetic data [20] and the sustainable source of coral tissue being used for active restoration of this threatened species.
Fig. 1.
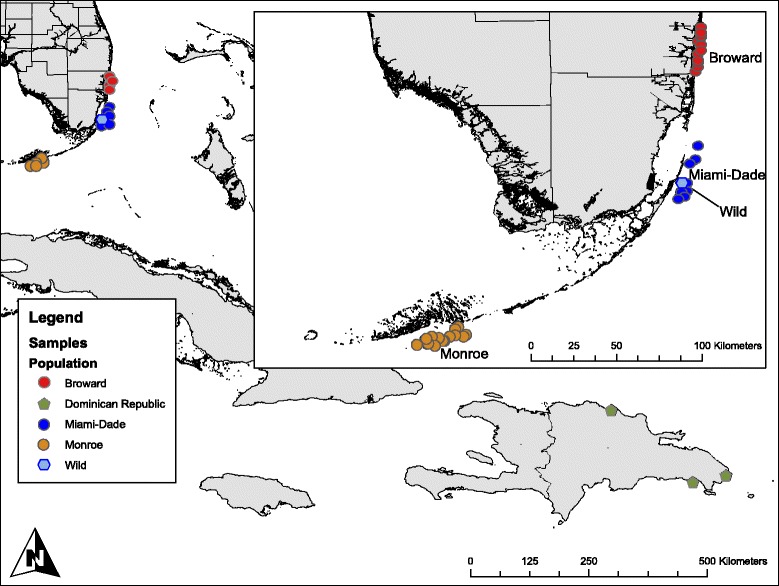
Locations of coral collection sites in Broward, Miami-Dade, and Monroe counties Florida and Dominican Republic. Wild was a single coral reef where all ten individuals were collected >50 m apart
Methods
Coral collections
A total of 77 samples were collected and analyzed: 66 individuals along the Florida Reef Tract and 11 individuals from around the Dominican Republic (Table 1). Dominican Republic corals were collected to serve as an out-group, enabling comparisons within the FRT to be considered relative to regional differences. Fifty-six FRT samples were received directly from a network of in situ nurseries harboring multiple corals originally collected from at least 500 m apart in separate reefs and tracked during propagation for active restoration efforts. Each of the FRT nursery corals was a unique genotype [21], which could potentially lead to an over-estimation of genetic diversity. An additional ten samples were collected from a single Miami-Dade county reef termed ‘Wild’ at 10-50 m intervals for a total of 66 colonies (Table 1) from 57 sites along the FRT. Nursery collections were used because they represent past wide sampling effort, making current collection efforts more efficient. Samples were considered to be A. cervicornis based on morphology and microsatellite tags, with the exception of the ‘Wild’ site, which was determined solely based on morphology. Other studies have discovered significant one-way introgression between A. palmata and A. cervicornis [15]; however the role of introgression is beyond the scope of the present study as the use of several thousand loci precludes the ability to compare individual genes to known A. palmata sequences. All corals were sampled by slicing a ~0.5 cm apical tip with a clean razor blade and placing the tip in 320uL of a chaotropic salt solution while in the field (4.5 M guanadinium thiocynate, 2 % N-lauroylsarcosine, 50 mM EDTA, 25 mM Tris–HCl pH 7.5, 0.2 % antifoam, 0.1 M β-mercaptoethanol) [22]. Samples were transported back to the University of Miami/RSMAS and stored at 4 °C prior to processing.
Table 1.
Sample collection locations for the 77 samples analyzed
| Region | Population | Samples Analyzed |
|---|---|---|
| Florida | Broward (NSU) | 23 |
| Miami-Dade (UM) | 10 | |
| Wild (Miami-Dade) | 10 | |
| Monroe (MOTE) | 23 | |
| Dom. Republic | Punta Cana (PCEF) | 11 |
| Total | 77 |
Parentheses indicate nursery management institution: Nova Southeastern University, University of Miami – Rosenstiel School of Marine and Atmospheric Science, Mote Marine Lab, Punta Cana Ecological Foundation
Collections were made under the following permits: Convention on International Trade in Endangered Species of Wild Fauna and Flora Permits 11US835702/9, United States Department of the Interior National Park Service Scientific Research and Collecting Permits BISC-2013-SCI-0010, NOAA Florida Keys National Marine Sanctuary Research Permit FKNMS-2011-150, and Florida Fish and Wildlife Conservation Commission Special Activity License SAL-13-1086-SCRP.
Genomic DNA and GBS
Genomic DNA was isolated using a silica column as described in [23]. Isolated DNA quality was assessed via gel electrophoresis and concentrations were quantified using Biotium AccuBlueTM High Sensitivity dsDNA Quantitative Solution according to manufacturer’s instructions. After quantification, 100 ng of DNA from each sample was dried down in a 96-well plate. Samples were then hydrated overnight with 5 ul of water before restriction enzyme digestion and further processing. GBS was preformed using the restriction enzyme ApeKI, unique barcoded adapters (0.4pmol/sample) and 50 ng of genomic DNA as described in [19]. A range of PCR cycles was used to optimize the amplification of restriction fragments using primers that anneal to the adapters. DNA from the 18-cycle run was pooled, and the GBS library was sequenced (Illumina Hi Seq 2500, 100 bp single end reads; Elim Biopharmaceuticals, Inc., Hayward, CA).
Data processing and analysis
Raw Illumina sequences were received from Elim Biopharmaceuticals and processed using the GBS analysis pipeline TASSEL 4.0 [24]. The TASSEL pipeline trims sequence reads to 64 bp and removes reads that do not contain a cut site and barcode (to remove barcode dimer sequences); reads that did not meet these requirements were discarded. Reads were then aligned to the A. digitifera genome (the only published acroporid genome) to prevent the inclusion of Symbiodinium DNA, which would be present in background levels in any coral sample. Aligning to a genome also enhances the identification of allelic SNPs at a specific locus because sequence reads that match two or more locations in the genome are discarded [24]. This selection of SNPs that align to one location is only possible with a reference genome. Alignment to the A. digitifera genome results in unique sequence tags, which are aligned, 64 bp sequence reads that have a unique genome location. The TASSEL pipeline with BWA and Bowtie was used to call SNPs with a minimum allele frequency of at least 5 % and a minimum of 5 reads per locus to reduce the impact of sequencing error (by ensuring minimum frequency and number of reads the likelihood of false polymorphism calls decreases). Only loci called by both alignment tools were used to produce a conservative selection of loci for analysis. Before downstream processing, SNPs were filtered using an iterative progression to select individuals with at least 70 % of the called loci and loci that were present in at least 90 % of samples for analyses. Arlequin v.3.11 [25] was used to test Hardy-Weinberg Equilibrium and calculate genetic diversity among coral collections by calculating the percentage of polymorphic SNPs, observed heterozygosity (HO), expected heterozygosity (HE) and fixation index (FST). Loci with significantly greater observed than expected heterozygosity (p < 0.01) were discarded from analysis, and loci with significant linkage-disequilibrium (D’ p-value < 0.01, or an r2 > 0.20) were identified using Tassel [24] with a 100 SNP sliding window (where the order of SNPs are defined by the A. digitifera genome) and removed.
For comparisons within populations, π, pairwise differences (different SNPs between samples/total SNPs*100), was calculated using the ‘ape’ package in R [26]. SNP π values were compared to more traditional measures of DNA sequence variation by correcting for the number of non-variable sites within each 64 bp sequence tag. Specifically, there are, on average, 1.4 SNP per 64 bp per sequence tag. Thus the average π * (1.4 SNP/64 bp sequence tag) provides an estimate of π when comparing DNA sequences with both invariable and polymorphic sites. For comparisons between populations, fixation index deviations from zero were tested by 10,000 permutations of alleles between individuals. To identify SNPs with FST outlier values (values larger than expected based on the observed data, [27]), the program LOSITAN [28] was used to generate 100,000 simulated SNPs, providing an expected neutral distribution of FST values and an estimate of P-values for each SNP. Structure [27] was used to identify the number of groups with similar allele frequencies (K). A model allowing admixture and correlated gene frequencies was used to carry out a total of 49 runs with seven independent runs for each K-value from 1–7. Ten-thousand permutations with 11,121 initial runs (burn-in) was used for each run. The K with the largest rate of change in the probability between groups was used to select the most parsimonious cluster [28]. RaXML was used to build a maximum likelihood tree [29] with 100 rapid bootstrap inferences. The best maximum likelihood tree (using a general time reversible model of nucleotide substitution and the Γ model of rate heterogeneity with ascertainment bias correction [30]) was selected and visualized using Dendroscope [31]. Discrimination analysis and comparisons of genetic and geographic distances (Mantel Test) were completed using ‘adegenet’ package in R [32, 33]. The Mantel Test was completed using a matrix of pair-wise differences in allele frequencies (Euclidean distances) and a matrix of geographic distances calculated from collection coordinates. Discrimination analyses (DAPC) was conducted in R using ‘adegenet’ [34]. DAPC was used in addition to Structure because it provides another metric of population differentiation, which does not assume un-relatedness, so potentially closely related individuals may be included.
Results and discussion
Coral samples
Table 1 lists the sample size for the 5 collections (Fig. 1). Along the Florida Reef Track, 56 individuals were sampled from separate reefs in Broward (Brwd), Miami-Dade (MD), and Monroe (Monr) Counties. We treated each county as a population. In addition to these three collections, 10 individuals were sampled from a single small reef in Miami-Dade (Wild), and 11 individuals were sampled from 11 locations in the Dominican Republic. Both Wild and the Dominican Republic were treated as separate populations. In previous studies [14, 15, 35] the FRT and DR would have been considered different regions, and the locations along the FRT were considered populations. In this study, the only difference is our treatment of the single reef with ten individuals sampled as a separate population (Wild). Although technically Wild is within Miami-Dade, the 10 individuals treated as a separate population provide insight not possible if merged with the 10 other individuals from separate reefs.
GBS samples and sequencing
Next-generation sequencing and identification of informative SNPs requires: i) filtering the data such that most SNPs occur in most individuals, ii) removing inappropriate SNPs that represent nucleotide differences between paralogs (different loci) versus polymorphisms between alleles and, iii) eliminating SNPs in linkage-disequilibrium [18, 19, 36–40]. The results of this filtering are shown and discussed below.
Sequencing data returned a total of 159,634,510 sequences and 91,643,894 (57.4 %) were retained because they contained both the barcode and cut site. Then, these retained sequences were aligned to the A. digitifera genome [41]. Alignment to the A. digitifera genome was used to remove Symbiodinium sequences. In total, 868,023 unique sequence tags (tags are aligned, 64 bp sequence reads that have a unique genome location) aligned to the published reference genome [41]. Sequence tags removed due to lack of alignment with the A. digitifera genome were not analyzed because of potential background symbiont or bacterial DNA contamination. After preliminary filtering, three individuals were discarded because they had less than 30 % of the sequence reads. It is possible that inefficiencies during library construction led to low numbers of reads in these individuals. The remaining 77 individuals had 400,000 to 2,300,000 reads per individual.
Short 64 bp sequences were aligned using two alignment tools, Bowtie [42] and BWA [43], to identify Single Nucleotide Polymorphisms (SNPs). Bowtie identified 306,643 SNPs and BWA identified 178,644 SNPs, of which 113,838 SNPs were called by both alignment tools. These 113,838 SNPs were iteratively filtered to meet two criteria: individuals with 70 % of all called SNPs and loci that were present in 90 % of individuals. These criteria produced a total of 5,230 SNPs. These differences in alignment tools affect allele frequency, and for these data, there was a substantial difference in the observed heterozygosity: BWA identified many more loci with large heterozygosity values. Only approximately 50 % of reads shared alignments with these two tools, which has been observed in other studies [44–46]. Taking a conservative approach to avoid errors due to alignment tools, only the 5,230 SNPs identified by both alignment tools with similar or identical allele frequencies were considered. An additional 466 SNPs were removed because of excessive observed heterozygosity or linkage to another SNP, leaving 4,764 SNPs. SNPs that had observed heterozygosity significantly greater than expected (i.e., not in Hardy-Weinberg equilibrium) were removed because they most likely represent alignments between different loci and not real polymorphisms at the same locus [47]. Eliminating these SNPs with excessive observed heterozygosity was done to reduce the technical error caused by mis-alignment. However this also could eliminate loci strongly affected by balancing selection. Thus, we err on the side of reduced technical error with the potential loss of SNPs affected by balancing selection. We also removed one of each pair of SNPs with significant linkage disequilibrium (D’ p-value < 0.01, or an r2 > 0.20) [48]. Removing SNPs in linkage-disequilibrium (LD) should not bias the measure of variance unless SNPs in LD have significantly greater or less variation. Strong LD is associated with background selection or directional selection, both of which would reduce variation. Thus, our SNP measurements may be conservative estimates of the variation present. After removing loci not in HWE and loci with significant linkage disequilibrium, 4,764 SNPs remained. These 4,764 SNPs were used for all analyses.
There is an additional concern that Symbiodinium may have conserved genes that could align to the A. digitifera genome. This would seem unlikely because if there were multiple types of Symbiodinium, these should have excessive observed heterozygosity and would be filtered out. Yet, to examine this possibility we used BLAST-N with all sequence tags that define the 4,764 SNP against the Symbiodinium minutum Mf 1.05b.01 (taxid:1280413) genome. Only one tag had an e-value < 10−5, and this sequence only matched 83 % of the genome for 49 of the 64 bp (63 % similarity for all 64 bp). Eight more tags had e-values < 1 %, but none of these matched more than half of the sequence tag. Thus, it seems unlikely that the alignments contain Symbiodinium sequences.
Genetic diversity within populations
The genetic diversity within a population can be represented by π, the nucleotide diversity or number of nucleotide differences among pairs of samples divided by the number of SNPs. Here, these data are represented as pairwise differences (π,) between samples. The π within each FRT population (Broward, Miami-Dade, Wild, and Monroe) averages 37 % with a range of 23.9–44.0 % (Table 2). This indicates that among all SNPs, an average of 37 % are different between any two individuals within a single population. The differences within a population are similar to the overall value when all 5 populations (including DR samples) are used. While only the ‘Wild’ site represented multiple individual colonies sampled from a single reef, π in Wild remained high (average 37 %, range 24.9 to 43.8 %) and in the range of π in the other populations (23.5 % to 44.0 %). Among the three FRT transects Broward and Monroe had an average π of 38.3 % and 39.5 %, respectively, but narrower range of π (35 % to 41.2 %, 36.2 to 43.8 %) than Miami-Dade. This was somewhat surprising because A. cervicornis in Broward County is growing at the edge of its spatial range, so high diversity may be unexpected. Importantly, within each population π measurements are comparable to π across populations (Table 3), indicating that genetic diversity starts at the local level, including single reefs. The measures presented here represent large enough differences between individuals that each individual collected represents a unique individual (minimum π: 23.5 %), including those from the single reef (‘Wild’).
Table 2.
Nucleotide diversity or pairwise differences (π) among individuals within a population
| π, Pairwise differences | |||
|---|---|---|---|
| Avg. | Min. | Max. | |
| All Pops | 0.392 | 0.235 | 0.440 |
| Broward | 0.383 | 0.350 | 0.412 |
| Miami-Dade | 0.383 | 0.235 | 0.416 |
| Monroe | 0.395 | 0.362 | 0.435 |
| Wild | 0.370 | 0.249 | 0.438 |
| Dom. Republic | 0.363 | 0.267 | 0.440 |
Average among all individuals and minimum and maximum differences for a pair of individuals within a population
Table 3.
Pairwise differences (π) between populations
| Broward | Miami-Dade | Monroe | Wild | Dom. Republic | |
|---|---|---|---|---|---|
| Broward | 0.383 | ||||
| Miami-Dade | 0.389 | 0.383 | |||
| Monroe | 0.392 | 0.393 | 0.395 | ||
| Wild | 0.392 | 0.395 | 0.398 | 0.370 | |
| Dom. Republic | 0.391 | 0.398 | 0.402 | 0.400 | 0.363 |
Average π among every individual from each population compared
Traditionally, A. cervicornis is thought to rely primarily on asexual propagation, so single reefs have been believed to be monotypic or have few genets [7, 49]. The observed level of diversity between individuals is also unexpected because previous genetic analyses of A. palmata indicate a high ramet/genet ratio where many reefs may be populated by one or a few genets [50]. Even though the dataset presented here only examines polymorphic sites, the similar levels of genetic variation within and among populations indicates that much diversity occurs among individuals. Additional evidence suggests that genetic diversity is occurring over even smaller spatial scales, where multiple colonies in close proximity (<5 m apart) show similar π and are likely unique genets (Drury, unpublished data). This level of genetic diversity is unlikely to be an artifact. All SNPs were called in 90 % of all individuals with a minimum allele frequency of 5 %. There was an average of 411 reads per SNP and 69 reads per SNP for the minor allele. Furthermore, 90 % of the minor alleles had more than 23 reads while the minimum number of reads for all SNPs was 8. Thus it seems unlikely that the detected SNPs are sequencing errors. Importantly, these measures are conservative because the sample size per population (n = 10 to 23) may overlook minor alleles and 5 % minimum allele frequency will underestimate the genetic divergence. Additionally, removing SNPs in LD and with excessive heterozygosity could reduce these estimates of genetic variation. Thus, our data are indicative of large standing genetic variation within populations, but they may be under-estimates.
Measures of π within A. cervicornis populations are similar to values in other GBS studies on stickleback and natural populations of Saccharomyces cerevisiae [51, 52]. Yet to compare our GBS measures directly to more traditional π values for complete gene sequences requires the frequency of SNPs and invariable sites within each tag. This value is estimated by dividing number of SNPs on a given tag by the length of reads; for all pooled polymorphic tags investigated here, there were an average of 1.45 SNPS per 64 bp equating to an adjusted π value of 0.9 %. This adjusted π value is similar to nucleotide diversity across Caribbean populations for the three nuclear genes in A. cervicornis [15], but nearly four times as large as π among Florida populations, estimated as 0.002 using the mitochondrial control region [17]. When compared across a wide array of taxa, π = 0.9 % is a substantial level of genetic variation relative to most animals [53].
The 57 individuals from the Florida nursery collections (Broward, Miami-Dade and Monroe, but not Wild, Table 1) were identified as unique genotypes by microsatellites within each population (Baums, unpublished data). Among these individuals there was an average π of 39.1 % (23.5 % to 43.5 %). Each of these samples was a different individual with the minimum π of 23.5 % between individuals (i.e., two individual were different at 23.5 % of SNPs). This non-random collection could inflate π within each of these three populations but should not inflate the variation among populations. Yet, π among these 57 individuals is similar to that of the single reef collection (Wild) and is similar to π among all populations (Table 2). Thus, within each of the three Florida transects, which were separate nursery collections, π was similar to that of a single reef and did not exceed π between individuals in different transects. This suggests that π is not greatly inflated by the selection of individuals with different microsatellite genotype. Although there may be a slight bias in π for the three FRT nurseries, our data indicate large, standing genetic variation among FRT corals and genetic variation within A. cervicornis populations along the FRT similar to the variation across all populations. The amount of variation discovered within and among A. cervicornis populations is similar to the genetic variation found in large outbred populations distributed over large geographic ranges [51–53]. Despite the dramatic decline in the census population, this decline did not result in substantial loss of genetic diversity, suggesting that remaining corals are i) old individuals sampled from a population with large diversity and a very large effective population size, ii) new recruits from a wide variety of parental inputs, or iii) are affected by non-neutral processes enhancing genetic diversity (e.g., divergent selection on different habitats/reefs).
Genetic diversity among populations
Substantial genetic diversity within populations is accompanied by significant divergence among populations and is illustrated by the relationships among individuals seen in the maximum-likelihood tree and Structure Plot (Fig. 2a, b). The maximum likelihood tree using 4,764 SNPs has six branches with over 70 % bootstrap support, including all individuals from the Dominican Republic in a single cluster with 100 % support on internal branches. Other branches with 100 % bootstrap support include pairs of individuals from Miami-Dade and two sets of colonies from the wild reef (with two and five individuals respectively) (Fig. 2a). Structure analysis suggests 3 or 4 groups of individuals have similar minimum mean likelihood Fig. 2b). Each column (Fig. 2b) represents an individual and the summary of allele frequencies for that individual. Based on rate-of-change likelihood, separation into 3 groups is the most parsimonious explanation for the data (Fig. 2c), with Broward, Miami-Dade, and Monroe populations sharing common allele frequencies while each is divergent from the Wild population and the Dominican Republic samples. Regional differences, i.e., Florida vs. Dominican Republic, agree with previous reports of Caribbean-scale population structure in A. cervicornis [16, 17]. Collections from the ‘Wild’ reef appear to have 50 % of individuals with divergent ancestry from other FRT (Fig. 2b) and share a well supported clade in the maximum-likelihood tree (Fig. 2a). The phylogeny and structure plots indicate differences within the FRT as a whole, but do not readily resolve differences within the three sub-regions.
Fig. 2.
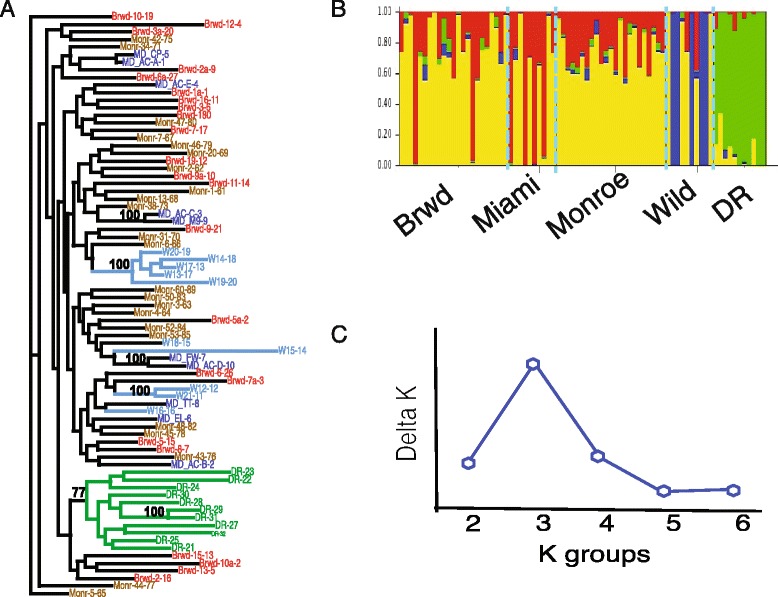
Maximum likelihood tree and Structure showing shared relationships among individuals based on 4.7 K SNP. a Maximum-likelihood tree with 100 bootstraps; only branches with >70 % support are enumerated. Taxa are color-coded (Broward: red, Miami-Dade: dark blue, Monroe: brown, Wild: light-blue and Dominican Republic: green). Clades for Wild have blue branches and the clade for Dominican Republic has green branches (b) Structure plot for five populations. Each individual is labeled with the color related to the predicted population. c Delta K; rate of change in the log probability of data between successive K values [28]
To further resolve differences among populations, we applied four different analyses: 1) a hierarchical AMOVA [54], 2) analysis of FST values across all loci, for individual loci and for outlier FST values [55], 3) a Mantel test and 4) discrimination analysis of principal components (DAPC).
An AMOVA of all 4,764 loci for two groups (Florida and DR) with four populations within Florida (Broward, Miami-Dade, Monroe and Wild) shows significant variation (p < 0.001) among groups, among populations within Florida and within populations (Table 4). The significant difference among groups confirms the regional differences between the Dominican Republic and Florida seen in the Structure analyses and maximum likelihood tree (Fig. 2). Greater than 90 % of the observed variance is within populations (Table 4), but there are also significant differences among the FRT nursery populations accounting for approximately 2 % of the variation. The genome-wide FST values (Table 5) are significant for all but Monroe and Broward pairwise comparisons and represent the first genetic structure formally resolved in Florida A. cervicornis. These FST values are not large (range: 0.016 to 0.092), so they may be more reflective of the statistical power of using many loci [56] and less ecologically relevant; nevertheless they represent a novel ability to distinguish between sub-regions of the Florida Reef Tract, which has been viewed as relatively homogenous in previous investigations [16, 17]. Interestingly, among the three Florida transects, Miami-Dade exhibits significant divergence in comparison to Monroe (~180 km) and Broward (~60 km) samples, but the latter transects are not significantly different despite larger spatial separation (~250 km).
Table 4.
AMOVA design and results based on 4,764 SNPs
| Source of variation | Sum of Squares | Variance components | Percent variation |
|---|---|---|---|
| Among Groups | 1916.1 | 37.685 | 4.35 |
| Among Pops within Groups | 3101.2 | 627.813 | 2.16 |
| Within Populations | 85576.0 | 664.813 | 93.49 |
| Total | 90593.3 | 664.813 |
Groups are FL and DR, with four populations within FL (Broward, Miami-Dade, Monroe and Wild)
Table 5.
FST and P-values
| Broward | Miami-Dade | Monroe | Wild | Dom. Republic | |
|---|---|---|---|---|---|
| Broward | ------ | 0.009 | 0.839 | 0.000 | 0.000 |
| Miami-Dade | 0.016 | ------ | 0.000 | 0.009 | 0.000 |
| Monroe | 0.003 | 0.012 | ------ | 0.000 | 0.000 |
| Wild | 0.041 | 0.045 | 0.034 | ------ | 0.000 |
| Dom. Republic | 0.056 | 0.069 | 0.050 | 0.092 | ------ |
FST values are based on all 4.7 K SNP. Below the diagonal are the FST values. Above the diagonal are the p-values for the specific comparisons; all comparisons are significant except Broward vs. Monroe
Among the three Florida transects (i.e., Broward, Miami-Dade and Monroe, without Wild), there are 300 SNPs with significant FST values (p < 0.01; Fig. 3, Table 6). Most SNPs with significant FST values among FRT transects are not physically close to each other (average distance is 88,235 bp), suggesting that most SNPs are evolving independently and the differences among populations are not due to one or a few linked loci. The average distance to the next closest SNP is > 25 kb (although many non-significant SNPs are often on the same 64 bp sequence tag). These close, non-significant SNPs indicate lack of linkage or selective sweeps and high, long-term standing genetic variation. The individuals sampled from the Miami-Dade nursery should have captured much of the local genetic variation as they were initially collected from a large area (~35 km span), collections were separated by at least 500 m, and all represent unique genets [57].
Fig. 3.
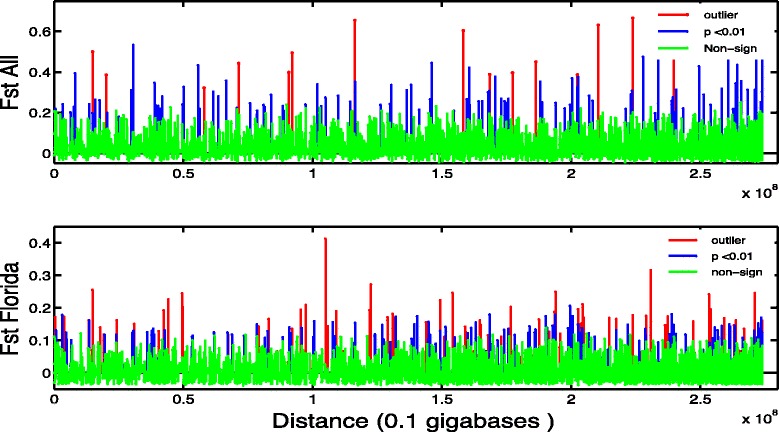
Locus specific genetic distance (FST value) by position relative to the A. digitifera genome. Significant FST values that are blue, and the subset that are outliers (potentially adaptive) are in red. SNPs with non-significant FST values are green. Distance along the X-axis is the sum of distances among the 4,765 scaffolds. a FST values for all five populations. b FST values for only the three Florida transects (i.e., Broward, Miami-Dade and Monroe, without Wild)
Table 6.
FST values and nucleotide distance for all five populations and three Florida transects
| FST all SNP | Significant FST | Outlier FST | Next non-significant FST | Distance (bp) among Significant FST SNP | Distance (bp) among Outlier FST SNP | Distance (bp) between significant and non-significant SNP | |
|---|---|---|---|---|---|---|---|
| All 5 populations | |||||||
| Counta | 4,762 | 141 | 17 | 108 | 23 | 0 | 108 |
| Average | 0.040 | 0.3018 | 0.4454 | 0.0989 | 75,876 | N/A | 31,576 |
| 95 % CIb | 0.0383, 0.0424 | 0.2835, 0.3200 | 0.3607, 0.5300 | 0.0780, 0.1198 | 2,338, 14,9414 | N/A | 22,578, 40,573 |
| Readc (range) | 411 (125:4,142) | 377 (198:1,097) | 367 (256:679) | 405 (251:841) | |||
| 3 Florida Populations (Broward, Miami-Dade, and Monroe) | |||||||
| Counta | 4,753 | 300 | 150 | 207 | 73 | 28 | 207 |
| Average | 0.012 | 0.1163 | 0.1165 | 0.0451 | 88,235 | 46,304 | 27,698 |
| 95 % CIb | 0.0099, 0.014 | 0.1097, 0.1229 | 0.1046, 0.1283 | 0.0363, 0.0539 | 55,264, 12,1207 | 4,627, 97,235 | 20,523, 34,872 |
| Readsc (range) | 411 (125:4,142) | 370 (125:967) | 377 (167:967) | 402 (208:947) | |||
aCounts refer to the number of polymorphic SNPs used in the analyses or (for distance) the pair of SNPs that shared the same scaffold. bCI is the 95 % confidence interval. cReads are the average number of 64 bp reads for each SNP. “Significant” and “non-siginficant” refers to SNPs with statistically significant FST values
To further parse differences between sub-regions, a Mantel Test was used to calculate the correlation between genetic and geographic distance. Here, 377 SNPs with significant FST values (from locus-specific FST values, Fig. 3) were compared to geographic coordinates from the original collection sites. Among all populations, there is a significant (p < 0.001) correlation between genetic variation and spatial distribution; this trend explains approximately 38 % of the genetic variation (R2 = 0.378) and is driven mainly by the differences between Florida and the Dominican Republic, supporting the regional structure previously reported [15]. When examining only the corals from the three Florida transects, the relationship is significant but explains much less variation (R2 = 0.104). Importantly, the genetic variation explained by geographic separation increases if either Broward or Monroe (the northernmost and southernmost Florida sub-regions, respectively) is excluded, because these two most distant sub-regions of the FRT are more similar to each other than either is to the spatially intermediate Miami-Dade corals. The results of the Mantel test support the pairwise FST values, which indicate significant structure between Miami-Dade and Monroe/Broward, but little genetic divergence between Monroe and Broward.
Using DAPC with 4,764 SNPs shows that the first discriminant function separates the Dominican Republic from the Florida corals, while the second discriminant function separates the four Florida populations (Fig. 4a). When only the four Florida populations are analyzed (Fig. 4b), there is clear discrimination among three of the four populations, with little difference between the Monroe and Broward (Fig. 4b), supporting conclusions in the Structure and AMOVA analyses. Although the Miami-Dade and ‘Wild’ individuals were collected from the same area, they are more readily distinguished from each other in comparison to the two most distant collections (Broward and Monroe). Despite significant FST value differences between the Miami-Dade nursery and Miami-Dade ‘Wild’ collections, π is similar in both collections (38.3 % vs. 37.0 %, respectively), suggesting a change in allele frequencies associated with the local environment. These data suggest there are many genetic differences among populations and genetic diversity is high in each of the three Florida transects. These data also support the conclusion that the differences among collections are not a linear function of geographic distance within Florida; geographically close collections may have more differences that geographically dispersed collections.
Fig. 4.
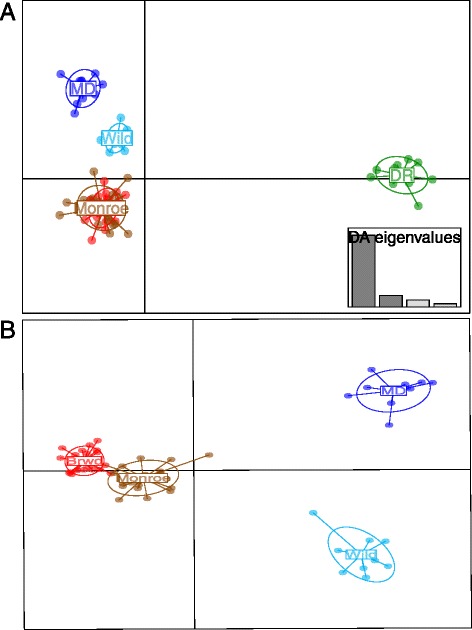
Discriminant analysis of populations. Discriminant analysis of principal components [34] was used to define the similarity and differences for (a) all five populations and (b) the four Florida populations. Populations are shown by different colors and inertia ellipses while dots represent individuals: Broward: Brwd-red, Miami-Dade: MD, dark blue, Monroe: brown and Dominican Republic: DR, green
Adaptive genes
Corals from the three Florida transects had 300 loci with significant FST values. Half or 150 of these SNPs were outliers. Outlier SNPs have FST values with changes in allele frequencies that are not found in 100,000 random permutations of the data and are thought to be due adaptive evolution [58, 59]. Although outlier tests suffer from both type I and II errors [60], a stepping-stone model of divergence is likely to be similar to the connectivity of the FRT populations, and thus the outlier test we used is unlikely to suffer from extensive type I errors [60]. Thus we conclude that some of the divergence among the FRT populations likely reflects adaptive evolution.
It has not escaped our attention that the Wild collection of 10 individuals within a single reef within Miami-Dade is different from the Miami-Dade transect collected from the surrounding area. While these individuals from Miami-Dade and Wild are phylogenetically similar to the other samples (they share internal branches in the phylogenetic tree), the comparison between these two populations shows 260 SNPs with significant FST values (average FST value = 0.232, range: 0.169 to 0.824). Each of these SNPs has an average of 120 reads among the twenty individuals from Miami-Dade and Wild populations (range: 45 to 1,048) and over 350 reads among all individuals. Two-hundred and thirty-four (89 %) of these SNPs with significant FST values are significant outliers [55]. The divergence between Wild and the Miami-Dade transect could represent local adaptation or could arise if the Wild individuals contained A. cervicornis – A. palmata hybrids. Unlike the other collections, which were identified based on morphology and microsatellite tags, the Wild collection was only identified by morphology. Although we have no reason to believe that the Wild samples were hybrids, the fine scale genetic divergence we found is similar to the rare, fine scale structure that was attributed to one-way introgression of A. palmata into A. cervicornis [15]. Thus although it is intriguing that natural selection is acting on a fine geographic scale, this conclusion may be premature until the species status of the Wild population is investigated.
Implications for restoration
Data presented here suggest that there is potentially much adaptive variation due to subtle environmental differences influencing coral distribution and growth, including temperature, water chemistry, light, nutrients, and sedimentation. This variation may occur over spatial scales as small as individual reefs. Due to the potential for high adaptive variation, introducing a broad range of genotypes along the FRT (such as those housed within nurseries) would enhance the frequency of adaptive genotypes and the subsequent rate of offspring survival. This is especially true if crossings during mass spawning events produce a larger range of genotypes able to take advantage of a large breadth of ecological niches. Thus, the best conservation and restoration strategy may be to increase genetic variation on all spatial scales (within reefs, among populations) as much as possible to provide diversity to cope with changing conditions [10].
This study found substantial genetic variation within existing staghorn populations being raised in coral nurseries. These nursery corals are presently used for coral propagation and outplanted to enhance population recovery of the threatened staghorn coral reefs. Greater than 90 % of the variation among all the samples is found within a nursery’s collection of corals, indicating that these nurseries have captured significant genetic diversity.
These GBS data indicate both large variation within populations and adaptive divergence among populations, and should help form policies that guide conservation efforts to restore staghorn coral reefs. We suggest that the caution against moving corals long distances during restoration [61] should be tempered, because genetic variation is very high within single reefs and among the three populations along Florida transects. Previous consideration of the implications of redistribution of corals during restoration suggests that moving corals beyond some ecologically relevant threshold may result in decreased fitness of a restored population due to founder effects, genetic swamping and inbreeding/outbreeding depression [61]. Here, we argue that the very high levels of diversity found within nursery source materials and on a single reef alleviate some concern. Very diverse assemblages on reefs targeted for restoration and in nursery source corals will likely not undergo fitness declines due to genetic swamping or outbreeding depression because there is much genetic variation within populations and no unique alleles in any of the Florida populations. Although there may be some reefs with one or few remaining colonies that have unique adaptive alleles, introducing genetically diverse corals would increase the genetic variation of any resulting coral larvae, and this diversity is needed for adaptation. Similarly, the potential for significant inbreeding depression would be decreased by the introduction of diverse coral assemblages. Outbreeding depression remains a concern. Yet in extant coral reefs, large genetic variation occurs within and among reefs. Thus, concerns about outbreeding depression for sexually produced coral larvae that will disperse long distances and face changing environments seems misplaced.
Conclusions
The GBS approach produced genotype frequencies for 4,764 SNPs that allowed for the resolution of population differences unavailable using other techniques [15–17]. Each SNP had an average of 411 reads/SNP with 69 reads/SNP for the minor allele, so genetic differences likely represent real nucleotide divergence and not sequencing error. However, there are imperfections to this approach, including the differences in heterozygosity produced by different SNP alignment tools. Despite the caveats with this method, GBS provides the ability to resolve previously undiscovered variation in populations of A. cervicornis. Here, we show, for the first time, population structure across the FRT and high diversity within populations, including within a single reef evidenced as the genetic structure between and among FRT populations. Previous work on A. cervicornis using mtDNA, a few nuclear genes, and microsatellites found no difference among the FRT coral populations [15–17, 62].
To further develop effective conservation and management plans for this species and other threatened corals considered as candidates for active propagation and restoration, it is essential to understand the extent of genetic variation within and among populations [15, 17]. Using a GBS approach, we highlight population differences by revealing many SNPs that have distinct allele frequencies among populations including one hundred and fifty SNPs, which have outlier FST values indicative of adaptive difference. There are also significant differences over small spatial scales, exemplified by differences between Wild and Miami-Dade individuals that were all collected within the same area (Fig. 1). The high genetic variation present in FRT A. cervicornis may allow this species to endure the interacting threats posed by local stressors and climate change factors such as temperature anomalies and acidification Additionally, π pair-wise differences) is large (37 %) for all collections and similar to GBS measures of π in large outbred populations of 3-spine stickleback or natural populations of yeast [51, 52].
The GBS methodology used here highlights the ability to discover subtle changes in populations by using thousands of loci and large numbers of individuals. Conservation genetics using these high throughput techniques provide a new lens for assessing management implications and population connectivity via important increases in resolution, but also in varied and specific genetic metrics such as population structure, nucleotide diversity, and loci that may be under selection. These data are particularly important to active restoration projects as they give a better understanding of population structure, how and where to relocate coral, and potential repercussions of active intervention. Furthermore, the ability to describe genetic diversity over local to regional distributions enables conservation practitioners to manage resources over appropriate scales, becoming more efficient and effective. GBS allows for increased restoration effectiveness through conservation genetics, while developing a more thorough understanding of threatened coral communities.
Availability of supporting data
Raw sequences have been submitted to NCBI as a Sequence Read Archive (SRA). The Acropora cervicornis hapmap (DOI: 10.6070/H4FB50XX) and sequence tags (DOI: 10.6070/H49K4872) are available at LabArchives.com. Sequence files are available at NCBI's BioSample database, accessions SAMN03295587 - SAMN03295662.
Acknowledgements
We thank the University of Miami and in particular Will Drennan and Gary Hitchcock from the Marine Science Program for supporting the undergraduate research, which developed most of these data. Additional support for the adaptor primers was from NSF MCB 1158241 and and IOS 1147042. We would like to thank Nova Southeastern University, University of Miami – RSMAS, Mote Marine Lab, and the Punta Cana Ecological Foundation for nursery samples. We thank Steve Vollmer for technical advice.
Abbreviations
- FRT
Florida reef tract
- GBS
Genotyping by sequencing
- SNP
Single nucleotide polymorphism
- DAPC
Discriminant analysis of principal components
Footnotes
K. E. Dale, J. M. Panlilio and S. V. Miller contributed equally to this work.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
CD composed and edited the final version of the manuscript. KED, JMP, and SVM contributed equally to the project and isolated gDNA, constructed GBS libraries and assisted in bioinformatic analyses and field collection. DL organized and directed fieldwork and along with CD, EAL, and EB were responsible for collecting and managing coral nurseries. DLC and MFO directed research and were responsible for statistical and bioinformatics analyses. All authors contributed to writing. All authors read and approved the final manuscript.
Contributor Information
C. Drury, Email: cdrury@rsmas.miami.edu
K. E. Dale, Email: k.dale1@umiami.edu
J. M. Panlilio, Email: panlilio@mit.edu
S. V. Miller, Email: smiller71@student.gsu.edu
D. Lirman, Email: lirman@rsmas.miami.edu
E. A. Larson, Email: goergen@nova.edu
E. Bartels, Email: ebartels@mote.org
D. L. Crawford, Email: dcrawford@rsmas.miami.edu
M. F. Oleksiak, Email: moleksiak@rsmas.miami.edu
References
- 1.Gardner TA, Côté IM, Gill JA, Grant A, Watkinson AR. Long-term region-wide declines in Caribbean corals. Sci. 2003;301(5635):958–960. doi: 10.1126/science.1086050. [DOI] [PubMed] [Google Scholar]
- 2.Hoegh-Guldberg O, Mumby PJ, Hooten AJ, Steneck RS, Greenfield P, Gomez E, et al. Coral reefs under rapid climate change and ocean acidification. Sci. 2007;318(5857):1737–1742. doi: 10.1126/science.1152509. [DOI] [PubMed] [Google Scholar]
- 3.Hughes TP. Catastrophes, phase shifts, and large-scale degradation of a Caribbean coral reef. Sci. 1994;265(5178):1547–1551. doi: 10.1126/science.265.5178.1547. [DOI] [PubMed] [Google Scholar]
- 4.Miller M, Bourque A, Bohnsack J. An analysis of the loss of acroporid corals at Looe Key, Florida, USA: 1983‚Äì2000. Coral Reefs. 2002;21(2):179–182. [Google Scholar]
- 5.Aronson RB, Precht WF: White-Band Disease and the Changing Face of Caribbean Coral Reefs. The Ecology and Etiology of Newly Emerging Marine Diseases. Netherlands: Springer; 2001. 25–38
- 6.Greenstein B, Curran H, Pandolfi J. Shifting ecological baselines and the demise of Acropora cervicornis in the western North Atlantic and Caribbean Province: a Pleistocene perspective. Coral Reefs. 1998;17(3):249–261. doi: 10.1007/s003380050125. [DOI] [Google Scholar]
- 7.Tunnicliffe V. Breakage and propagation of the stony coral acropora-cervicornis. P Natl Acad Sci-Biol. 1981;78(4):2427–2431. doi: 10.1073/pnas.78.4.2427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Highsmith RC. Reproduction by fragmentation in corals. Mar Ecol Prog Ser Oldendorf. 1982;7(2):207–226. doi: 10.3354/meps007207. [DOI] [Google Scholar]
- 9.Young CN, Schopmeyer SA, Lirman D. A review of reef restoration and coral propagation using the threatened genus acropora in the Caribbean and Western Atlantic. Bull. Mar. Sci. 2012;88(4):1075–1098. doi: 10.5343/bms.2011.1143. [DOI] [Google Scholar]
- 10.Hoffmann AA, Sgro CM. Climate change and evolutionary adaptation. Nat. 2011;470(7335):479–485. doi: 10.1038/nature09670. [DOI] [PubMed] [Google Scholar]
- 11.Hofmann GE, Todgham AE. Living in the now: physiological mechanisms to tolerate a rapidly changing environment. Annu Rev Physiol. 2010;72(1):127–145. doi: 10.1146/annurev-physiol-021909-135900. [DOI] [PubMed] [Google Scholar]
- 12.Palumbi SR, Barshis DJ, Traylor-Knowles N, Bay RA. Mechanisms of reef coral resistance to future climate change. Sci (NY) 2014;344(6186):895–898. doi: 10.1126/science.1251336. [DOI] [PubMed] [Google Scholar]
- 13.Parmesan C, Yohe G. A globally coherent fingerprint of climate change impacts across natural systems. Nat. 2003;421(6918):37–42. doi: 10.1038/nature01286. [DOI] [PubMed] [Google Scholar]
- 14.Baums IB, Miller MW, Hellberg ME. Regionally isolated populations of an imperiled Caribbean coral, Acropora palmata. Mol Ecol. 2005;14(5):1377–1390. doi: 10.1111/j.1365-294X.2005.02489.x. [DOI] [PubMed] [Google Scholar]
- 15.Vollmer SV, Palumbi SR. Restricted gene flow in the Caribbean staghorn coral Acropora cervicornis: implications for the recovery of endangered reefs. J Hered. 2007;98(1):40–50. doi: 10.1093/jhered/esl057. [DOI] [PubMed] [Google Scholar]
- 16.Baums IB, Johnson ME, Devlin-Durante MK, Miller MW. Host population genetic structure and zooxanthellae diversity of two reef-building coral species along the Florida Reef Tract and wider Caribbean. Coral Reefs. 2010;29(4):835–842. doi: 10.1007/s00338-010-0645-y. [DOI] [Google Scholar]
- 17.Hemond EM, Vollmer SV. Genetic Diversity and Connectivity in the Threatened Staghorn Coral Acropora cervicornis in Florida. Plos One. 2010;5(1):e8652. doi: 10.1371/journal.pone.0008652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Narum SR, Buerkle CA, Davey JW, Miller MR, Hohenlohe PA. Genotyping-by-sequencing in ecological and conservation genomics. Mol Ecol. 2013;22(11):2841–2847. doi: 10.1111/mec.12350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Elshire RJ, Glaubitz JC, Sun Q, Poland JA, Kawamoto K, Buckler ES, et al. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS One. 2011;6(5):e19379. doi: 10.1371/journal.pone.0019379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schopmeyer SA, Lirman D, Bartels E, Byrne J, Gilliam DS, Hunt J, et al. In situ coral nurseries serve as genetic repositories for coral reef restoration after an extreme cold-water event. Restoration Ecol. 2012;20(6):696–703. doi: 10.1111/j.1526-100X.2011.00836.x. [DOI] [Google Scholar]
- 21.Baums IB, Devlin-Durante K, Brown L, Pinzon JH. Nine novel, polymorphic microsatellite markers for the study of threatened Caribbean acroporid corals. Mol Ecol Resour. 2009;9(4):1155–1158. doi: 10.1111/j.1755-0998.2009.02581.x. [DOI] [PubMed] [Google Scholar]
- 22.Dayan DI, Crawford DL, Oleksiak MF. Phenotypic plasticity in gene expression contributes to divergence of locally adapted populations of Fundulus heteroclitus. Mol Ecol. 2015;24(13):3345–59. doi: 10.1111/mec.13188. [DOI] [PubMed] [Google Scholar]
- 23.Ivanova NV, Dewaard JR, Hebert PDN. An inexpensive, automation-friendly protocol for recovering high-quality DNA. Mol Ecol Notes. 2006;6(4):998–1002. doi: 10.1111/j.1471-8286.2006.01428.x. [DOI] [Google Scholar]
- 24.Bradbury PJ, Zhang Z, Kroon DE, Casstevens TM, Ramdoss Y, Buckler ES. TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics. 2007;23(19):2633–2635. doi: 10.1093/bioinformatics/btm308. [DOI] [PubMed] [Google Scholar]
- 25.Excoffier L, Laval G, Schneider S. Arlequin (version 3.0): An integrated software package for population genetics data analysis. Evol Bioinform. 2005;1:47–50. [PMC free article] [PubMed] [Google Scholar]
- 26.Paradis E, Claude J, Strimmer K. APE: analyses of phylogenetics and evolution in R language. Bioinformatics. 2004;20(2):289–290. doi: 10.1093/bioinformatics/btg412. [DOI] [PubMed] [Google Scholar]
- 27.Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genet. 2000;155(2):945–959. doi: 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Evanno G, Regnaut S, Goudet J. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol. 2005;14(8):2611–2620. doi: 10.1111/j.1365-294X.2005.02553.x. [DOI] [PubMed] [Google Scholar]
- 29.Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30(9):1312–3. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tavaré S. Some probabilistic and statistical problems in the analysis of DNA sequences. Lectures Math Life Sci. 1986;17:57–86. [Google Scholar]
- 31.Huson DH, Scornavacca C. Dendroscope 3: an interactive tool for rooted phylogenetic trees and networks. Syst Biol. 2012;61(6):1061–1067. doi: 10.1093/sysbio/sys062. [DOI] [PubMed] [Google Scholar]
- 32.Jombart T. adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics. 2008;24(11):1403–1405. doi: 10.1093/bioinformatics/btn129. [DOI] [PubMed] [Google Scholar]
- 33.Jombart T: An introduction to adegenet 1.41. http://adegenet.r-forge.r-project.org/documentation.html. 2014.
- 34.Jombart T, Devillard S, Balloux F. Discriminant analysis of principal components: a new method for the analysis of genetically structured populations. BMC Genetics. 2010;11(1):94. doi: 10.1186/1471-2156-11-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Baums IB, Hughes CR, Hellberg ME. Mendelian microsatellite loci for the Caribbean coral Acropora palmata. Mar Ecol Prog Ser. 2005;288:115–127. doi: 10.3354/meps288115. [DOI] [Google Scholar]
- 36.Chen N, Van Hout CV, Gottipati S, Clark AG. Using Mendelian inheritance to improve high-throughput SNP discovery. Genetics. 2014;198(3):847–857. doi: 10.1534/genetics.114.169052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Glaubitz JC, Casstevens TM, Lu F, Harriman J, Elshire RJ, Sun Q, et al. TASSEL-GBS: a high capacity genotyping by sequencing analysis pipeline. PLoS One. 2014;9(2):e90346. doi: 10.1371/journal.pone.0090346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hohenlohe PA, Bassham S, Etter PD, Stiffler N, Johnson EA, Cresko WA. Population Genomics of Parallel Adaptation in Threespine Stickleback using Sequenced RAD Tags. PLoS Genet. 2010;6(2):e1000862. doi: 10.1371/journal.pgen.1000862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McClure MC, Bickhart D, Null D, Vanraden P, Xu L, Wiggans G, et al. Bovine exome sequence analysis and targeted SNP genotyping of recessive fertility defects BH1, HH2, and HH3 reveal a putative causative mutation in SMC2 for HH3. PLoS One. 2014;9(3):e92769. doi: 10.1371/journal.pone.0092769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Oliver RE, Tinker NA, Lazo GR, Chao S, Jellen EN, Carson ML, et al. SNP discovery and chromosome anchoring provide the first physically-anchored hexaploid oat map and reveal synteny with model species. PLoS One. 2013;8(3):e58068. doi: 10.1371/journal.pone.0058068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shinzato C, Shoguchi E, Kawashima T, Hamada M, Hisata K, Tanaka M, et al. Using the Acropora digitifera genome to understand coral responses to environmental change. Nature. 2011;476(7360):320–323. doi: 10.1038/nature10249. [DOI] [PubMed] [Google Scholar]
- 42.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9(4):357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li H, Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics. 2009;25(14):1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nielsen R, Mattila DK, Clapham PJ, Palsboll PJ. Statistical approaches to paternity analysis in natural populations and applications to the North Atlantic humpback whale. Genetics. 2001;157(4):1673–1682. doi: 10.1093/genetics/157.4.1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.O’Rawe J, Jiang T, Sun G, Wu Y, Wang W, Hu J, et al. Low concordance of multiple variant-calling pipelines: practical implications for exome and genome sequencing. Genome Med. 2013;5(3):1–18. doi: 10.1186/gm432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bayer T, Aranda M, Sunagawa S, Yum LK, DeSalvo MK, Lindquist E, et al. Symbiodinium transcriptomes: Genome insights into the dinoflagellate symbionts of reef-building corals. Plos One. 2012;7(4):e35269. doi: 10.1371/journal.pone.0035269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hosking L, Lumsden S, Lewis K, Yeo A, McCarthy L, Bansal A, et al. Detection of genotyping errors by Hardy-Weinberg equilibrium testing. Eur J Hum Genet. 2004;12(5):395–399. doi: 10.1038/sj.ejhg.5201164. [DOI] [PubMed] [Google Scholar]
- 48.Anderson CA, Pettersson FH, Clarke GM, Cardon LR, Morris AP, Zondervan KT. Data quality control in genetic case–control association studies. Nat Protoc. 2010;5(9):1564–1573. doi: 10.1038/nprot.2010.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Neigel JE, Avise JC. Clonal diversity and population structure in a reef-building coral, acropora cervicornis: self-recognition analysis and demographic interpretation. Evol. 1983;37(3):437–453. doi: 10.2307/2408259. [DOI] [PubMed] [Google Scholar]
- 50.Baums IB, Miller MW, Hellberg ME. Geographic variatin in clonal structure in a reef-buildn Caribbena coral. Acropora Palmata Ecol M. 2006;76(4):503–519. [Google Scholar]
- 51.Catchen J, Bassham S, Wilson T, Currey M, O'Brien C, Yeates Q, et al. The population structure and recent colonization history of Oregon threespine stickleback determined using restriction-site associated DNA-sequencing. Mol Ecol. 2013;22(11):2864–2883. doi: 10.1111/mec.12330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hyma KE, Fay JC. Mixing of vineyard and oak-tree ecotypes of Saccharomyces cerevisiae in North American vineyards. Mol Ecol. 2013;22(11):2917–2930. doi: 10.1111/mec.12155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Leffler EM, Bullaughey K, Matute DR, Meyer WK, Ségurel L, Venkat A, et al. Revisiting an Old Riddle: What Determines Genetic Diversity Levels within Species? PLoS Biol. 2012;10(9):e1001388. doi: 10.1371/journal.pbio.1001388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Excoffier L, Smouse PE, Quattro JM. Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA. Genetics. 1992;131:479–491. doi: 10.1093/genetics/131.2.479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Beaumont MA, Balding DJ. Identifying adaptive genetic divergence among populations from genome scans. Mol Ecol. 2004;13(4):969–980. doi: 10.1111/j.1365-294X.2004.02125.x. [DOI] [PubMed] [Google Scholar]
- 56.Willing E-M, Dreyer C, van Oosterhout C. Estimates of genetic differentiation measured by FST do not necessarily require large sample sizes when using many SNP markers. PLoS One. 2012;7(8):e42649. doi: 10.1371/journal.pone.0042649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lirman D, Schopmeyer S, Galvan V, Drury C, Baker AC, Baums IB. Growth dynamics of the threatened caribbean staghorn coral acropora cervicornis: influence of host genotype, symbiont identity, colony size, and environmental setting. PloS One. 2014;9(9) doi: 10.1371/journal.pone.0107253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Antao T, Lopes A, Lopes R, Beja-Pereira A, Luikart G. LOSITAN: A workbench to detect molecular adaptation based on a Fst-outlier method. BMC Bioinformatics. 2008;9(1):323. doi: 10.1186/1471-2105-9-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Beaumont M, Nichols R. Evaluating loci for use in the genetic analysis of population structure. Proc R Soc B. 1996;363:1619–1626. doi: 10.1098/rspb.1996.0237. [DOI] [Google Scholar]
- 60.Lotterhos KE, Whitlock MC. Evaluation of demographic history and neutral parameterization on the performance of FST outlier tests. Mol Ecol. 2014;23(9):2178–2192. doi: 10.1111/mec.12725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Baums IB. A restoration genetics guide for coral reef conservation. Mol Ecol. 2008;17(12):2796–2811. doi: 10.1111/j.1365-294X.2008.03787.x. [DOI] [PubMed] [Google Scholar]
- 62.Vollmer SV, Palumbi SR. Hybridization and the evolution of reef coral diversity. Sci. 2002;296(5575):2023–2025. doi: 10.1126/science.1069524. [DOI] [PubMed] [Google Scholar]