

ABSTRACT
Research into the molecular genetics and pathomechanisms of ichthyoses have advanced considerably, resulting in the identification of several causative genes and molecules underlying the disease. In 2009, the First Ichthyosis Consensus Conference was held to establish a consensus for the nomenclature and classification of inherited ichthyoses, by which an international consensus for the classification of inherited ichthyosis was achieved. In this review, the pathogeneses of various ichthyoses are summarized based on their revised classification and terminology. Skin barrier defects are involved in the pathogenesis of various types of ichthyosis. The known causative molecules underlying ichthyosis include ABCA12, lipoxygenase-3, 12R-lipoxygenase, CYP4F22, ichthyin and steroid sulfatase, all of which are thought to be related to the intercellular lipid layers. ABCA12 is a known keratinocyte lipid transporter associated with lipid transport in lamellar granules and a loss of ABCA12 function leads to defective lipid transport in the keratinocytes, resulting in the most severe, harlequin ichthyosis phenotype. Other causative molecules for ichthyoses are transglutaminase 1, keratins and filaggrin. Transglutaminase 1 plays a role in cornified cell envelope formation. Keratins 1, 10 and 2 are involved in the keratin network of suprabasal keratinocytes and filaggrin is essential for the formation of keratohyalin granules. It is important to obtain information concerning genetic defects and to elucidate ichthyotic disease pathomechanisms for the establishment of an effective therapy and beneficial genetic counseling, including a prenatal diagnosis for families affected by ichthyotic disease.
Key Words: ABCA12, Congenital ichthyosiform erythroderma, Epidermolytic ichthyosis, Harlequin ichthyosis, Lamellar ichthyosis, Prenatal diagnosis
INTRODUCTION
The ichthyoses form a large, clinically and etiologically heterogeneous group of cornification disorders that typically affect all or most of the skin surface.1) Six major distinct clinical subtypes are known in hereditary non-syndromic ichthyoses. Starting with the most severe form, they are: harlequin ichthyosis (HI, MIM#242500); lamellar ichthyosis (LI, MIM#242300); congenital ichthyosiform erythroderma (CIE, MIM#242100); epidermolytic ichthyosis (EI, MIM #113800); recessive X-linked ichthyosis (RXLI, MIM#308100); to the mildest form of ichthyosis vulgaris (IV, MIM#146700).2) Superficial epidermolytic ichthyosis (SEI, MIM #146800) is an additional subtype similar to EI. For a long time, the pathomechanisms and underlying genetic defects of ichthyoses were unknown, although significant progress has recently been made in our understanding of the molecular basis of human epidermal keratinization processes.
In 1978, the causative abnormality underlying RXLI was identified as a steroid sulfatase deficiency caused by genetic defects in the steroid sulfatase gene (STS).3,4) In 1992, mutations in the keratin 1 gene (KRT1) and keratin 10 gene (KRT10) were detected as a cause of EI.5-7) Since transglutaminase (TGase) 1 gene (TGM1) mutations were identified as the cause of LI in 1995,8,9) mutations in several other genes have also been identified in severe autosomal recessive congenital ichthyoses (ARCI).10)
In 2005, a loss-of-function mutations in the ABCA12 gene were reported to underlie HI, the most severe type of ichthyosis.11,12) In 2006, null mutations in the gene coding filaggrin (FLG) were detected as the causative defects leading to IV.13)
To date, the number of genes identified and demonstrated to cause ichthyosis in human patients has reached eleven; they are shown in Table 1, i.e., FLG,13)KRT1, KRT10,5-7)KRT2,14-16)TGM1,8,9)ABCA12,11,17,18) two lipoxygenase genes, ALOXE3 and ALOX12B,19)NIPAL420) and FLJ39501.21) Most ichthyosis phenotypes mentioned above exhibit a primary abnormality associated with a barrier function in the stratum corneum as their underlying pathogenetic mechanisms.1) The skin barrier of the stratum corneum has three major components, i.e., intercellular lipid layers, a cornified cell envelope and keratin/filaggrin degradation products (Fig. 1)
Table 1.
Essential components of stratum cornuem barrier and causative molecules/genes for ichthyoses (modified from Ref. No. 1)
| Stratum corneum barrier components | Molecule | Gene (locus) | Mode of inheritance | Type of Mutations | Phenotype |
|---|---|---|---|---|---|
| Intercellular lipid layers | ABCA12 | ABCA12 (2q34) | AR | truncation/deletion (rarely missense) | HI |
| ABCA12 | ABCA12 (2q34) | AR | missense/missense or missense/truncation | LI or CIE | |
| lipoxygenase-3 | ALOXE3 (17p13.1) | AR | missense/truncation | LI or CIE | |
| 12R-lipoxygenase | ALOX12B (17p13.1) | AR | missense/truncation | LI or CIE | |
| CYP4F22 | FLJ39501 (19P12) | AR | missense/truncation | LI | |
| NIPAL4 | NIPAL4 (5q33) | AR | missense/truncation | CIE or LI | |
| Steroid sulfatase | STS (Xp22.32) | X-LR | mostly large deletion | RXLI | |
| Cornified cell envelope | TGase 1 | TGM1 (14q11.2) | AR | missense/truncation/ deletion/insertion | LI or CIE |
| Keratin network and keratohyalin granules | keratin 1 | KRT1 (12q12-q13) | AD | missense | EI |
| keratin 10 | KRT10 (17q21) | AD (rarely AR) | missense (rarely nonsense) | EI | |
| keratin 2 | KRT2 (12q11-q13) | AD | missense | SEI | |
| Filaggrin (profilaggrin) | FLG (1q21.3) | ASD | truncation | IV |
AD, autosomal dominant; AR, autosomal recessive; ASD, autosomal semidominant; X-LR, X-linked recessive; CIE, congenital ichthyosiform erythroderma; EI, epidermolytic ichthyosis; HI, harlequin ichthyosis; IV, ichthyosis vulgaris; LI, lamellar ichthyosis; RXLI, recessive X-linked ichthyosis; SEI, superficial epidermolytic ichthyosis
Fig. 1.
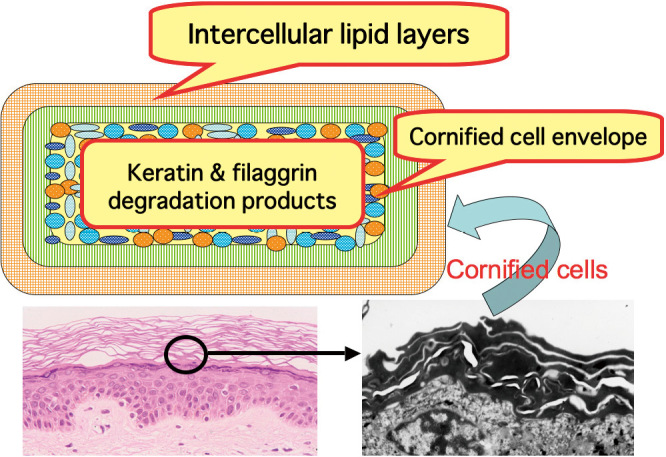
Major components of skin barrier in stratum corneum consist of intercellular lipid layers, cornified cell envelope and keratin/filaggrin degradation products. Figure modified from Ref. No. 1.
In 2009, the First Ichthyosis Consensus Conference was held to establish a consensus for the nomenclature and classification of inherited ichthyoses and an international consensus for the classification of inherited ichthyosis was successfully achieved.2) The new classification and nomenclature should prove useful to all clinicians and can serve as a reference point for future research. In this updated review, based on the revised classification and terminology of ichthyoses, the pathogeneses of various ichthyoses are described in association with descriptions of specific defects in the essential components comprising the epidermal skin barrier, highlighting a few crucial diseases and mechanisms. In addition, I briefly mentioned the prenatal diagnosis of severe congenital ichthyosis at the end of the present review.
MAJOR ICHTHYOSIS SUBTYPES AND THEIR CAUSATIVE MOLECULES
Harlequin ichthyosis (HI)
Formation of the intercellular lipid layers is essential for epidermal barrier function, and the defective formation of those layers is thought to result in a serious loss of barrier function, and to lead to extensive hyperkeratosis.22) Formation of the intercellular lipid layers involves a highly complex series of processes that include the transport of lipids into the lamellar granules, and a multi-step metabolism of this lipid content within lamellar granules. ABCA12 has been highlighted, because it was recognized as a key molecule in keratinocytes lipid transport.11,23)
Among the severe ARCI diseases, HI is the most devastating congenital ichthyosis, with affected newborns showing large, thick, plate-like scales over the whole body with severe ectropion, eclabium and flattened ears (Fig. 2A).10) In 2005, we revealed that ABCA12 is a keratinocyte lipid transporter, and demonstrated that ABCA12 mutations lead to the HI phenotype.11) Another group independently reported that ABCA12 mutations underlie HI by linkage analysis.12)ABCA12 mutations were also found to underlie LI and CIE cases.17,18) ABCA12 is a member of a large superfamily of the ATP-binding cassette (ABC) transporters that bind and hydrolyze ATP to transport various molecules across a limiting membrane or into a vesicle.24) All ABCA subfamily members are thought to be lipid transporters.25) ABCA12 is a keratinocyte transmembrane lipid transporter protein associated with lipid transport in lamellar granules to the apical surface of granular layer keratinocytes.11)
Fig. 2.
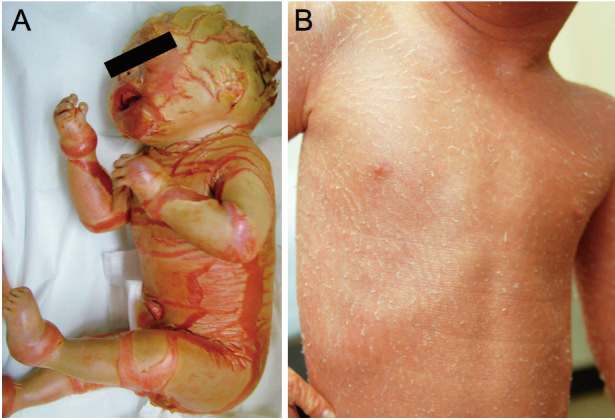
Clinical features of ichthyosis. (A) An HI patient harboring a homozygous ABCA12 splice site mutation. Thick, plate-like scales are seen on the whole body. Figure modified from Ref. No. 11. (B) A CIE patient carrying compound heterozygous ABCA12 nonsense and missense mutations. Fine, whitish scales are observed on erythrodermic skin. Figure modified from Ref. No. 42.
Ultrastructurally, lamellar granule abnormalities are apparent in HI patient epidermis.26) Several morphologic abnormalities have been reported, e. g., abnormal lamellar granules in the granular layer keratinocytes and a lack of extracellular lipid lamellae in the stratum corneum. They reflect defective lipid transport by lamellar granules and the malformation of intercellular lipid layers in the stratum corneum in HI.26) In addition, cultured epidermal keratinocytes from an HI patient carrying ABCA12 mutations demonstrated defective glucosylceramide transport, and this phenotype was recoverable by an in vitro ABCA12 corrective gene transfer.11) Based on these findings, we were able to shed light on the pathomechanisms of HI with the underlying ABCA12 mutations leading to a loss of ABCA12 function. Lamellar granules are lipid transporting and secreting granules in the epidermal kerationcytes. Mutations in the lipid transporter protein ABCA12 cause defective lipid accumulations into lamellar granules,11,27) resulting in malformation of the intercellular lipid layers of the stratum corneum.11) The fact that ABCA3 (a member of the same protein superfamily as ABCA12) functions in pulmonary surfactant lipid secretion via the production of similar lamellar-type granules within lung alveolar type II cells28,29) further supports this concept.
We subsequently transplanted cultured keratinocytes from patients with HI and succeeded in reconstituting HI skin lesions in immunodeficient mice.27) These reconstructed HI lesions showed similar changes to those observed in HI patients’ skin. In addition, we generated Abca12 disrupted (Abca12-/-) mice that reproduced the human HI phenotype, showing marked hyperkeratosis with eclabium and skin fissures.30) Lamellar granule abnormalities and defective ceramide distribution were noteworthy in the epidermis. Skin permeability assays of Abca12-/- mouse fetuses revealed a severe skin barrier dysfunction after the initiation of keratinization. Another group independently developed Abca12-/- mice, which also confirmed the clinical features of HI.31) A mouse strain carrying a homozygous spontaneous missense mutation was also reported to exhibit skin manifestations similar to HI.32)
HI patients often die in the first one or two weeks of life. However, once they survive beyond the neonatal period, HI survivors’ phenotypes improve within several weeks after birth. In order to clarify the mechanisms of phenotype recovery, we studied grafted skin and keratinocytes from Abca12-disrupted (Abca12-/-) mice and found that, during maturation, Abca12-/- epidermal keratinocytes regain their normal differentiation processes, although the exact mechanisms of this restoration are still unknown.33)
Congenital ichthyosiform erythroderma (CIE) and lamellar ichthyosis (LI)
The formation of a 15-nm-thick layer of protein called the cornified cell envelope (CCE) on the inner surface of the cell membrane is essential for the skin barrier function.1) CCE is assembled by the accumulation of several precursor proteins including involucrin, small proline-rich proteins and loricrin.34) TGases in the epidermis are thought to be responsible at least in part, for the assembly of cornified cell envelope precursor proteins that form the cornified cell envelope.35) TGase 1, the major subtype of the three TGases expressed in the epidermis,36,37) is a membrane-associated TGase of about 92 kD. Since the identification of TGase 1 gene (TGM1) mutations in a number of families with LI in 1995,8,9) further TGM1 mutations have been reported in LI families. In addition, TGM1 mutations were reported to underlie the CIE phenotype.38,39)
There is little doubt that the defective formation of the stratum corneum intercellular lipid layers is caused by abnormal keratinocyte lipid metabolism, transport, and/or secretion, constituting one of the major pathogenetic mechanisms underlying congenital ichthyosis. Several critical molecules causing ichthyosis are thought to be involved in the formation of the stratum corneum intercellular lipid layers.1)
In 2003, a keratinocyte lipid transporter ABCA12 was reported to be the causative molecule in type 2 LI (OMIM #601277) (see “harlequin ichthyosis” section above).17) Type 2 LI is a subtype of LI which links to 2q33-35. Several genotype/phenotype correlations with ABCA12 mutations have been elucidated as follows.40) Combinations of missense mutations resulting in only one amino acid alteration underlie the LI phenotype.17) In contrast, most mutations in HI are truncation or deletion mutations which lead to more severe changes, such as a loss of the ABCA12 peptide function affecting important nucleotide-binding fold domains and/or transmembrane domains. In HI, so far at least one mutation on each allele must be a truncation or deletion mutation within a conserved region that seriously impacts the ABCA12 function.11,40,41) In the Japanese population, CIE patients harboring ABCA12 mutations as the causative genetic defect are not rare (Fig. 2B).18,42,43) Further accumulations of data on ABCA12 mutations and their effects on the protein function/structure together with specific mutation sites are needed to better elucidate genotype/phenotype correlations that help to predict HI patient prognosis.
In 2002, mutations in two lipoxygenase genes, ALOXE3 and ALOX12B, coding lipoxygenase-3 and 12(R)-lipoxygenase, respectively, were reported to underlie ARCI.19) Although their exact functions are unknown, lipoxygenase-3 and 12(R)-lipoxygenase are non-heme iron-containing dioxygenases expressed in the epidermis.44,45) They may be associated with the lipid metabolism of lamellar granule contents and/or intercellular lipid layers in the epidermis. 12(R)-lipoxygenase knockout mice have exhibited a severe impairment of the skin barrier function.46) That loss of barrier function was associated with a perturbance of the assembly/extrusion of lamellar granules. Cornified cell envelopes from the skin of 12(R)-lipoxygenase deficient mice showed increased fragility.46) Lipid analysis revealed a disordered composition of ceramides, especially a decrease in th ester-bound ceramide species.46) Based on these findings, the 12(R)-lipoxygenase-lipoxygenase-3 pathway was thought to play a key role in the process of epidermal barrier formation by affecting lipid metabolism.46) In fact, partially disturbed lamellar granule secretion has been reported in the epidermis of a CIE patient with ALOX12B mutations.47)
NIPAL4 (ichthyin) defects have also been reported to underlie certain cases of LI or CIE phenotypes.20) NIPAL4 is a protein with several transmembrane domains, which belongs to a new family of proteins with an unknown function. NIPAL4-like proteins are localized in the plasma membrane, and share homologies to both transporters and G-protein coupled receptors.20) NIPAL4 was suggested to be a membrane receptor for certain ligands (trioxilins A3 and B3) from the hepoxilin pathway,20) although the underlying mechanisms of exactly how NIPAL4 mutations lead to an ichthyotic phenotype remain to be clarified.
Mutations in FLJ39501 were identified as causative genetic defects in lamellar ichthyosis type 3 (MIM 604777).21)FLJ39501 encodes a cytochrome P450, family 4, subfamily F, polypeptide 2 homolog of the leukotriene B4-omega-hydroxylase (CYP4F22). The exact function of CYP4F22 has not yet been elucidated yet, but it is thought to catalyze the 20-hydroxylation of trioxilin A3 form the 12(R)-lipoxygenase pathway. Further oxidation of this substrate would lead to 20-carboxy-(R)–trioxilin A3, a compound suspected to be involved in skin hydration, and would be an essential product lacking in various forms of ARCI.
Epidermolytic ichthyosis (EI) and superficial epidermolytic ichthyosis (SEI)
A normal keratin filament network is an important structure for keratohyalin granule formation and for maintaining the integrity and dimensions of the cornified cell cytoplasm. In this context, the keratin-network would be essential to normal skin barrier formation. Mutations in differentiation-specific keratins are known to result in ichthyosis phenotypes.
EI is caused by mutations in either the keratin 1 gene (KRT1) or the keratin 10 gene (KRT10).5-7) Most of the causative mutations are missense mutations that reside within the beginning or at the end of the rod domain segments of keratin peptides, which are called helix initiation and helix termination motifs. Those motifs are highly conserved regions of approximately 20 amino acids, which have been implicated in molecular overlapping interactions as part of the formation of 10 nm intermediate filaments from dimers comprising both type I acidic and a type II basic-neutral keratins.48) Single amino acid alterations in these essential helix boundary motifs frequently lead to a significant disease phenotype in the majority of keratin diseases.
EI is a severe congenital ichthyosis that exhibits from birth widespread blisters and erosions on a background of erythrodermic skin.1,2) After the perinatal period, blister formation ceases and generalized hyperkeratosis becomes apparent. Histologically, a predominant vacuolization of the cells is observed in the middle and upper spinous and granular layers of the epidermis. The vacuolated keratinocytes show large and irregularly shaped cytoplasmic granules. Ultrastructurally, irregularly shaped, abnormal, clumped keratin filaments are seen in the keratinocytes from the upper spinous to the granular layers.49) EI generally exhibits an autosomal dominant inheritance, although only a few families showing recessive inheritance traits have been reported.50,51) In such families, the causative mutations are nonsense mutations. As for genotype-phenotype correlations in EI, palmoplantar keratoderma exists in patients with KRT1 mutations, but not in those with KRT10 mutations.52)
SEI is also an autosomal dominant ichthyosis which shows similar, but slightly milder clinical features than those of EI.53) Keratin 2 gene (KRT2) mutations underlie SEI patients.14-16) Occasionally, cases with SEI can be difficult to clinically differentiate from EI, so that molecular genetic studies would be essential for a more definite diagnosis.54) In the human epidermis, keratin 1 and 10 expressions occur in the suprabasal layers, replacing keratins 5 and 14 as the cells differentiate. Keratin 2 is expressed somewhat later than keratins 1 and 10 in keratinocyte differentiation as the keratinocytes approach the granular layer. Thus, consistent with the restricted keratin 2 expression sites, in SEI, clumped keratin filaments were restricted to the cytoplasm of granular layer cells and the uppermost spinous layer cells, leading to granular degeneration only in the uppermost spinous and granular layers of the patient’s epidermis. Such restricted granular degeneration results in milder clinical manifestations and the presence of superficial denuded areas (the mauserung phenomenon) that are characteristic of SEI.
Ichthyosis vulgaris (IV)
IV is a common genetic keratinization disorder, clinically characterized by scaling, especially on the flexor limbs, and with palmoplantar hyperlinearity. The epidermis of IV patients shows a decrease in their size and numbers or, complete absence of keratohyalin granules.55)
The degradation products of keratohyalin granules occupy the cytoplasm of keratinized cells in the stratum corneum and play important roles in the skin barrier function. Keratohyalin granules in the granular layer of the epidermis are predominantly composed of large (>400-kDa) profilaggrin polyproteins.56,57) Upon the terminal differentiation of keratinocytes, profilaggrin is cleaved into 10-12 essentially identical 37-kDa filaggrin peptides. The liberated filaggrin aggregates the keratin filaments,56) causing a collapse of the granular cells into a flattened squame-shape. In addtion, the degradation products of filaggrin contribute to moisture retention in the cornified layers. Thus, filaggrin, a major component of keratohyalin granules, is indispensable to the normal, intact, skin barrier function. In this context, a loss or reduction in filaggrin expression results in excessively dry skin and impaired barrier function, which leads to clinical features of IV.
In 2006, FLG mutations were identified in IV patients in European populations13) and have been shown to be major predisposing factors for atopic dermatitis.58) Subsequently, FLG mutations were identified in Japanese, Chinese, Taiwanese and Korean populations.59,60) Based on the information about population-specific FLG mutations, numbers of cohort studies of atopic dermatitis for FLG mutations have been performed confirming that about 25–50% of patients with atopic dermatitis were demonstrated to harbor FLG mutations as a predisposing factor. It was demonstrated that FLG mutations are also strongly associated with atopic dermatitis in the Japanese population.59,61) Skin barrier defects due to FLG mutations are thought to play important roles in the pathogenesis of atopic diseases including atopic dermatitis, allergic rhinitis and asthma.62)
Recessive X-linked ichthyosis (RXLI)
Genetic defects in the steroid sulfatase gene (STS) were reported to underlie RXLI.3,4) Most STS mutations underlying RXLI are large deletions and, nowadays, fluorescence in situ hybridization (FISH) techniques are a useful tool in detecting causative STS mutations.63) The hyperkeratosis and scaling observed in RXLI are associated with an abnormal accumulation of cholesterol sulfate in the stratum corneum.64) Steroid sulfatase is concentrated in lamellar granules and then secreted into the intercellular spaces of the stratum corneum, along with other lamellar granule-derived lipid hydrolases.65) In those spaces, steroid sulfatase degrades cholesterol sulfate, generating some cholesterol for the barrier. Furthermore, the progressive decline in cholesterol sulfate permits corneodesmosome degradation leading to intact desquamation.65) Thus, two molecular pathways contribute to disease pathogenesis in RXLI. Steroid sulfatase deficiency leads to both malformation of the intercellular lipid barrier, and a delay in corneodesmosome degradation, resulting in corneocyte retension.65) In addition, increased Ca2+ in the intercellular space of the stratum corneum in X-linked ichthyosis has been reported to contribute to corneocyte retention by increasing corneodesmosomes and interlamellar cohesion.65)
PRENATAL DIAGNOSIS OF SEVERE CONGENITAL ICHTHYOSES
The quality of life of patients with severe congenital ichthyoses is seriously affected in some cases, so that parental requests for prenatal diagnosis cannot be easily ignored. Due to the recent advances in our understanding of the genetic defects underlying severe congenital ichthyosis, it has become possible to make DNA-based prenatal diagnoses for congenital ichthyosis families by sampling chorionic villus or amniotic fluid in the earlier stages of pregnancy. That lowers the risk to fetal health and reduces the burden on mothers compared with prenatal diagnoses by fetal skin biopsy.1)
In cases of HI, before identification of ABCA12 as the causative gene, prenatal diagnoses had been performed by fetal skin biopsy and electron microscopic observation at the later stages of pregnancies, 19–23 weeks estimated gestational age, for more than 20 years.66-69) When a fetus was diagnosed as affected, any interruption at the late stage of pregnancy posed a serious problem.
After the identification of ABCA12 as the causative gene for HI, it became feasible to perform DNA-based prenatal diagnoses for HI by chorionic villus or amniotic fluid sampling at a much earlier stage of pregnancy. That significantly lowered risk to fetal health and reduced the burden on mothers, as in the case of other severe genetic disorders.70) Indeed, prenatal diagnoses and the exclusion of HI by DNA testing have been performed in our laboratory.70,71)
Prenatal diagnosis of LI by the ultrastructural observation of fetal skin samples involves a somewhat high-risk, since LI patients can exhibit regional, individual, and familial variability in their disease phenotypes.72) In LI families with TGM1 mutations, successful prenatal DNA-base diagnoses and prenatal exclusions of LI have been reported.73,74) Prenatal diagnosis by mutation analysis in lipoxygenase-3, 12(R)-lipoxygenase and ABCA12, etc. is theoretically available in LI and CIE families with previously identified mutations on a case-by-case basis.
Successful prenatal diagnosis of EI by fetal skin biopsy was reported in the 1980s75) and, at present, prenatal diagnosis by mutation analysis has become feasible for EI in families whose causative mutations have been elucidated.76,77)
CONCLUSION AND REMARKS
As summarized above in this updated review, our knowledge of the molecular genetics and pathogenesis of ichthyosis has advanced dramatically in the last couple of decades. In addition, we now have several powerful tools for the treatment of genetic disorders, such as siRNA gene-silencing technology, read-through compounds to read through nonsense mutations, and improved corrective gene transfer techniques. Fortunately, the skin is the most easily accessible organ for these novel treatment approaches. Thus, based on our knowledge of the pathomechanisms of various ichthyoses described in the present review, I am sanguine about the development of novel, highly effective therapeutic methods in the near future.
REFERENCES
- 1).Akiyama M, Shimizu H. An update on molecular aspects of the non-syndromic ichthyoses. Exp Dermatol, 2008; 17: 373–382. [DOI] [PubMed]
- 2).Oji V, Tadini G, Akiyama M, Blanchet-Bardon C, Bodemer C, Bourrat E, Coudiere P, DiGiovanna JJ, Elias P, Fischer J, Fleckman P, Gina M, Harper J, Hashimoto T, Hausser I, Hennies HC, Hohl D, Hovnanian A, Ishida-Yamamoto A, Jacyk WK, Leachman S, Leigh I, Mazereeuw-Hautier J, Milstone L, Morice-Picard F, Paller AS, Richard G, Schmuth M, Shimizu H, Sprecher E, Van Steensel M, Taïeb A, Toro JR, Vabres P, Vahlquist A, Williams M, Traupe H. Revised nomenclature and classification of inherited ichthyoses: results of the First Ichthyosis Consensus Conference in Sorèze 2009. J Am Acad Dermatol, 2010; 63: 607–641. [DOI] [PubMed]
- 3).Shapiro LJ, Weiss R, Webster D, France JT. X-linked ichthyosis due to steroid-sulfatase deficiency. Lancet, 1978; I: 70–72. [DOI] [PubMed]
- 4).Koppe JG, Marinkovic-Ilsen A, Rijken Y, de Groot WP, Jobsis AC. X-linked ichthyosis. A sulfatase deficiency. Arch Dis Child, 1978; 53: 803–806. [DOI] [PMC free article] [PubMed]
- 5).Chipev CC, Korge BP, Markova N, Bale SJ, DiGiovanna JJ, Compton JG, Steinert PM. A leucine > proline mutation in the H1 subdomain of keratin 1 causes epidermolytic hyperkeratosis. Cell, 1992; 70: 821–828. [DOI] [PubMed]
- 6).Cheng J, Syder AJ, Yu QC, Letai A, Paller AS, Fuchs E. The genetic basis of epidermolytic hyperkeratosis: a disorder of differentiation-specific epidermal keratin genes. Cell, 1992; 70: 811–819. [DOI] [PubMed]
- 7).Rothnagel JA, Dominey AM, Dempsey LD, Longley MA, Greenhalgh DA, Gagne TA, Huber M, Frenk E, Hohl D, Roop DR. Mutations in the rod domains of keratins 1 and 10 in epidermolytic hyperkeratosis. Science, 1992; 257: 1128–1130. [DOI] [PubMed]
- 8).Huber M, Rettler I, Bernasconi K, Frenk E, Lavrijsen SP, Ponec M, Bon A, Lautenschlager S, Schorderet DF, Hohl D. Mutations of keratinocyte transglutaminase in lamellar ichthyosis. Science, 1995; 267: 525–528. [DOI] [PubMed]
- 9).Russell LJ, DiGiovanna JJ, Rogers GR Steinert PM, Hashem N, Compton JG, Bale SJ. Mutations in the gene for transglutaminase 1 in autosomal recessive lamellar ichthyosis. Nat Genet, 1995; 9: 279–283. [DOI] [PubMed]
- 10).Akiyama M. Harlequin ichthyosis and other autosomal recessive congenital ichthyoses: the underlying genetic defects and pathomechanisms. J Dermatol Sci, 2006; 42: 83–89. [DOI] [PubMed]
- 11).Akiyama M, Sugiyama-Nakagiri Y, Sakai K McMillan JR, Goto M, Arita K, Tsuji-Abe Y, Tabata N, Matsuoka K, Sasaki R, Sawamura D, Shimizu H. Mutations in ABCA12 in harlequin ichthyosis and functional rescue by corrective gene transfer. J Clin Invest, 2005; 115: 1777–1784. [DOI] [PMC free article] [PubMed]
- 12).Kelsell DP, Norgett EE, Unsworth H, Teh MT, Cullup T, Mein CA, Dopping-Hepenstal PJ, Dale BA, Tadini G, Fleckman P, Stephens KG, Sybert VP, Mallory SB, North BV, Witt DR, Sprecher E, Taylor AE, Ilchyshyn A, Kennedy CT, Goodyear H, Moss C, Paige D, Harper JI, Young BD, Leigh IM, Eady RA, O’Toole EA. Mutations in ABCA12 underlie the severe congenital skin disease harlequin ichthyosis. Am J Hum Genet, 2005; 76: 794–803. [DOI] [PMC free article] [PubMed]
- 13).Smith FJD, Irvine AD, Terron-Kwiatkowski A, Sandilands A, Campbell LE, Zhao Y, Liao H, Evans AT, Goudie DR, Lewis-Jones S, Arseculeratne G, Munro CS, Sergeant A, O’Regan G, Bale SJ, Compton JG, DiGiovanna JJ, Presland RB, Fleckman P, McLean WH. Loss-of-function mutations in the gene encoding filaggrin cause ichthyosis vulgaris. Nat Genet, 2006; 38: 337–342. [DOI] [PubMed]
- 14).Rothnagel JA, Traupe H, Wojcik S, Huber M, Hohl D, Pittelkow MR, Saeki H, Ishibashi Y, Roop DR. Mutations in the rod domain of keratin 2e in patients with ichthyosis bullosa of Siemens. Nat Genet, 1994; 7: 485–490. [DOI] [PubMed]
- 15).McLean WHI, Morley SM, Lane EB, Eady RA, Griffiths WA, Paige DG, Harper JI, Higgins C, Leigh IM. Ichthyosis bullosa of Siemens-a disease involving keratin 2e. J Invest Dermatol, 1994; 103: 277–281. [DOI] [PubMed]
- 16).Kremer H, Zeeuwen P, McLean WHI, Mariman EC, Lane EB, van de Kerkhof CM, Ropers HH, Steijlen PM. Ichthyosis bullosa of Siemens is caused by mutations in the keratin 2e gene. J Invest Dermatol, 1994; 103: 286–289. [DOI] [PubMed]
- 17).Lefèvre C, Audebert S, Jobard F, Bouadjar B, Lakhdar H, Boughdene-Stambouli O, Blanchet-Bardon C, Heilig R, Foglio M, Weissenbach J, Lathrop M, Prud‘homme JF, Fischer J. Mutations in the transporter ABCA12 are associated with lamellar ichthyosis type 2. Hum Mol Genet, 2003; 12: 2369–2378. [DOI] [PubMed]
- 18).Natsuga K, Akiyama M, Kato N, Sakai K, Sugiyama-Nakagiri Y, Nishimura M, Hata H, Abe M, Arita K, Tsuji-Abe Y, Onozuka T, Aoyagi S, Kodama K, Ujiie H, Tomita Y, Shimizu H. Novel ABCA12 mutations identified in two cases of non-bullous congenital ichthyosiform erythroderma associated with multiple skin malignant neoplasia. J Invest Dermatol, 2007; 127; 2669–2673. [DOI] [PubMed]
- 19).Jobard F, Lefèvre C, Karaduman A, Blanchet-Bardon C, Emre S, Weissenbach J, Ozgüc M, Lathrop M, Prud’homme JF, Fischer J. Lipoxygenase-3 (ALOXE3) and 12(R)-lipoxygenase (ALOX12B) are mutated in non-bullous congenital ichthyosiform erythroderma (NCIE) linked to chromosome 17p13.1. Hum Mol Genet, 2002; 11: 107–113. [DOI] [PubMed]
- 20).Lefèvre C, Bouadjar B, Karaduman A, Jobard F, Saker S, Ozguc M, Lathrop M, Prud’homme JF, Fischer J. Mutations in ichthyin a new gene on chromosome 5q33 in a new form of autosomal recessive congenital ichthyosis. Hum Mol Genet, 2004; 13: 2473–2482. [DOI] [PubMed]
- 21).Lefèvre C, Bouadjar B, Ferrand V, Tadini G, Mégarbané A, Lathrop M, Prud’homme JF, Fischer J. Mutations in a new cytochrome P450 gene in lamellar ichthyosis type 3. Hum Mol Genet, 2006; 15: 767–776. [DOI] [PubMed]
- 22).Akiyama M. Pathomechanisms of harlequin ichthyosis and ABCA transporters in human diseases. Arch Dermatol, 2006; 142: 914–918. [DOI] [PubMed]
- 23).Sakai K, Akiyama M, Sugiyama-Nakagiri Y, McMillan JR, Sawamura D, Shimizu H. Localization of ABCA12 from Golgi apparatus to lamellar granules in human upper epidermal keratinocytes. Exp Dermatol, 2007; 16: 920–926. [DOI] [PubMed]
- 24).Borst P, Elferink RO. Mammalian ABC transporters in health and disease. Annu Rev Biochem, 2002; 71: 537–592. [DOI] [PubMed]
- 25).Peelman F, Labeur C, Vanloo B, Roosbeek S, Devaud C, Duverger N, Denèfle P, Rosier M, Vandekerckhove J, Rosseneu M. Characterization of the ABCA transporter subfamily: identification of prokaryotic and eukaryotic members, phylogeny and topology. J Mol Biol, 2003; 325: 259–274. [DOI] [PubMed]
- 26).Akiyama M, Dale BA, Smith LT, Shimizu H, Holbrook KA. Regional difference in expression of characteristic abnormality of harlequin ichthyosis in affected fetuses. Prenat Diagn, 1998; 18: 425–436. [PubMed]
- 27).Yamanaka Y, Akiyama M, Sugiyama-Nakagiri Y, Sakai K, Goto M, McMillan JR, Ota M, Sawamura D, Shimizu H. Expression of the keratinocyte lipid transporter ABCA12 in developing and reconstituted human epidermis. Am J Pathol, 2007; 171: 43–52. [DOI] [PMC free article] [PubMed]
- 28).Yamano G, Funahashi H, Kawanami O, Zhao LX, Ban N, Uchida Y, Morohoshi T, Ogawa J, Shioda S, Inagaki N. ABCA3 is a lamellar body membrane protein in human lung alveolar type II cells. FEBS Lett, 2001; 508: 221–225. [DOI] [PubMed]
- 29).Shulenin S, Nogee LM, Annilo T, Wert SE, Whitsett JA, Dean M. ABCA3 gene mutations in newborns with fatal surfactant deficiency. N Engl J Med, 2004; 350: 1296–1303. [DOI] [PubMed]
- 30).Yanagi T, Akiyama M, Nishihara H, Sakai K, Nishie W, Tanaka S, Shimizu H. Harlequin ichthyosis model mouse reveals alveolar collapse and severe fetal skin barrier defects. Hum Mol Genet, 2008; 17: 3075–3083. [DOI] [PubMed]
- 31).Zuo Y, Zhuang DZ, Han R, Isaac G, Tobin JJ, McKee M, Welti R, Brissette JL, Fitzgerald ML, Freeman MW. ABCA12 maintains the epidermal lipid permeability barrier by facilitating formation of ceramide linoleic esters. J Biol Chem, 2008; 283: 36624–36635. [DOI] [PMC free article] [PubMed]
- 32).Smyth I, Hacking DF, Hilton AA, Mukhamedova N, Meikle PJ, Ellis S, Slattery K, Collinge JE, de Graaf CA, Bahlo M, Sviridov D, Kile BT, Hilton DJ. A mouse model of harlequin ichthyosis delineates a key role for Abca12 in lipid homeostasis. PLoS Genet, 2008; 4: e1000192. [DOI] [PMC free article] [PubMed]
- 33).Yanagi T, Akiyama M, Nishihara H, Ishikawa J, Sakai K, Miyamura Y, Naoe A, Kitahara T, Tanaka S, Shimizu H. 2010. Self-improvement of keratinocyte differentiation defects during skin maturation in ABCA12 deficient harlequin ichthyosis model mice. Am J Pathol, 2010; 177: 106–118. [DOI] [PMC free article] [PubMed]
- 34).Steinert PM, Marekov LN. The proteins elafin, filaggrin, keratin intermediate filaments, loricirn, and small proline-rich proteins 1 and 2 are isodipeptide cross-linked components of the human epidermal cornified cell envelope. J Biol Chem, 1995; 270: 17702–17711. [DOI] [PubMed]
- 35).Greenberg CS, Birckbichler PJ, Rice RH. Transglutaminases: multifunctional cross-linking enzyme that stabilizes tissues. FASEB J, 1991; 5: 3071–3077. [DOI] [PubMed]
- 36).Kim I-G, McBride OW, Wang M, Kim S-Y, Idler WW, Steinert PM. Structure and organization of the human transglutaminase 1 gene. J Biol Chem, 1992; 267: 7710–7717. [PubMed]
- 37).Yamanishi K, Inazawa J, Liew F-M, Nonomura K, Ariyama T, Yasuno H, Abe T, Doi H, Hirano J, Fukushima S. Structure of the gene for human transglutaminase 1. J Biol Chem, 1992; 267: 17858–17863. [PubMed]
- 38).Laiho E, Ignatius J, Mikkola H, Yee VC, Teller DC, Niemi KM, Saarialho-Kere U, Kere J, Palotie A. Transglutaminase 1 mutations in autosomal recessive congenital ichthyosis: private and recurrent mutations in an isolated population. Am J Hum Genet, 1997; 61: 529–538. [DOI] [PMC free article] [PubMed]
- 39).Akiyama M, Takizawa Y, Kokaji T, Shimizu H. Novel mutations of TGM1 in a child with congenital ichthyosiform erythroderma. Br J Dermatol, 2001; 144: 401–407. [DOI] [PubMed]
- 40).Akiyama M. ABCA12 mutations and autosomal recessive congenital ichthyosis: a review of genotype/phenotype correlations and of pathogenetic concepts. Hum Mutat, 2010; 31: 1090–1096. [DOI] [PubMed]
- 41).Akiyama M, Sakai K, Sugiyama-Nakagiri Y, Yamanaka Y, McMillan JR, Sawamura D, Niizeki H, Miyagawa S, Shimizu H. Compound heterozygous mutations including a de novo missense mutation in ABCA12 led to a case of harlequin ichthyosis with moderate clinical severity. J Invest Dermatol, 2006; 126: 1518–1523. [DOI] [PubMed]
- 42).Akiyama M, Sakai K, Hatamochi A, Yamazaki S, McMillan JR, Shimizu H. Novel compound heterozygous nonsense and missense ABCA12 mutations lead to non-bullous congenital ichthyosiform erythroderma. Br J Dermatol, 2008; 158: 864–867. [DOI] [PubMed]
- 43).Sakai K, Akiyama M, Yanagi T, McMillan JR, Suzuki T, Tsukamoto K, Sugiyama H, Hatano Y, Hayashitani M, Takamori K, Nakashima K, Shimizu H. ABCA12 is a major causative gene for non-bullous congenital ichthyosiform erythroderma. J Invest Dermatol, 2009; 129: 2306–2309. [DOI] [PubMed]
- 44).Krieg P, Marks F, Fürstenberger G. A gene cluster encoding human epidermis-type lipoxygenases at chromosome 17p13.1: cloning, physical mapping, and expression. Genomics, 2001; 73: 300–323. [DOI] [PubMed]
- 45).Yu Z, Schneider C, Boeglin WE, Marnett LJ, Brash AR. The lipoxygenase gene ALOXE3 implicated in skin differentiation encodes a hydroperoxide isomerase. Proc Natl Acad Sci U S A, 2003; 100: 9162–9167. [DOI] [PMC free article] [PubMed]
- 46).Epp N, Fürstenberger G, Müller K, de Juanes S, Leitges M, Hausser I, Thieme F, Liebisch G, Schmitz G, Krieg P. 12R-lipoxygenase deficiency disrupts epidermal barrier function. J Cell Biol, 2007; 177: 173–182. [DOI] [PMC free article] [PubMed]
- 47).Akiyama M, Sakai K, Yanagi T, Tabata N, Yamada M, Shimizu H. Partially disturbed lamellar granule secretion in mild congenital ichthyosiform erythroderma with ALOX12B mutations. Br J Dermatol, 2010; 163: 201–204. [DOI] [PubMed]
- 48).Steinert PM, Yang JM, Bale SJ, Compton JG. Concurrence between the molecular overlap regions in keratin intermediate filaments and the locations of keratin mutations in genodermatoses. Biochem Biophys Res Commun, 1993: 197: 840–848. [DOI] [PubMed]
- 49).Ishida-Yamamoto A, McGrath JA, Judge MR, Leigh IM, Lane E, Eady RAJ. Selective involvement of keratins K1 and K10 in the cytoskeletal abnormality of epidermolytic hyperkeratosis (bullous congenital ichthyosiform erythroderma). J Invest Dermatol, 1992; 99: 19–26. [DOI] [PubMed]
- 50).Müller FB, Huber M, Kinaciyan T, Hausser I, Schaffrath C, Krieg T, Hohl D, Korge BP, Arin MJ. A human keratin 10 knockout causes recessive epidermolytic hyperkeratosis. Hum Mol Genet, 2006; 15: 1133–1141. [DOI] [PubMed]
- 51).Tsubota A, Akiyama M, Kanitakis J, Sakai K, Nomura T, Claudy A, Shimizu H. Mild recessive bullous congenital ichthyosiform erythroderma due to a novel homozygous keratin 10 nonsense mutation. J Invest Dermatol, 2008; 128: 1648–1652. [DOI] [PubMed]
- 52).DiGiovanna JJ, Bale SJ. Epidermolytic hyperkeratosis: applied molecular genetics. J Invest Dermatol, 1994; 102: 390–394. [DOI] [PubMed]
- 53).Traupe H, Kolde G, Hamm H, Happle R. Ichthyosis bullosa of Siemens: a unique type of epidermolytic hyperkeratosis. J Am Acad Dermatol, 1986; 14: 1000–1005. [DOI] [PubMed]
- 54).Akiyama M, Tsuji-Abe Y, Yanagihara M, Nakajima K, Kodama H, Yaosaka M, Abe M, Sawamura D, Shimizu H. Ichthyosis bullosa of Siemens: its correct diagnosis facilitated by molecular genetic testing. Br J Dermatol, 2005; 152: 1353–1356. [DOI] [PubMed]
- 55).Sybert VP, Dale BA, Holbrook KA. Ichthyosis vulgaris: identification of a defect in synthesis of filaggrin correlated with an absence of keratohyaline granules. J Invest Dermatol, 1985; 84: 191–194. [DOI] [PubMed]
- 56).Steinert PM, Cantieri JS, Teller DC, Lonsdale-Eccles JD, Dale BA. Characterization of a class of cationic proteins that specifically interact with intermediate filaments. Proc Natl Acad Sci U S A, 1981; 78: 4097–4101. [DOI] [PMC free article] [PubMed]
- 57).Dale BA, Resing KA, Lonsdale-Eccles JD. Filaggrin: a keratin filament associated protein. Ann NY Acad Sci, 1985; 455: 330–342. [DOI] [PubMed]
- 58).Palmer CNA, Irvine AD, Terron-Kwiatkowski A, Zhao Y, Liao H, Lee SP Goudie DR, Sandilands A, Campbell LE, Smith FJ, O’Regan GM, Watson RM, Cecil JE, Bale SJ, Compton JG, DiGiovanna JJ, Fleckman P, Lewis-Jones S, Arseculeratne G, Sergeant A, Munro CS, El Houate B, McElreavey K, Halkjaer LB, Bisgaard H, Mukhopadhyay S, McLean WH. Common loss-of-function variants of the epidermal barrier protein filaggrin are a major predisposing factor for atopic dermatitis. Nat Genet, 2006; 38: 441–446. [DOI] [PubMed]
- 59).Nomura T, Sandilands A, Akiyama M, Sakai K, Ota M, Sugiura H, Yamamoto K, Sato H, Smith FJD, McLean WHI, Shimizu H. Unique mutations in the filaggrin gene in Japanese patients with ichthyosis vulgaris and atopic dermatitis. J Allergy Clin Immunol, 2007; 119: 434–440. [DOI] [PubMed]
- 60).Akiyama M. FLG mutations in ichthyosis vulgaris and atopic eczema; spectrum of mutations and population genetics. Br J Dermatol, 2010; 162: 472–477. [DOI] [PubMed]
- 61).Nemoto-Hasebe I, Akiyama M, Nomura T, Sandilands A, McLean WHI, Shimizu H. FLG mutation p.Lys4021X in the C-terminal imperfect filaggrin repeat in Japanese patients with atopic eczema. Br J Dermatol, 2009; 161: 1387–1390. [DOI] [PubMed]
- 62).Osawa R, Konno S, Akiyama M, Nemoto-Hasebe I, Nomura T, Nomura Y, Abe R, Sandilands A, McLean WHI, Hizawa N, Nishimura M, Shimizu H. Japanese-specific filaggrin gene mutations in Japanese patients suffering from atopic eczema and asthma. J Invest Dermatol, 2010; 130: 2834–2836. [DOI] [PubMed]
- 63).Valdes-Flores M, Kofman-Alfaro SH, Jimenez-Vaca AL, Cuevas-Covarrubias SA. Carrier identification by FISH analysis in isolated cases of X-linked ichthyosis. Am J Med Genet, 2001; 102: 146–148. [DOI] [PubMed]
- 64).Williams ML, Elias PM. Stratum corneum lipids in disorders of cornification. I. increased cholesterol sulfate content of stratum corneum in recessive x-linked ichthyosis. J Clin Invest, 1981; 68: 1404–1410. [DOI] [PMC free article] [PubMed]
- 65).Elias PM, Crumrine D, Rassner U, Hachem JP, Menon GK, Man W, Choy MH, Leypoldt L, Feingold KR, Williams ML. Basis for abnormal desquamation and permeability barrier dysfunction in RXLI. J Invest Dermatol, 2004; 122: 314–319. [DOI] [PubMed]
- 66).Blanchet-Bardon C, Dumez Y, Labbé F, Lutzner MA, Puissant A, Henrion R, Bernheim A. Prenatal diagnosis of harlequin fetus. Lancet, 1983; I: 132. [DOI] [PubMed]
- 67).Akiyama M, Kim D-K, Main DM, Otto CE, Holbrook KA. Characteristic morphologic abnormality of harlequin ichthyosis detected in amniotic fluid cells. J Invest Dermatol, 1994; 102: 210–213. [DOI] [PubMed]
- 68).Akiyama M, Suzumori K, Shimizu H. Prenatal diagnosis of harlequin ichthyosis by the examinations of keratinized hair canals and amniotic fluid cells at 19 weeks’ estimated gestational age. Prenat Diagn, 1999; 19: 167–171. [PubMed]
- 69).Shimizu A, Akiyama M, Ishiko A, Yoshiike T, Suzumori K, Shimizu H. Prenatal exclusion of harlequin ichthyosis; potential pitfalls in the timing of the fetal skin biopsy. Br J Dermatol, 2005; 153: 811–814. [DOI] [PubMed]
- 70).Akiyama M, Titeux M, Sakai K, McMillan JR, Tonasso L, Calvas P, Jossic F, Hovnanian A, Shimizu H. DNA-based prenatal diagnosis of harlequin ichthyosis and characterization of ABCA12 mutation consequences. J Invest Dermatol, 2007; 127: 568–573. [DOI] [PubMed]
- 71).Yanagi T, Akiyama M, Sakai K, Nagasaki A, Ozawa N, Kosaki R, Sago H, Shimizu H. DNA-based prenatal exclusion of harlequin ichthyosis. J Am Acad Dermatol, 2008b; 58: 653–656. [DOI] [PubMed]
- 72).Holbrook KA, Dale BA, Williams ML, Perry TB, Hoff MS, Hamilton EF, Fisher C, Senikas V. The expression of congenital ichthyosiform erythroderma in second trimester fetuses of the same family: morphologic and biochemical studies. J Invest Dermatol, 1988; 91: 521–531. [DOI] [PubMed]
- 73).Schorderet DF, Huber M, Laurini RN, Von Moos G, Gianadda B, Délèze G, Hohl D. Prenatal diagnosis of lamellar ichthyosis by direct mutational analysis of the keratinocyte transglutaminase gene. Prenat Diagn, 1997; 17: 483–486. [DOI] [PubMed]
- 74).Bichakjian CK, Nair RP, Wu WW, Goldberg S, Elder JT. Prenatal exclusion of lamellar ichthyosis based on identification of two new mutations in the transglutaminase 1 gene. J Invest Dermatol, 1998; 110: 179–182. [DOI] [PubMed]
- 75).Golbus MS, Sagebiel RW, Filly RA, Gindhart TD, Hall YG. Prenatal diagnosis of congenital bullous ichthyosiform erythroderma (epidermolytic hyperkeratosis) by fetal skin biopsy. N Eng J Med, 1980; 302: 93–95. [DOI] [PubMed]
- 76).Rothnagel JA, Longley MA, Holder RA, Küster W, Roop DR. Prenatal diagnosis of epidermolytic hyperkeratosis by direct gene sequencing. J Invest Dermatol, 1994; 102: 13–16. [DOI] [PubMed]
- 77).Tsuji-Abe Y, Akiyama M, Nakamura H, Takizawa Y, Sawamura D, Matsunaga K, Suzumori K, Shimizu H. DNA-based prenatal exclusion of bullous congenital ichthyosiform erythroderma at the early stage, 10–11 weeks’ of pregnancy in two consequent siblings. J Am Acad Dermatol, 2004; 51: 1008–1011. [DOI] [PubMed]