

Background and Purpose
The synthetic peptide PnPP‐19 has been studied as a new drug candidate to treat erectile dysfunction. However, PnTx2–6, the spider toxin from which the peptide was designed, induces hyperalgesia. Therefore, we intended to investigate the role of PnPP‐19 in the nociceptive pathway.
Experimental Approach
Nociceptive thresholds were measured by paw pressure test. PnPP‐19 was administered intraplantarly alone or with selective cannabinoid or opioid receptor antagonists. The hydrolysis of PnPP‐19 by neutral endopeptidase (NEP) (EC 3.4.24.11), an enzyme that cleaves enkephalin, was monitored by HPLC and the cleavage sites were deduced by LC–MS. Inhibition by PnPP‐19 and Leu‐enkephalin of NEP enzyme activity was determined spectrofluorimetrically.
Key Results
PnPP‐19 (5, 10 and 20 μg per paw) induced peripheral antinociception in rats. Specific antagonists of μ opioid receptors (clocinnamox), δ opioid receptors (naltrindole) and CB1 receptors (AM251) partly inhibited the antinociceptive effect of PnPP‐19. Inhibition of fatty acid amide hydrolase by MAFP or of anandamide uptake by VDM11 enhanced PnPP‐19‐induced antinociception. NEP cleaved PnPP‐19 only after a long incubation, and K i values of 35.6 ± 1.4 and 14.6 ± 0.44 μmol·L−1 were determined for PnPP‐19 and Leu‐enkephalin respectively as inhibitors of NEP activity.
Conclusions and Implications
Antinociception induced by PnPP‐19 appears to involve the inhibition of NEP and activation of CB1, μ and δ opioid receptors. Our data provide a greater understanding of the antinociceptive effects of PnPP‐19. This peptide could be useful as a new antinociceptive drug candidate.
Abbreviations
- Ki
inhibitory constant
- NEP
neutral endopeptidase
Tables of Links
| TARGETS |
|---|
| GPCRs a |
| CB1 receptors |
| CB2 receptors |
| δ receptors |
| κ receptors |
| μ receptors |
| Enzymes b |
| FAAH, fatty acid amide hydrolase |
| NEP, neutral endopeptidase |
| LIGANDS |
|---|
| AM251 |
| AM630 |
| MAFP |
| Naltrindole |
| Nor‐binaltorphimine |
| Naloxone |
| PGE2 |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (a,bAlexander et al., 2015a, 2015b).
Introduction
Animal venoms have been used as sources of new compounds with specific pharmacological properties, constituting potential tools for neurobiological studies and also potential new drug candidates. Particularly, the venom of the Brazilian armed spider Phoneutria nigriventer contains a large range of peptide toxins, which have several activities in biological systems. Toxins from this venom have been described to be acting on many different targets, such as sodium, calcium and potassium voltage‐gated ion channels (Matavel et al., 2002; de Lima et al., 2015; Silva et al., 2015). Some of these toxins are also able to induce an increase in vascular permeability (Marangoni et al., 1993) and potentiate penile erection (Nunes et al., 2008) Furthermore, other venom components can inhibit glutamate uptake by brain synaptosomes (Mafra et al., 1999), while others inhibit NMDA‐evoked currents in rat hippocampal neurons (Figueiredo et al., 2001).
Interestingly, two toxins isolated from the venom of P. nigriventer have been suggested as potential drug sources for pain treatment. These toxins, PnTx3–3 and PnTx3–6, inhibit voltage‐activated calcium channels and induce antinociceptive effect (Souza et al., 2008; Dalmolin et al., 2011). However, to the best of our knowledge, none of the toxins from the venom of this spider have been assessed for analgesic activity due to activation of opioid or cannabinoid receptors.
Recently, our group has shown the involvement of the opioid and endocannabinoid systems in the mechanism of action of different substances, such as ketamine and xylazine (Romero et al., 2013b; Pacheco et al., 2014). There are two types of cannabinoid receptors, CB1 receptors that are expressed primarily in central and peripheral neurons and CB2 receptors mainly found in immune cells (see Pertwee, 2006). For the opioid receptors, there are three types, μ, δ and κ receptors, and all of them are expressed in both central and peripheral nervous system (Peng et al., 2012). In addition, our group has demonstrated that the opioidergic and endocannabinoidergic systems are strongly linked and the activation of one pathway is mediated by the other (Pacheco et al., 2008; Pacheco et al., 2009; Reis et al., 2009).
The toxin PnTx2–6 was isolated from PhTx2 fraction of the P. nigriventer venom (Cordeiro et al., 1992), and induces a different range of biological effects from those of PnTx3–3 and PnTx3–6. The toxin PnTx2–6 causes neuronal depolarization by slowing down the inactivation of Na+ channels (Matavel et al., 2002), and it also induces penile erection (Nunes et al., 2008). When this toxin was injected s.c. into the rat hind paw (0.57 nmol per paw) and the nociceptive threshold measured by the paw pressure test (Randall and Selitto, 1957), it induced hyperalgesia in both the toxin‐treated and the saline‐injected hind paw. For this reason, the authors concluded that this toxin, PnTx2–6, had a systemic nociceptive effect, even when administered at low doses (K P Nunes, unpublished data).
The peptide PnPP‐19 represents a discontinuous epitope of the primary structure of the toxin PnTx2–6 and it was proposed as the most likely region of the toxin to interact with its molecular target, the sodium channel (Silva et al., 2015). Our group has synthesized the peptide PnPP‐19 and has shown that, similar to the native toxin PnTx2–6, PnPP‐19 potentiates erectile function in rats. However, it no longer acts on any sodium channel subtypes (Silva et al., 2015).
Given the potential use of PnPP‐19 as a drug to treat erectile dysfunction and the lack of information concerning its effect in the nociceptive pathway, the present work aimed to determine the effects of PnPP‐19 on nociception. Our results have shown that the peptide exhibited antinociceptive activity, mediated by activation of both opioid and cannabinoid receptors in the peripheral nervous system. We also demonstrated the involvement of the endocannabinoid system, because inhibitors of anandamide metabolism, by fatty acid amide hydrolase (FAAH), and of its uptake potentiated the antinociceptive effect induced by the peptide. In addition, PnPP‐19 inhibited the neutral endopeptidase (NEP) (EC 3.4.24.11), which is responsible for the cleavage of many endogenous peptides, among them, the opioid enkephalin (see Roques et al., 1993).
Methods
Animals
All animal care and experimental protocols were approved by the local Ethics Committee on Animal Experimentation (CETEA) of UFMG (Protocol number: 131/2014) and are reported in accordance with ARRIVE guidelines (Kilkenny et al., 2010; McGrath & Lilley, 2015). Efforts were made to minimize suffering and reduce the number of animals used in the experiments.
Male Wistar rats (170–220 g) provided by the CEBIO (The Animal Centre) of Universidade Federal de Minas Gerais (UFMG) were used in the experiments. The rats were housed in groups of a maximum of four animals per cage at a temperature‐controlled room (23 ± 1°C) on an automatic 12 h light/dark cycle (06:00–18:00 h of light phase). All testing was carried out during the light phase (08:00–15:00 h). Food and water were freely available until the onset of the experiments. In this work, all the tested groups comprised 4 animals and a total of 152 animals were used to provide all the data.
Algesimetric method
Rats were injected with PGE2 (2 μg) in the plantar surface (s.c.) of the right hind paw and measured by the paw‐pressure test described by Randall and Selitto (1957). An analgesimeter (Ugo‐Basile, Italy) with a cone‐shaped paw presser with a rounded tip was used to apply a linearly increasing force to the hind paw. The weight in grams required to elicit the nociceptive response, paw withdrawal, was determined as the nociceptive threshold. A cut‐off value of 300 g was used to prevent damage to the paws. The nociceptive threshold was measured in the right paw and determined by the average of three consecutive trials recorded before (zero time) and 3 h after PGE2 injection (peak of effect). The results were calculated by the difference between these two averages (Δ of nociceptive threshold) and expressed as grams. To reduce stress, the rats were habituated to the apparatus for one day prior to the experiments.
Experimental protocol
Dose–response curves were obtained by injecting the peptide PnPP‐19 (50 μL) 3 h after local administration of PGE2 (100 μL) into the hind paw and the nociceptive response was measured every 5 min, for 30 min. In the protocol used to determine whether the drug was acting outside the injected paw, PGE2 (100 μL) was injected into both hind paws (left and right), while PnPP‐19 (50 μL) was administered into the right paw; only in this experiment, were both right and left paws assessed. After determination of the dose and the peak of action of PnPP‐19, the next experiments were carried out using the injection of the peptide, concomitantly with opioid or cannabinoid antagonists as follows: PnPP‐19 (50 μL) was administered s.c. in the right hind paw 2:55 h after local injection of PGE2 (100 μL). Naloxone, clocinnamox, naltrindole or nor‐BNI (50 μL) was intraplantarly injected into the right hind paw, 35 min prior to the measurement of hyperalgesia (3 h). AM251, AM630, MAFP and VDM11 (50 μL) were intraplantarly injected 15 min prior to the measurement of hyperalgesia (3 h). The nociceptive threshold was always measured in the right hind paw. This protocol was assessed in pilot experiments and published data was used to determine the dose and optimal time point for the injection of each substance (Pacheco et al., 2008; Reis et al., 2009; Galdino et al., 2014; Veloso et al., 2014).
Hydrolysis of PnPP‐19 by NEP
A recombinant soluble form of human NEP was prepared as previously described (Lemay et al., 1989; Fossiez et al., 1992), and it was kindly donated by Dr. Guy Boileau from the University of Montreal (Montreal, Canada). PnPP‐19 (20 μmol·L−1) was incubated with recombinant NEP (0.2875 nmol·L−1) in Tris–HCl (25 mmol·L−1) buffer containing 0.1 mol·L−1 NaCl, pH 7.0, 37°C. Aliquots from the incubated solutions were taken at appropriate time points (15 min, 30 min, 1 h and overnight), and the reaction was stopped in 5% v/v TFA (trifluoroacetic acid) solution. The samples were analysed by HPLC (Shimadzu CBM‐20 A) with UV detection at 220 and 280 nm, using RP‐18E column [C18‐Hewlett Packard (Palo Alto, CA, USA)]. The column was eluted with a two‐solvent system: solvent A, TFA/H2O (1:1000, v/v) and solvent B, TFA/acetonitrile/H2O (1:900:100, v/v/v), at a flow rate of 1 mL·min−1 with 10–80% gradient of solvent B over 20 min.
Determination of PnPP‐19 cleavage sites
For the determination of PnPP‐19 cleavage sites, a HPLC system connected to a MS detector (LC/MS), model LCMS 2010 EV (Shimadzu Inc, Nakagyo‐ku, Kyoto, Japan) was used. Electrospray ionization probe was used for data analysis. Fragments resulting from NEP hydrolysis of PnPP‐19 were isolated by HPLC using a C18 column (CLC – ODS Shimadzu 4, 6 × 150 mm). The column was eluted with the two‐solvent system: solvent A, TFA/H2O (1:1000, v/v) and solvent B, TFA/ acetonitrile /H2O (1:900:100, v/v/v) at a flow rate of 1 mL·min−1 with 10–80% gradient of solvent B over 20 min.
Determination of the inhibitory activity of PnPP‐19 and Leu‐enkephalin towards NEP
The inhibition of NEP activity by PnPP‐19 and Leu‐enkephalin was assessed by determining the inhibitory constant (K i), using the selective NEP fluorogenic substrate Abz‐(d)Arg‐Gly‐Leu‐Eddnp (Barros et al., 2007) and appropriate concentrations of PnPP‐19 or Leu‐enkephalin, as the inhibitors. The hydrolysis was monitored using a spectrofluorimeter (Shimadzu‐RF‐5301pc) calibrated with wavelengths of λem = 420 nm and λex = 320 nm. The assays were carried out in Tris–HCl (25 mmol·L−1) buffer containing NaCl (100 mmol·L−1), pH 7.0 at 37°C. The enzyme concentration used was 0.23 nmol·L−1 and the FRET substrate concentration was 4.95 μmol·L−1. The substrate solution was kept in a thermostatic chamber at 37°C for 5 min before the addition of NEP. After the determination of NEP activity in the absence of inhibitors, cumulative concentrations of PnPP‐19 or Leu‐enkephalin were added every 1 min during the experiment in order to induce a decrease of the hydrolysis rate of the fluorescent substrate. The fluorescence was continuously followed, and the apparent inhibition constant (K iapp) values were obtained using the equation:
where v o and v i are the velocity of less than 2% of substrate hydrolysis in absence and in presence of different inhibitor concentrations [I]. The assays were performed in duplicate, and the K i parameters were obtained from the equation:
The K i values for the NEP inhibitors were calculated by the tight‐binding titration data analysis grafit version 5 programme (Erithacus Software Ltd., Horley, UK).
Data and statistical analysis
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). Results are presented as means ± SEM, unless otherwise stated. Statistical analysis was carried out using graphprism software. Our data were distributed normally and analysed statistically by one‐way anova with post hoc Bonferroni's test for multiple comparisons. Probabilities less than 5% (P < 0.05) were considered to show statistically significant differences between means.
Materials
The following drugs and chemicals were used: PGE2 (Enzo Life Science, Farmingdale, NY, USA); PnPP‐19 (synthesized by ChinaPeptides, Shanghai, China); naloxone (Sigma, USA); clocinnamox (Tocris, Ellisville, MO, USA); naltrindole (Tocris); nor‐binaltorphimine dihydrochloride (Nor‐BNI) (Tocris), N‐(piperidin‐1‐yl)‐5‐(4‐iodophenyl)‐1‐(2,4‐dichlorophenyl)‐4‐methyl‐1 H‐pyrazole‐3‐carboxamide (AM251) (Tocris), [6‐iodo‐2‐methyl‐1‐(2‐morpholin‐4‐ylethyl)indol‐3‐yl]‐(4‐methoxyphenyl)methanone (AM630) (Tocris), (5Z,8Z,11Z,14Z)‐5,8,11,14‐eicosatetraenyl‐methyl ester phosphonofluoridic acid (MAFP) (Tocris) and (5Z,8Z,11Z,14Z)‐N‐(4‐hydroxy‐2‐methylphenyl)‐5,8,11,14‐eicosatetraenamide (VDM11) (Tocris). The drugs were dissolved as follows: PGE2 (ethanol 2% in saline); naloxone, clocinnamox, naltrindole and nor‐BNI (saline); AM251 and AM630 (12% DMSO in saline), MAFP and VDM11 (10% DMSO in saline) and injected in a volume of 50 μL per paw. The FRET substrate Abz‐(d)RGL‐EDDnp, containing ortho‐aminobenzoyl (Abz) and N‐(2,4‐dinitrophenyl)ethylenediamine (EDDnp) as donor/acceptor pair, was purchased from Amino Tech (São Paulo, Brazil).
Results
PnPP‐19 exhibited peripheral antinociceptive effects
First, to investigate the role of PnPP‐19 in nociception, the peptide was injected (5, 10 and 20 μg per paw) into rat paws‐that were hyperalgesic following the administration of PGE2. PnPP‐19 induced a dose‐dependent antinociceptive response, with the maximal effects at 10 or 20 μg per paw (Figure 1A). Further assays showed that the antinociceptive effect of PnPP‐19 (10 μg per paw) was peripheral, because its effect was restricted to the peptide‐treated paw (right paw; Figure 1B).
Figure 1.
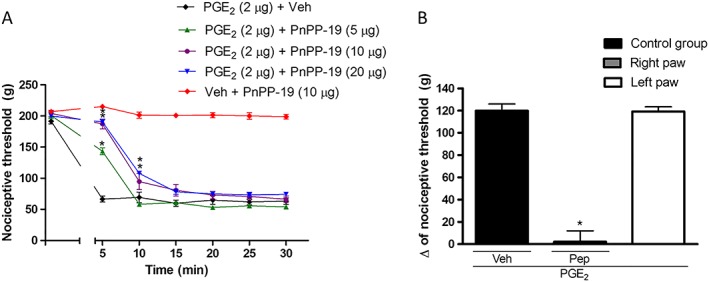
Peripheral antinociceptive effect of PnPP‐19 on PGE2 – induced hyperalgesia in rats. (A) PnPP‐19 (5, 10 and 20 μg) was administered 3 h after local administration of PGE2 (2 μg per paw), and the nociceptive or antinociceptive response was measured every 5 min, for 30 min. Injection of 10 μg of PnPP‐19 alone did not affect the nociceptive threshold. (B) PGE2 (2 μg) was administered in both right and left hind paws, followed by an injection of PnPP‐19 (Pep; 10 μg) only into the right paw (only in this experiment, both right and left paw were measured). Both peptide and saline (as vehicle; Veh) were administered at 2 h and 55 min after local administration of PGE2. The nociceptive or antinociceptive response was followed in both paws. The response in both assays was measured through the paw pressure test, as described in Material and Methods. Data shown are the means ± SEM (n = 4). *P < 0.05 compared with PGE2 + Veh (ANOVA + Bonferroni's test). Veh: saline.
PnPP‐19‐induced peripheral antinociception is mediated by μ and δ opioid receptors
The bioinformatic assay chemoinformatic similarity ensemble approach (SEA) data bank was used as a tool to predict a molecular target for the PnPP‐19 peptide. This programme generated many possible targets. However, the enzyme NEP was shown as the most likely target for the peptide with the opioid receptors also among the first group of possibilities. In addition, because the opioid pathway is associated with the mechanism of action of various analgesic drugs, we investigated the participation of this pathway in the antinociceptive response induced by PnPP‐19.
Intraplantar administration of the non‐specific opioid receptor antagonist naloxone (100 and 200 μg per paw; Figure 2A), the μ receptor antagonist clocinnamox (40 and 80 μg per paw; Figure 2B) or the δ receptor antagonist naltrindole (60 and 120 μg per paw; Figure 2C) partly inhibited the antinociceptive effect of PnPP‐19 (10 μg per paw). On the other hand, administration of the κ receptor antagonist nor‐BNI (100 μg per paw; Figure 2D) did not modify the antinociception elicited by the peptide. The effect of the highest effective dose of all tested antagonists did not differ from the hyperalgesic control (2 μg per paw of PGE2 + vehicle; Figure 2A–D).
Figure 2.
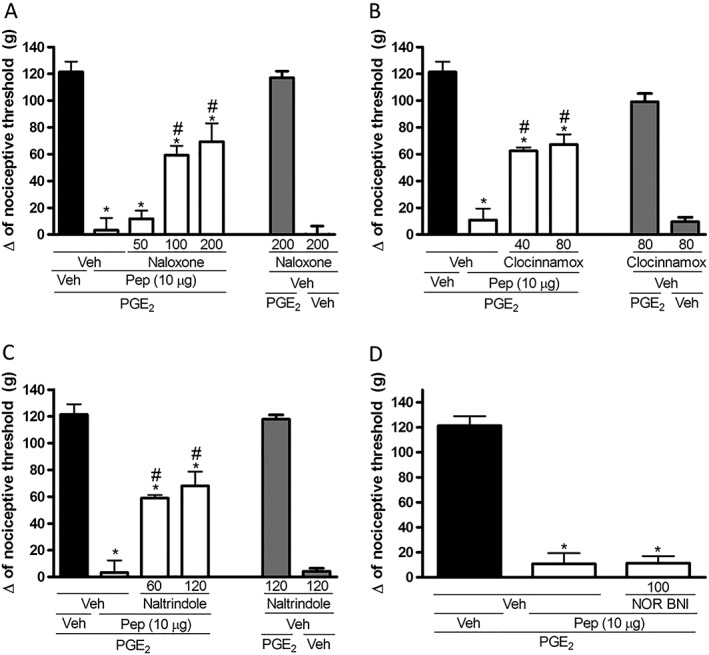
Effect of s.c. administration of opioid receptor antagonists on the peripheral antinociception produced by PnPP‐19. (A) Non‐specific opioid receptor antagonist naloxone (50, 100, 200 μg per paw), (B) μ‐opioid receptor antagonist clocinnamox (40 and 80 μg per paw), (C) δ‐opioid receptor antagonist naltrindole (60 and 120 μg per paw) and (D) κ opioid receptor antagonist nor‐binaltorphimine (100 μg per paw) were administered 30 min before the injection of PnPP‐19 (10 μg per paw). PnPP‐19 was administered at 2 h and 55 min after local administration of PGE2 (2 μg per paw). The response was measured by the paw‐pressure test. Data are shown as the mean ± SEM (n = 4); *P < 0.05 compared with PGE2 + Veh + Veh and # P < 0.05 compared with PGE2 + Veh + PnPP‐19 (10 μg per paw) (ANOVA + Bonferroni's test). Veh: saline; Pep: PnPP‐19.
NEP enzymatic activity over PnPP‐19
Following the results from the SEA data bank, we investigated whether PnPP‐19 could act as a substrate or as an inhibitor of NEP. When the peptide was incubated with recombinant NEP for 1 h, the enzyme did not cleave PnPP‐19 at any site (data not shown). On the other hand, when the PnPP‐19 was incubated with NEP for a longer period (overnight), the enzyme cleaved the peptide at six different sites (Figure 3), all of them with a hydrophobic amino acid residue at the P1’ position (according to the Schechter and Berger nomenclature – Schechter and Berger, 1968).
Figure 3.

Neutral endopeptidase (NEP) cleavage sites (arrows) in PnPP‐19. PnPP‐19 (20 μmol·L−1) was incubated overnight at 37°C with NEP (0.2875 nmol·L−1). Samples were analysed by LCMS for the determination of cleavage sites.
Next, we determined the inhibitory constant (K i) of Leu‐enkephalin and PnPP‐19 as inhibitors of NEP catalytic activity using the fluorogenic synthetic substrate Abz‐(d)RGL‐EDDnp (Figure 4A and B). The K i values obtained for Leu‐enkephalin and PnPP‐19 were 14.6 ± 0.44 and 35.6 ± 1.4 μmol·L−1 respectively.
Figure 4.

Determination of the inhibitory constant (K i) of Leu‐enkephalin (A) or PnPP‐19 (B) on the hydrolytic activity of neutral endopeptidase (NEP). The assays were performed using the FRET‐substrate Abz‐(d)Arg‐Gly‐Leu‐Eddnp. Inset: Residual activity in the presence of different inhibitors concentrations.
The cannabinoid CB1 receptor is involved in the peripheral antinociception induced by PnPP‐19
Because the opioid and cannabinoid pathways are known to interact (Befort, 2015), we investigated whether the activation of cannabinoid receptors was also involved in the antinociceptive response induced by PnPP‐19 (10 μg per paw). Intraplantar administration of the CB1 receptor antagonist AM251 (80 and 160 μg per paw) partly inhibited PnPP‐19‐induced peripheral antinociception (Figure 5A). However, the CB2 receptor antagonist AM630 (100 μg per paw) did not modify the peripheral antinociceptive effects of PnPP‐19 (Figure 5B). The antagonists by themselves did not significantly modify the nociceptive threshold of the control groups when injected together with PGE2 or vehicle.
Figure 5.
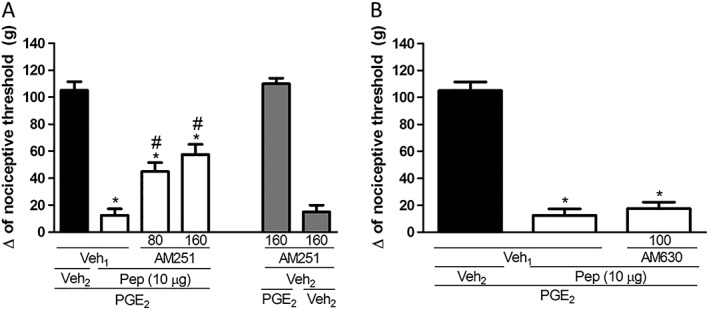
Effect induced by intraplantar administration of AM251 (A) or AM630 (B) on the peripheral antinociception produced by PnPP‐19. AM251 (80 and 160 μg per paw) or AM630 (100 μg per paw) were administered 10 min prior to injection of PnPP‐19 (10 μg per paw). PnPP‐19 was administered at 2 h and 55 min after local administration of PGE2 (2 μg per paw). The response was measured by the paw pressure test. Data are shown as the mean ± SEM (n = 4); *P < 0.05 compared with PGE2 + Veh1 + Veh2 and # P < 0.05 compared with PGE2 + Veh1 + PnPP‐19 (10 μg per paw) (ANOVA + Bonferroni's test). Veh1: 12% DMSO in saline; veh2: saline; pep: PnPP‐19.
Increase of PnPP‐19‐induced antinociception by MAFP and VDM11
Because PnPP‐19 induces activation of CB1 receptors and the endogenous cannabinoid anandamide is slightly selective for these receptors (Lin et al., 1998), we used MAFP, an inhibitor of the major anandamide metabolizing enzyme, fatty acid amide hydrolase (FAAH) and the anandamide uptake inhibitor VDM11 to confirm the potentiation of the effects of PnPP‐19 on the nociceptive pathway. Both MAFP (2 and 4 μg per paw; Figure 6A) and VDM11 (20 μg per paw; Figure 6B) enhanced the antinociception induced by a low dose of PnPP‐19 (5 μg per paw). MAFP and VDM11 given alone did not induce any effect.
Figure 6.
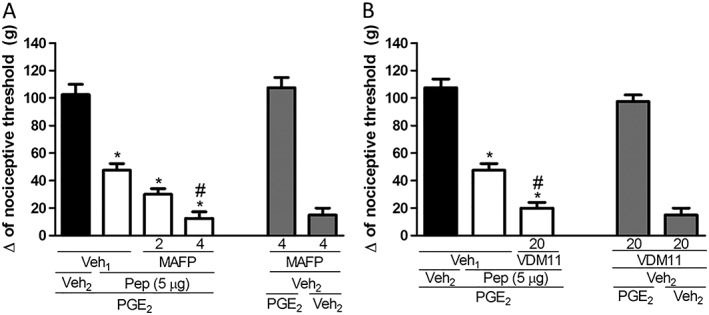
Potentiation of PnPP‐19‐induced antinociception by the FAAH inhibitor MAFP and anandamide uptake inhibitor VDM11. The MAFP (2 and 4 μg per paw) and VDM11 (20 μg per paw) were administered 10 min prior to PnPP‐19 (5 μg per paw). PnPP‐19 was administered at 2 h and 55 min after local administration of PGE2 (2 μg per paw). The response was measured by the paw pressure test. Data are expressed the mean ± SEM (n = 4); *P < 0.05 compared with PGE2 + Veh1 + Veh2 and # P < 0.05 compared with PGE2 + Veh1 + PnPP‐19 (5 μg per paw) (ANOVA + Bonferroni's test). Veh1: 10% DMSO in saline; veh2: saline; pep: PnPP‐19.
Discussion and conclusions
Because PnPP‐19 has been suggested as a treatment for erectile dysfunction (Silva et al., 2015) and also taking into account that PnTx2–6 (the toxin used as a model to obtain PnPP‐19) showed nociceptive effects in rats (K Nunes, unpublished data), we decided to investigate if PnPP‐19 could induce a hyperalgesic response, similar to the native toxin. However, instead of eliciting pain, PnPP‐19 induced a dose‐dependent antinociception in our rat model.
Initially, the ability of PnPP‐19 to induce peripheral antinociception was investigated. To achieve this, we decided to use PGE2 to induce hyperalgesia. PGs are considered as a prototype of potent direct sensitizers in animal models by stimulating a decrease of primary sensory neurons resting potential through activation of Gs protein‐coupled receptors. The activation of such receptors sensitizes sodium and calcium channels and suppresses outward potassium currents (Meves, 2006). According to Ferreira (1972), a single injection of PGE2 is capable of sensitizing nociceptors to mechanical and chemical stimuli. The use of such substance as an inducer of hyperalgesia presents, over other models of hyperalgesic induction, such as the use of the inflammatory molecule carrageenan, the advantage of eliminating the possibility that the peripheral effects of the tested compound are the results of its interaction and modulation of the mediators produced during the inflammatory process. Our results are in agreement with previous studies, which demonstrate that PGE2 produces an intense nociceptive effect when administered peripherally, at a dose of 2 μg per paw (Pacheco et al., 2008; Veloso et al., 2014). Therefore, using PGE2 to induce hyperalgesia in our model, we demonstrated that PnPP‐19 produced a peripheral antinociceptive effect, in a dose‐dependent manner.
Among the venomous animals, spiders comprise the group containing the largest number of species (Platnick, 1997). Many spider toxins induce antinociceptive effects, mainly by the interaction with ion channels. However, some toxins exert their antinociceptive activity by affecting glutamatergic neurotransmission or by inhibiting P2X3 receptors (see Gazerani and Cairns, 2014). On the other hand, up to date, none of the spider toxins have been described as interacting with opioid or cannabinoid systems.
The involvement of opioid receptors in central and peripheral antinociception has been extensively studied over the last few decades. Only a few animal toxins are known to induce an antinociceptive effect due to activation of the opioid system, including a neurotoxin from the venom of the king cobra (Ophiophagus hannah), the crude venom of the snake Micrurus lemniscatus and two scorpion toxins, AmmVIII and LqqIT2 (Pu et al., 1995; Martin‐Eauclaire et al., 2010; Leite dos Santos et al., 2012).
The opioid receptors belong to the superfamily of GPCRs and they are coupled to Gi/Go proteins. Many studies have focused on elucidating the molecular mechanisms triggered by opioid receptor signalling. These include the reduction of neuronal excitability by inhibition of EPSCs evoked by NMDA receptors, calcium channels and adenyl cyclase activity, in conjunction with a stimulation of potassium channels (see Law et al., 2000). Therefore, opioid peptides inhibit the sensitization of primary afferent neurons promoted by PGE2 through activation of those receptors. Several molecules, which do not bind to opioid receptors, are still able to induce antinociception, indirectly, via activation of this pathway. Examples of the indirect analgesics are xylazine, an agonist at the α2‐adrenoceptor, and ketamine, a NMDA receptor antagonist (Romero et al., 2013b; Pacheco et al., 2014).
In this work, the SEA data bank suggested that the opioid pathway and NEP would be the main targets for PnPP‐19. None of the spider toxins described to elicit pain relief act on these receptors nor is there any spider toxin known to interact with NEP (Gazerani and Cairns, 2014). In agreement with the results generated from the SEA data bank, we found that the antinociceptive effects of PnPP‐19‐ were partly due to the activation of μ and δ opioid receptors. It is well established that these two types of receptors will form heterodimers and the activation of one receptor of the heterodimer can affect the signalling pathway of the other, which is in accordance with our results (Gupta et al., 2010; Gomes et al., 2011). Interestingly, sildenafil, a drug currently used to treat erectile dysfunction, also induces antinociception through the activation of the same receptors (Yoon et al., 2008).
We also found that PnPP‐19 inhibited NEP, an enzyme responsible for the cleavage of many endogenous peptides, among them, the opioid peptide enkephalin (see Roques et al., 1993). The inhibitory constants of PnPP‐19 and Leu‐enkephalin towards NEP catalytic activity were similar. However, NEP only cleaved PnPP‐19 after a long period of incubation (overnight). Thus, although PnPP‐19 is a substrate for NEP, it might have a low catalytic constant (k cat). Therefore, we suggest that when PnPP‐19 is administered in vivo, it competes with the endogenous Leu‐enkephalin for the catalytic site of NEP, thereby increasing the levels of the endogenous opioid and causing the antinociceptive response. Leu‐enkephalin is known to activate both μ and δ receptors (Hruby, 2002), the receptors that appeared to be involved in the peripheral antinociception induced by PnPP‐19. In addition, NEP is a zinc metallopeptidase, which has specificity for cleaving substrates containing hydrophobic aliphatic or aromatic amino acids in the P1’ position (Turner et al., 1985; Hersh and Morihara, 1986). In agreement with this specificity, we found the NEP to cleave PnPP‐19 at six different sites, all of them close to hydrophobic amino acid residues.
The endogenous inhibitor of NEP in humans is called opiorphin (Wisner et al., 2006), and the one found in rats (Rattus norvegicus) is called sialorphin (Rougeot et al., 2003). Both of these endogenous inhibitors exhibit antinociceptive effects mediated by activation of μ and δ receptors (Rougeot et al., 2003; Wisner et al., 2006), as observed with PnPP‐19. In addition, the gene expression of opiorphin is down‐regulated in patients reporting erectile dysfunction (Tong et al., 2007; Tong et al., 2008). It reinforces our previous results showing that PnPP‐19 potentiates erectile function (Silva et al., 2015) and also highlights the role of NEP on this pathway.
The interaction of cannabinoid and opioid pathways has been extensively reported. The close vicinity of CB1 receptors with μ or δ receptors at the neuronal level has been shown (Befort, 2015), and the heterodimerization of cannabinoid and opioid receptors has been described (Rios et al., 2006; Bushlin et al., 2012). In addition, activation of cannabinoid receptors stimulates the release of endogenous opioid peptides (Ibrahim et al., 2005). Our group demonstrated that the central and peripheral antinociceptive effect induced by the exogenous μ receptor agonist, morphine, was mediated by activation of CB1 receptors (Pacheco et al., 2008; Pacheco et al., 2009). There is also evidence that the antinociception elicited by anandamide is mediated by activation of opioid receptors (Reis et al., 2009). In this study, the observed interaction between both systems could also explain part of the mechanism of action of PnPP‐19 on the nociceptive pathway, because the peptide PnPP‐19 induced peripheral antinociception partly through the activation of μ and δ receptors and CB1 receptors. We also investigated the possible involvement of CB2 receptors in PnPP‐19‐induced antinociception. Administration of a high dose (100 μg per paw) of the selective CB2 receptor antagonist AM630 (Romero et al., 2013a) did not inhibit the effect of the peptide. Because AM251, a selective CB1 receptor antagonist, partly inhibited the antinociception induced by the peptide, and that cannabinoid peripheral antinociception is mainly mediated by activation of CB1 receptors (Agarwal et al., 2007), we concluded that the activation of CB2 receptors might not be required for the antinociception elicited by PnPP‐19.
The CB1 receptor is expressed both in central and peripheral nervous systems (Herkenham et al., 1990; Hohmann et al., 1999; Fox et al., 2001) and it is the main target for endocannabinoids and exogenous cannabinoids in the peripheral nervous system (Agarwal et al., 2007). Interestingly, besides the analgesic effect of cannabinoids, they are also involved in erectile function. For instance, cannabinoids are involved in priapism (Matta et al., 2014), and the endogenous cannabinoid anandamide induced relaxation of cavernosal tissue (Ghasemi et al., 2006). Among the cannabinoid receptors, only CB1 receptors are expressed in rat corpus cavernosum tissue (Ghasemi et al., 2006).
The involvement of endocannabinoids in pain modulation might be assessed indirectly by administration of pharmacological agents that inhibit endocannabinoid uptake or metabolism (Hohmann and Suplita, 2006) and such inhibitors have been used as a pharmacological strategy to maximize the effects of the endogenously released cannabinoids. The endogenous cannabinoid anandamide is an agonist of both CB1 and CB2 receptors although it presents marginally greater affinity for CB1 receptors (K i: 61.0 nM) than for CB2 receptors (K i: 1930 nM) (Lin et al., 1998). In addition, the peripheral antinociception induced by anandamide injected into the rat hind paw is mainly elicited by activation of CB1 and not by CB2 receptors (Reis et al., 2009). It has been proposed that the biological action of anandamide is rapidly terminated by a re‐uptake system, the anandamide membrane transporter, which transports anandamide into the cell where it is hydrolyzed (Di Marzo et al., 1994). The enzyme primarily responsible for the hydrolysis of anandamide is FAAH (Hohmann and Suplita, 2006). In this study we administered an inhibitor of this enzyme, as well as a potent and selective inhibitor of the anandamide membrane transporter, in order to evaluate the involvement of endogenous cannabinoids in the peripheral antinociceptive effect induced by an injection of a low dose of PnPP‐19 (5 μg/paw). Inhibition of FAAH and of the anandamide membrane transporter potetntiated the peripheral antinociception produced by PnPP‐19. These data suggest that the peripheral antinociceptive effect of PnPP‐19 is associated with anandamide release, which then could activate CB1 receptors. These findings are in accordance with our data that show the involvement of only the CB1 receptor in the peripheral antinociception induced by PnPP‐19.
Among the analgesic animal toxins described so far, there is one peptide, crotalphine, obtained from the venom of the South American rattlesnake Crotalus durissus terrificus, which induces antinociception due to the activation and interaction of both opioid and cannabinoid systems (Konno et al., 2008; Machado et al., 2014). This is very comparable with what was found for PnPP‐19. However, the exact pathways involved in the action of crotalphine and its molecular target are still not well understood.
The data presented here reveal at least part of the mechanism of action underlying the peripheral antinociceptive effect induced by the synthetic peptide PnPP‐19. Our results suggest that such effects were due to activation of CB1, μ and δ opioid receptors. In addition to that, the peptide could inhibit the enzyme NEP, which would increase enkephalin levels, potentiating the activation of these opioid receptors. However, further studies are required to test whether PnPP‐19 acts as an exogenous agonist of opioid or cannabinoid receptors, and if so, to determine its affinity for these receptors. Moreover, the release of the endogenous cannabinoid anandamide may modulate the peripheral antinociceptive effect induced by the peptide. Experiments to evaluate the possible role of PnPP‐19 in the CNS are being developed.
Our current data are useful for the analysis of antinociceptive effects induced by inhibition of NEP, interactions between opioid and cannabinoid systems and for a better understanding of the role of PnPP‐19 on erectile function and nociceptive pathways. In addition, our results may contribute to the consideration of PnPP‐19 as a potential lead compound for the development of new drug candidates.
Author contributions
A.C.N.F., D.F.P. and M.F.M.M. performed the research. M.E. de L., D.F.P. and I.D.D. designed the research study. M.E. de L.,I.D. D. and A.K.C. contributed essential reagents or tools. A.C.N.F., D.F.P., M.F.M.M., A.K.C., I.D.D. and M.E. de L. analysed the data and reviewed the manuscript. A.C.N.F. and D.F.P. wrote the paper.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organizations engaged with supporting research.
Acknowledgements
Fellowships were awarded by the Brazilian Agencies FAPEMIG (Fundação de Amparo à Pesquisa do Estado de Minas Gerais), CAPES (Coordenação de Aperfeiçoamento de Pessoal de Nível Superior), CNPq (Conselho Nacional de Desenvolvimento Científico e Tecnológico) and INCTTOX (Instituto de Ciência e Tecnologia em Toxinas).
Freitas, A. C. N. , Pacheco, D. F. , Machado, M. F. M. , Carmona, A. K. , Duarte, I. D. G. , and de Lima, M. E. (2016) PnPP‐19, a spider toxin peptide, induces peripheral antinociception through opioid and cannabinoid receptors and inhibition of neutral endopeptidase. British Journal of Pharmacology, 173: 1491–1501. doi: 10.1111/bph.13448.
References
- Agarwal N, Pacher P, Tegeder I, Amaya F, Constantin CE, Brenner GJ, et al. (2007). Cannabinoids mediate analgesia largely via peripheral type 1 cannabinoid receptors in nociceptors. Nat Neurosci 10: 870–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE, et al. (2015a). The Concise Guide to PHARMACOLOGY 2015/16: G Protein‐Coupled Receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE, et al. (2015b). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barros NM, Campos M, Bersanetti PA, Oliveira V, Juliano MA, Boileau G, et al. (2007). Neprilysin carboxydipeptidase specificity studies and improvement in its detection with fluorescence energy transfer peptides. Biol Chem 388: 447–455. [DOI] [PubMed] [Google Scholar]
- Befort K (2015). Interactions of the opioid and cannabinoid systems in reward: Insights from knockout studies. Front Pharmacol 6: 6 http://doi.org/10.3389/fphar.2015.00006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushlin I, Gupta A, Stockton SD, Miller LK, Devi LA (2012). Dimerization with cannabinoid receptors allosterically modulates delta opioid receptor activity during neuropathic pain. PLoS One 7: e49789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordeiro MON, Diniz CR, Valentim AOC, Von Eickstedt VR, Gilroy J, Richardson M (1992). The purification and amino acid sequences of four Tx2 neurotoxins from the venom of the Brazilian ‘armed’ spider Phoneutria nigriventer (Keys). FEBS Lett 310: 153–156. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SPH, Giembycz MA, et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP . Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalmolin GD, Silva CR, Rigo FK, Gomes GM, Cordeiro MON, Richardson M, et al. (2011). Antinociceptive effect of Brazilian armed spider venom toxin Tx3‐3 in animal models of neuropathic pain. Pain 152: 2224–2232. [DOI] [PubMed] [Google Scholar]
- de Lima ME, Figueiredo SG, Matavel A, Nunes KP, Silva CN, Almeida FDM, et al. (2015). Phoneutria nigriventer venom and toxins: a review In: Gopalakrishnakone P, Corzo GA, Diego‐Garcia E, de Lima ME. (eds). Spider Venoms. Springer: Netherlands, pp. 1–24. [Google Scholar]
- Di Marzo V, Fontana A, Cadas H, Schinelli S, Cimino G, Schwartz JC, et al. (1994). Formation and inactivation of endogenous cannabinoid anandamide in central neurons. Nature 372: 686–691. [DOI] [PubMed] [Google Scholar]
- Ferreira SH (1972). Prostaglandins, aspirin‐like drugs and analgesia. Nat New Biol 240: 200–203. [DOI] [PubMed] [Google Scholar]
- Figueiredo SG, de Lima ME, Nascimento Cordeiro M, Diniz CR, Patten D, Halliwell RF, et al. (2001). Purification and amino acid sequence of a highly insecticidal toxin from the venom of the Brazilian spider Phoneutria nigriventer which inhibits NMDA‐evoked currents in rat hippocampal neurones. Toxicon 39: 309–317. [DOI] [PubMed] [Google Scholar]
- Fossiez F, Lemay G, Labonté N, Parmentier‐Lesage F, Boileau G, Crine P (1992). Secretion of a functional soluble form of neutral endopeptidase‐24.11 from a baculovirus‐infected insect cell line. Biochem J 284 (Pt 1): 53–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox A, Kesingland A, Gentry C, McNair K, Patel S, Urban L, et al. (2001). The role of central and peripheral Cannabinoid1 receptors in the antihyperalgesic activity of cannabinoids in a model of neuropathic pain. Pain 92: 91–100. [DOI] [PubMed] [Google Scholar]
- Galdino G, Romero TR, Silva JF, Aguiar DC, De Paula AM, Cruz JS, et al. (2014). The endocannabinoid system mediates aerobic exercise‐induced antinociception in rats. Neuropharmacology 77: 313–324. [DOI] [PubMed] [Google Scholar]
- Gazerani P, Cairns BE (2014). Venom‐based biotoxins as potential analgesics. Expert Rev Neurother 14: 1261–1274. [DOI] [PubMed] [Google Scholar]
- Ghasemi M, Sadeghipour H, Mani AR, Tavakoli S, Hajrasouliha AR, Ebrahimi F, et al. (2006). Effect of anandamide on nonadrenergic noncholinergic‐mediated relaxation of rat corpus cavernosum. Eur J Pharmacol 544: 138–145. [DOI] [PubMed] [Google Scholar]
- Gomes I, IJzerman AP, Ye K, Maillet EL, Devi LA (2011). G Protein‐Coupled Receptor Heteromerization: A Role in Allosteric Modulation of Ligand Binding. Mol Pharmacol 79: 1044–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta A, Mulder J, Gomes I, Rozenfeld R, Bushlin I, Ong E, et al. (2010). Increased abundance of opioid receptor heteromers after chronic morphine administration. Sci Signal 3: ra54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herkenham M, Lynn AB, Little MD, Johnson MR, Melvin LS, de Costa BR, et al. (1990). Cannabinoid receptor localization in brain. Proc Natl Acad Sci U S A 87: 1932–1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hersh LB, Morihara K (1986). Comparison of the subsite specificity of the mammalian neutral endopeptidase 24.11 (enkephalinase) to the bacterial neutral endopeptidase thermolysin. J Biol Chem 261: 6433–6437. [PubMed] [Google Scholar]
- Hohmann AG, Briley EM, Herkenham M (1999). Pre‐ and postsynaptic distribution of cannabinoid and mu opioid receptors in rat spinal cord. Brain Res 822: 17–25. [DOI] [PubMed] [Google Scholar]
- Hohmann AG, Suplita RL (2006). Endocannabinoid mechanisms of pain modulation. AAPS J 8: E693–E708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hruby VJ (2002). Designing peptide receptor agonists and antagonists. Nat Rev Drug Discov 1: 847–858. [DOI] [PubMed] [Google Scholar]
- Ibrahim MM, Porreca F, Lai J, Albrecht PJ, Rice FL, Khodorova A, et al. (2005). CB2 cannabinoid receptor activation produces antinociception by stimulating peripheral release of endogenous opioids. Proc Natl Acad Sci U S A 102: 3093–3098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). NC3Rs Reporting Guidelines Working Group. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konno K, Picolo G, Gutierrez VP, Brigatte P, Zambelli VO, Camargo AC, et al. (2008). Crotalphine, a novel potent analgesic peptide from the venom of the South American rattlesnake Crotalus durissus terrificus . Peptides 29: 1293–1304. [DOI] [PubMed] [Google Scholar]
- Law PY, Wong YH, Loh HH (2000). Molecular mechanisms and regulation of opioid receptor signaling. Annu Rev Pharmacol Toxicol 40: 389–430. [DOI] [PubMed] [Google Scholar]
- Leite dos Santos GG, Casais e Silva LL, Pereira Soares MB, Villarreal CF (2012). Antinociceptive properties of Micrurus lemniscatus venom. Toxicon 60: 1005–1012. [DOI] [PubMed] [Google Scholar]
- Lemay G, Waksman G, Roques BP, Crine P, Boileau G (1989). Fusion of a cleavable signal peptide to the ectodomain of neutral endopeptidase (EC 3.4.24.11) results in the secretion of an active enzyme in COS‐1 cells. J Biol Chem 264: 15620–15623. [PubMed] [Google Scholar]
- Lin S, Khanolkar AD, Fan P, Goutopoulos A, Qin C, Papahadjis D, et al. (1998). Novel analogues of arachidonylethanolamide (anandamide): affinities for the CB1 and CB2 cannabinoid receptors and metabolic stability. J Med Chem 41: 5353–5361. [DOI] [PubMed] [Google Scholar]
- Machado FC, Zambelli VO, Fernandes AC, Heimann AS, Cury Y, Picolo G (2014). Peripheral interactions between cannabinoid and opioid systems contribute to the antinociceptive effect of crotalphine. Br J Pharmacol 171: 961–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mafra RA, Figueiredo SG, Diniz CR, Cordeiro MN, Cruz JD, De Lima ME (1999). PhTx4, a new class of toxins from Phoneutria nigriventer spider venom, inhibits the glutamate uptake in rat brain synaptosomes. Brain Res 831: 297–300. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marangoni RA, Antunes E, Brain SD, de Nucci G (1993). Activation by Phoneutria nigriventer (armed spider) venom of tissue kallikrein‐kininogen‐kinin system in rabbit skin in vivo. Br J Pharmacol 109: 539–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin‐Eauclaire MF, Abbas N, Sauze N, Mercier L, Berge‐Lefranc JL, Condo J, et al. (2010). Involvement of endogenous opioid system in scorpion toxin‐induced antinociception in mice. Neurosci Lett 482: 45–50. [DOI] [PubMed] [Google Scholar]
- Matavel A, Cruz JS, Penaforte CL, Araújo DA, Kalapothakis E, Prado VF, et al. (2002). Electrophysiological characterization and molecular identification of the Phoneutria nigriventer peptide toxin PnTx2‐6. FEBS Lett 523: 219–223. [DOI] [PubMed] [Google Scholar]
- Matta A, Tandra PK, Berim L (2014). Priapism in a patient with sickle cell trait using marijuana. BMJ Case Rep. doi:10.1136/bcr-2014-204199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meves H (2006). The action of prostaglandins on ion channels. Curr Neuropharmacol 4: 41–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunes KP, Costa‐Gonçalves A, Lanza LF, Cortes SF, Cordeiro MN, Richardson M, et al. (2008). Tx2‐6 toxin of the Phoneutria nigriventer spider potentiates rat erectile function. Toxicon 51: 1197–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacheco D, Klein A, De Castro PA, Da Fonseca Pacheco CM, De Francischi JN, Duarte ID (2008). The mu‐opioid receptor agonist morphine, but not agonists at delta‐ or kappa‐opioid receptors, induces peripheral antinociception mediated by cannabinoid receptors. Br J Pharmacol 154: 1143–1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacheco DF, Klein A, Perez AC, Pacheco CM, de Francischi JN, Reis GM, et al. (2009). Central antinociception induced by mu‐opioid receptor agonist morphine, but not delta‐ or kappa‐, is mediated by cannabinoid CB1 receptor. Br J Pharmacol 158: 225–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacheco DF, Romero TR, Duarte ID (2014). Central antinociception induced by ketamine is mediated by endogenous opioids and μ‐ and δ‐opioid receptors. Brain Res 1562: 69–75. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SPH, Buneman OP, et al, NC‐IUPHAR(2014). The IUPHAR/BPS Guide to PHARMACOLOGY: an expert‐driven knowledge base of drug targets and their ligands. Nucl. Acids Res. 42 (Database Issue): D1098–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng J, Sarkar S, Chang SL (2012). Opioid receptor expression in human brain and peripheral tissues using absolute quantitative real‐time RT‐PCR. Drug Alcohol Depend 124: 223–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertwee RG (2006). Cannabinoid pharmacology: the first 66 years. Br J Pharmacol 147 (Suppl 1): S163–S171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platnick NI (1997). Advances in spider taxonomy, 1992–1995: with redescriptions 1940–1980. New York Entomological Society & The American Museum of Natural History.
- Pu XC, Wong PT, Gopalakrishnakone P (1995). A novel analgesic toxin (hannalgesin) from the venom of king cobra (Ophiophagus hannah). Toxicon 33: 1425–1431. [DOI] [PubMed] [Google Scholar]
- Randall LO, Selitto JJ (1957). A method for measurement of analgesia activity on inflamed tissue. Arch Int Pharmacodyn 111: 209–219. [PubMed] [Google Scholar]
- Reis GM, Pacheco D, Perez AC, Klein A, Ramos MA, Duarte ID (2009). Opioid receptor and NO/cGMP pathway as a mechanism of peripheral antinociceptive action of the cannabinoid receptor agonist anandamide. Life Sci 85: 351–356. [DOI] [PubMed] [Google Scholar]
- Rios C, Gomes I, Devi LA (2006). mu opioid and CB1 cannabinoid receptor interactions: reciprocal inhibition of receptor signaling and neuritogenesis. Br J Pharmacol 148: 387–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero TR, Resende LC, Guzzo LS, Duarte ID (2013a). CB1 and CB2 cannabinoid receptor agonists induce peripheral antinociception by activation of the endogenous noradrenergic system. Anesth Analg 116: 463–472. [DOI] [PubMed] [Google Scholar]
- Romero TR, Pacheco DAF, Duarte ID (2013b). Xylazine induced central antinociception mediated by endogenous opioids and μ‐opioid receptor, but not δ‐or κ‐opioid receptors. Brain Res 1506: 58–63. [DOI] [PubMed] [Google Scholar]
- Roques BP, Noble F, Daugé V, Fournié‐Zaluski MC, Beaumont A (1993). Neutral endopeptidase 24.11: structure, inhibition, and experimental and clinical pharmacology. Pharmacol Rev 45: 87–146. [PubMed] [Google Scholar]
- Rougeot C, Messaoudi M, Hermitte V, Rigault AG, Blisnick T, Dugave C, et al. (2003). Sialorphin, a natural inhibitor of rat membrane‐bound neutral endopeptidase that displays analgesic activity. Proc Natl Acad Sci U S A 100: 8549–8554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schechter I, Berger A (1968). On the active site of proteases. 3. Mapping the active site of papain; specific peptide inhibitors of papain. Biochem Biophys Res Commun 32: 898–902. [DOI] [PubMed] [Google Scholar]
- Silva CN, Nunes KP, Torres FS, Cassoli JS, Santos DM, Almeida Fde M, et al. (2015). PnPP‐19, a synthetic and non toxic peptide designed from a P. nigriventer toxin, potentiates erectile function via NO/cGMP. J Urol. doi:10.1016/j.juro.2015.06.081 [DOI] [PubMed] [Google Scholar]
- Souza AH, Ferreira J, Cordeiro MN, Vieira LB, De Castro CJ, Trevisan G, et al. (2008). Analgesic effect in rodents of native and recombinant Ph alpha 1beta toxin, a high‐voltage‐activated calcium channel blocker isolated from armed spider venom. Pain 140: 115–126. [DOI] [PubMed] [Google Scholar]
- Tong Y, Tar M, Melman A, Davies K (2008). The opiorphin gene (ProL1) and its homologues function in erectile physiology. BJU Int 102: 736–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong Y, Tar M, Monrose V, DiSanto M, Melman A, Davies KP (2007). hSMR3A as a marker for patients with erectile dysfunction. J Urol 178: 338–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner AJ, Matsas R, Kenny AJ (1985). Are there neuropeptide‐specific peptidases? Biochem Pharmacol 34: 1347–1356. [DOI] [PubMed] [Google Scholar]
- Veloso CEC, Rodrigues VG, Ferreira RC, Duarte LP, Klein A, Duarte ID, et al. (2014). Tingenone, a pentacyclic triterpene, induces peripheral antinociception due to opioidergic activation. Planta Med 80: 1615–1621. [DOI] [PubMed] [Google Scholar]
- Wisner A, Dufour E, Messaoudi M, Nejdi A, Marcel A, Ungeheuer MN, et al. (2006). Human Opiorphin, a natural antinociceptive modulator of opioid‐dependent pathways. Proc Natl Acad Sci U S A 103: 17979–17984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon MH, Kim WM, Lee HG, Kim YO, Huang LJ, An TH (2008). Roles of opioid receptor subtypes on the antinociceptive effect of intrathecal sildenafil in the formalin test of rats. Neurosci Lett 441: 125–128. [DOI] [PubMed] [Google Scholar]