

Abstract
Adrenaline is a hormone that has profound actions on the cardiovascular system and is also a mediator of the fight‐or‐flight response. Adrenaline is now increasingly recognized as an important metabolic hormone that helps mobilize energy stores in the form of glucose and free fatty acids in preparation for physical activity or for recovery from hypoglycaemia. Recovery from hypoglycaemia is termed counter‐regulation and involves the suppression of endogenous insulin secretion, activation of glucagon secretion from pancreatic α‐cells and activation of adrenaline secretion. Secretion of adrenaline is controlled by presympathetic neurons in the rostroventrolateral medulla, which are, in turn, under the control of central and/or peripheral glucose‐sensing neurons. Adrenaline is particularly important for counter‐regulation in individuals with type 1 (insulin‐dependent) diabetes because these patients do not produce endogenous insulin and also lose their ability to secrete glucagon soon after diagnosis. Type 1 diabetic patients are therefore critically dependent on adrenaline for restoration of normoglycaemia and attenuation or loss of this response in the hypoglycaemia unawareness condition can have serious, sometimes fatal, consequences. Understanding the neural control of hypoglycaemia‐induced adrenaline secretion is likely to identify new therapeutic targets for treating this potentially life‐threatening condition.
Abbreviations
- ASNA
adrenal sympathetic nerve activity
- CART
cocaine‐ and amphetamine‐regulated transcript
- CVLM
caudal ventrolateral medulla
- IML
intermediolateral cell column
- PeH
perifornical hypothalamic
- RVLM
rostroventrolateral medulla
- SPNs
sympathetic preganglionic neurons
Tables of Links
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (a,bAlexander et al., 2015a, 2015b).
Introduction
The path to the discovery of adrenaline began when Oliver and Schafer described the pressor effect of extracts of the adrenal gland (Oliver and Schafer, 1895). Although they neither isolated the active principle nor gave it a name, they concluded that it was confined to the adrenal medulla and not the cortex. At the turn of the 20th century, purification of a similar extract was achieved by Abel in Baltimore and independently by Takamine in New York working under the auspices of the Parke‐Davis Company: Takamine referred to the new compound as ‘adrenalin’ (Takamine, 1902), while Abel preferred ‘epinephrin’ (Abel, 1898). As pointed out by Davenport, the Merck Index lists as many as 35 names for adrenaline including ‘adrenine’ (Merck, 1968; Davenport, 1982). In 1849, Addison noted that the adrenal glands were necessary for life; and, for a short time, some thought that the essential principle, which we now know to be cortisol, was the pressor substance isolated from the adrenal medulla (Addison, 1855; Davenport, 1982).
In this review, we examine the importance of adrenaline as a metabolic hormone that mobilizes energy stores in the form of glucose and free fatty acids during the counter‐regulatory response to hypoglycaemia (Cryer, 1981).
Pharmacology of adrenaline
Adrenaline is best known to pharmacologists as a substance that has profound effects on the cardiovascular system. In general, the effects of exogenously administered adrenaline on the cardiovascular system are similar to that of sympathetic nerve stimulation. This was noted by Walter B. Cannon when he attempted to prove that adrenaline was the sympathetic neurotransmitter or, as he termed it, ‘sympathin’ (Cannon and Rosenblueth, 1935). However, as Cannon discovered, there are differences between the pharmacological effects of adrenaline and the sympathetic neurotransmitter. Firstly, the transmitter released at the sympathetic vascular neuroeffector junction is the N‐demethylated catecholamine, noradrenaline (von Euler, 1946); and secondly, adrenaline and noradrenaline have different potencies at α‐ and β‐adrenoceptors (Ahlquist, 1948; Westfall and Westfall, 2011). Finally, Cannon's attempts to identify the sympathetic transmitter (Cannon and Rosenblueth, 1935) were confounded by contamination of the preparations of adrenaline that Cannon had available to him by variable amounts of noradrenaline (von Euler, 1966; Davenport, 1982).
Adrenaline has powerful, dose‐dependent effects on the cardiovascular system. Intravenous injections of adrenaline rapidly produce a powerful vasopressor effect as a result of profound vasoconstriction, an increase in the rate and force of contraction of the heart, increased myocardial cell automaticity, bronchodilatation, increased respiratory rate and redistribution of blood towards the brain, heart and skeletal muscle and away from skin, kidneys and gut (Goldstein, 1999; Westfall and Westfall, 2011). Ahlquist (1948) first described the differences in the rank order of potency of adrenaline, noradrenaline and isoprenaline in a variety of peripheral tissues. These observations led to the proposal that the differences in the actions of the catecholamines could be explained by the existence of distinct receptors that ultimately were termed α‐ and β‐adrenoceptors (Ahlquist, 1948). The development of more selective agonists and antagonists, as well as molecular cloning of G‐protein coupled receptors, led to further sub‐classification of the adrenoceptors as α1A‐, α1B‐, α1D‐, α2A‐, α2B‐ and α2C‐adrenoceptors as well as β1‐, β2‐ and β3‐adrenoceptors (Alexander et al., 2015a). Despite its profound effects on cardiovascular function, the contribution of adrenaline to normal cardiovascular regulation or to the development of essential hypertension is probably minimal because plasma levels of adrenaline are not consistently elevated in this condition (Goldstein, 1983). Circulating adrenaline can be taken up and released by sympathetic nerves, but the contribution of such a mechanism to essential hypertension is far from clear (Esler, 1993).
Biosynthesis and catabolism of adrenaline
The biosynthesis and catabolism of adrenaline are depicted in Figure 1. Tyrosine is hydroxylated in the meta position to form dihydroxyphenylalanine (DOPA) by tyrosine hydroxylase, the rate‐limiting step in catecholamine biosynthesis (Kuhar et al., 1999). DOPA is subsequently decarboxylated by DOPA decarboxylase (AADC) to produce dopamine, which is subsequently hydroxylated at the β‐carbon by dopamine β‐hydroxylase (DBH) to produce noradrenaline. The final step of adrenaline biosynthesis occurs when noradrenaline is N‐methylated by PNMT (Axelrod, 1966). When released into the circulation from the adrenal medulla, adrenaline and noradrenaline are O‐methylated by catecholamine methyltransferase (COMT) to produce metanephrine and normetanephrine, which are then deaminated by MAO to form 3‐methoxy‐4‐hydroxy‐mandelic acid (Axelrod, 1966).
Figure 1.
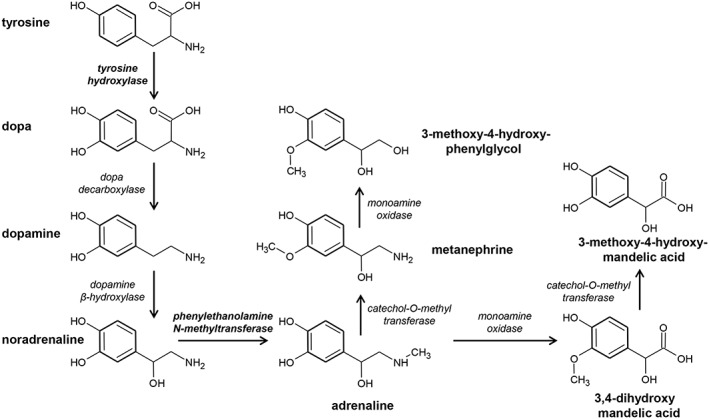
Biosynthesis and catabolism of adrenaline. Adrenaline biosynthesis begins with the hydroxylation of phenylalanine to tyrosine. Tyrosine hydroxylase catalyses the conversion of tyrosine to DOPA (dihydroxyphenylalanine), the rate‐limiting step in catecholamine biosynthesis. The final step in adrenaline biosynthesis is the methylation of noradrenaline by PNMT.
Disorders of catecholamine metabolism, such as dopamine β‐hydroxylase deficiency, are very rare with perhaps 20 or so cases described in the world. Because dopamine β‐hydroxylase catalyses the conversion of dopamine to noradrenaline, individuals with this disorder have defective sympathetic nerve function manifested by absent noradrenaline spillover into the circulation and impaired adrenal catecholamine synthesis (Rea et al., 1990; Thompson et al., 1995). The symptoms of the disease include postural hypotension, low blood pressure, difficulty in maintaining body temperature, ptosis, exercise intolerance and importantly, hypoglycaemia. Microneurographic studies in these patients indicate elevated sympathetic nerve traffic and normal baroreflex inhibition. Therefore, noradrenaline released from sympathetic nerves is critical for the maintenance of resting arterial blood pressure as well as a range of other functions. In contrast, the cardiovascular effects of depletion of peripheral adrenaline in response to treatment with inhibitors of PNMT are much less dramatic. Thus, peripheral adrenaline is not essential for maintenance of hypertension or centrally evoked hypertension in stroke‐prone hypertensive rats (Rogers et al., 1991). Similarly, PNMT inhibitors, such as LY13406, which has minimal adrenoceptor blocking activity, do not alter blood pressure in rats with deoxycorticosterone acetate–salt‐induced hypertension despite pronounced PNMT inhibition (Biollaz et al., 1984).
Adrenaline biosynthesis is strongly influenced by glucocorticoids released from the adrenal cortex (Wurtman et al., 1972). Glucocorticoids are transported at a high concentration by the intra‐adrenal portal vascular system directly to the chromaffin cells to induce the synthesis of PNMT (Wurtman and Axelrod, 1965, 1966). The dependence of PNMT synthesis on the presence of glucocorticoids is present in most mammals including humans and rats. In species in which the chromaffin tissue is not surrounded entirely by the steroid‐secreting adrenal cortex (e.g. the rabbit), little adrenaline is present in chromaffin cells not in contact with the adrenal cortical cells (Coupland, 1956). In other species where the steroid‐secreting cells and catecholamine‐secreting cells are located in independent glands (e.g. the dogfish), no adrenaline is synthesized (Coupland, 1953).
Innervation of the adrenal gland
The adrenal gland is often regarded as a modified sympathetic ganglion in which the postganglionic neurons are represented by the adrenal chromaffin cells that synthesize the catecholamines adrenaline and noradrenaline. These are secreted into the circulation rather than being released as a neurotransmitter, as is the case for conventional sympathetic postganglionic neurons. Sympathetic postganglionic neurons express nicotinic acetylcholine receptors, and ganglionic transmission is blocked by nicotinic receptor antagonists, such as hexamethonium and mecamylamine (McIsaac and Millerschoen, 1966). Similarly, adrenal chromaffin cells secrete catecholamines in response to acetylcholine acting at nicotinic acetylcholine receptors (Wakade, 1981) and to a lesser extent at muscarinic receptors (Wakade and Wakade, 1983). The unique innervation of the adrenal gland consists of axons (i) from cholinergic sympathetic preganglionic neurons (SPNs) that innervate the adrenal chromaffin cells directly (Kesse et al., 1988) and (ii) from noradrenergic sympathetic postganglionic neurons that innervate blood vessels in the adrenal cortex and medulla (Parker et al., 1990; Carlsson et al., 1993; Toth et al., 1997) as well as spinal and vagal afferent axons (Mohamed et al., 1988; Coupland et al., 1989; Niijima, 1992) (Figure 2). Chromaffin cells synthesize either adrenaline or noradrenaline, and the proportion of adrenaline‐synthesizing cells exceeds those that synthesize noradrenaline. In the rat, ~70–80% of all chromaffin cells synthesize adrenaline (Verhofstad et al., 1985). Whether the chromaffin cells synthesize adrenaline or noradrenaline is governed by whether or not the cells express PNMT, because this enzyme catalyses the N‐methylation of noradrenaline to produce adrenaline (Figure 1). Adrenal SPNs are found in the intermediolateral cell column (IML) between thoracic spinal segments T4 and T12 in the rat (Strack et al., 1988) and between thoracic segment 2 and lumbar segment 2 in the rabbit (Jensen et al., 1992). While all adrenal SPNs release acetylcholine as their primary transmitter, some adrenal SPNs also express cocaine‐ and amphetamine‐regulated transcripts (Fenwick et al., 2006), and these project to the noradrenaline‐synthesizing chromaffin cells (Gonsalvez et al., 2010). In contrast, SPNs that innervate the adrenaline‐synthesizing chromaffin cells express the neuropeptide enkephalin (Holgert et al., 1995). In the cat, a proportion of the adrenal SPNs express the calcium‐binding protein calretinin, and calretinin‐immunoreactive terminals are associated with noradrenergic chromaffin cells in the adrenal medulla (Edwards et al., 1996).
Figure 2.
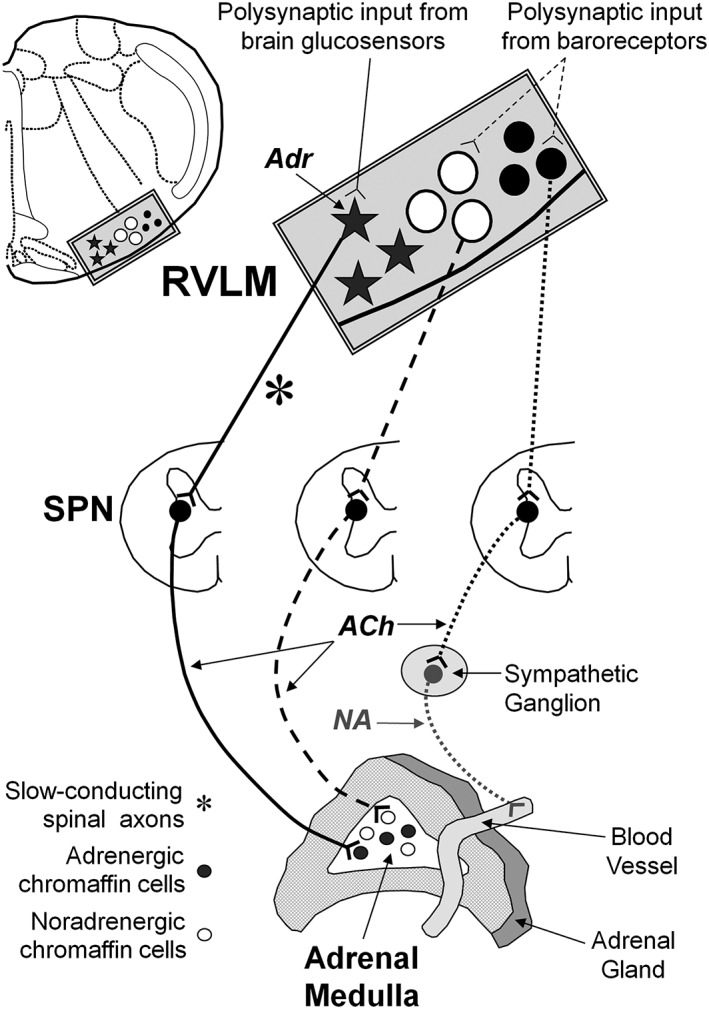
Innervation of the adrenal gland. The adrenal gland is innervated by preganglionic cholinergic neurons that target the adrenal chromaffin cells exclusively. Postganglionic noradrenergic neurons target only the vasculature of the entire gland. ‘Adrenaline’ chromaffin cells receive input from preganglionic neurons that receive premotor input from the rostral ventrolateral medulla (RVLM) that, in turn, receive input from glucose‐sensing neurons located elsewhere in the brain and the periphery. ‘Noradrenaline’ chromaffin cells receive input from preganglionic neurons that receive premotor input from barosensitive neurons in the RVLM.
Tracing studies using neurotropic viruses have identified and labelled premotor cell groups in supraspinal regions that project to adrenal SPNs located in the IML of the spinal cord (Wesselingh et al., 1989; Strack et al., 1989a, 1989b; Westerhaus and Loewy, 2001, 1999; Geerling et al., 2003; Kerman et al., 2007). Labelled neurons were found in the rostroventrolateral medulla (RVLM), caudal raphé nuclei, ventromedial medulla, the A5 cell group and the paraventricular hypothalamic nucleus and the perifornical hypothalamic area (PeH). In the rabbit, adrenal SPNs receive input from serotonergic neurons as well as from RVLM C1 adrenergic neurons (Li et al., 1992; Jensen et al., 1995). These same groups of hindbrain neurons are labelled after injection of viral tracer into the kidney as well as other sympathetic vascular tissue (Ding et al., 1993; Schramm et al., 1993; Sly et al., 1999; Toth et al., 2008a, 2008b). In rats, adrenal SPNs express Fos in response to glucoprivation (2‐DG), and these neurons receive close appositions from varicosities of PNMT‐immunoreactive axons (Figure 3). Data from Fos and viral tracing studies suggest that the adrenergic input to adrenal SPNs arises from C1 neurons in the RVLM and/or C3 neurons in and around the dorsal medullary midline (Ritter et al., 1998; Ritter et al., 2001; Card et al., 2006; Menuet et al., 2014). These findings are consistent with the view that RVLM C1 neurons that are modulated by central glucose sensors innervate adrenal SPNs (Verberne and Sartor, 2010).
Figure 3.
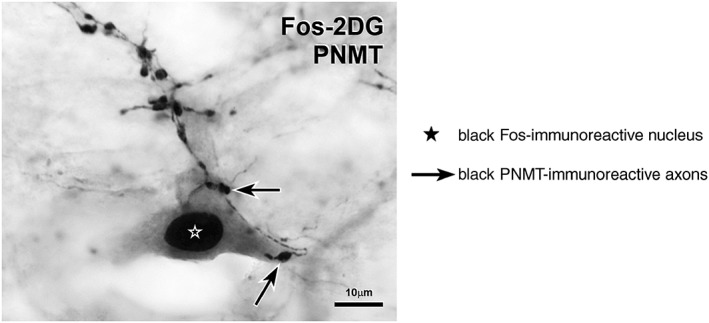
Neuroglucoprivation activates a sympathetic preganglionic neuron (SPN) with PNMT inputs. This choline acetyltransferase‐immunoreactive (grey cytoplasmic staining) SPN from a rat that had received 2‐DG (400 mg·kg−1. i.p.) has a black Fos‐immunoreactive nucleus and receives close appositions from varicosities of black PNMT‐immunoreactive axons. The presence of Fos‐immunoreactivity indicates neuronal activation.
Studies that have used two different neurotropic viral tracers to identify premotor neurons that control both the heart and adrenal catecholamine secretion (Strack et al., 1989a; Standish et al., 1994) are of limited value because there is a high probability that the viruses have labelled the one feature that the heart and adrenal gland have in common – the sympathetic postganglionic innervation of the blood supply to each organ. Unfortunately, no method has been devised for selectively labelling the adrenal chromaffin cell innervation with neurotropic viral tracers, as opposed to the vascular innervation of the adrenal gland.
Differential control of adrenal catecholamine secretion
There is physiological and anatomical evidence that the chromaffin cells that synthesize adrenaline or noradrenaline are controlled differentially. Thus, hypoglycaemia selectively increases adrenaline release, whereas cold exposure selectively increases noradrenaline release (Vollmer et al., 1992). However, the increase in noradrenaline secretion in response to cold probably arises from increased release from sympathetic nerves in order to restore normotension. The hypotensive response to haemorrhage is unaffected by adrenalectomy or adrenal denervation in awake rabbits (Schadt and Gaddis, 1988), suggesting that the recovery from haemorrhage depends largely on sympathetic vasomotor activation and vasopressin release. Contrary to the view that adrenal secretion acts in concert with sympathetic vasomotor activation, there is substantial evidence indicating that adrenal catecholamine release is activated by nerve pathways distinctly separate from those regulating the vasomotor system. The prevailing view that the vasomotor and adrenal catecholamine secretory systems act in concert arises, at least in part, from the limitations of the experimental approaches that have been used to study them. Thus, electrical or chemical stimulation of the RVLM produces pressor responses accompanied by adrenaline secretion and hyperglycaemia because premotor sympathetic vasomotor neurons and the premotor neurons that drive adrenaline secretion are intermingled (Verberne et al., 1999; Kerman et al., 2007; Verberne and Sartor, 2010). Interestingly, prior removal of the adrenal glands did not alter the magnitude of the pressor response to stimulation of the RVLM (Verberne and Sartor, 2010). Stimulation of the splanchnic sympathetic nerve evokes sympathetically mediated vasoconstriction in the splanchnic vascular bed along with adrenal catecholamine secretion, but the secreted catecholamines appear to contribute little to the vasoconstrictor response (Reed et al., 1971; Edwards, 1982). Both responses occur simply because the splanchnic sympathetic nerve contains postganglionic nerve fibres that innervate vascular smooth muscle as well as preganglionic fibres that innervate adrenal medullary chromaffin cells (Celler and Schramm, 1981; Sapru et al., 1982).
Morrison and colleagues recorded from rat SPNs that were antidromically activated by stimulation of the adrenal nerve (Morrison and Cao, 2000). They showed that these neurons could be subdivided on the basis of their axonal conduction velocities and their response to stimulation of premotor sympathetic neurons in the RVLM, as well as their response to baroreceptor activation or systemic glucoprivation. Thus, ‘adrenaline’ adrenal SPNs are activated by glucoprivation produced by systemic administration of the glucoprivic agent 2‐deoxyglucose (2‐DG). Furthermore, they are insensitive to baroreflex activation and instead receive input from slow‐conducting RVLM neurons. This slow‐conducting pathway corresponds to slow‐conducting, baroinsensitive neurons in RVLM that are activated by glucoprivation and are most probably C1 neurons (Verberne and Sartor, 2010). In contrast, ‘noradrenaline’ adrenal SPNs are barosensitive, activated by RVLM stimulation over a fast‐conducting pathway and were insensitive to glucoprivation (Morrison and Cao, 2000).
Several studies indicate that electrical stimulation of sites in the hypothalamus elicits selective increases in plasma adrenaline or noradrenaline (Folkow and Von Euler, 1954; Tsuchimochi et al., 2010). These findings suggest that adrenaline and noradrenaline secretion from the adrenal gland can be controlled independently. This view is supported by observations that insulin‐induced hypoglycaemia per se or neuroglucoprivation induced by 2‐deoxyglucose is not accompanied by marked sympathetic vasomotor changes (Bardgett et al., 2010; Verberne and Sartor, 2010; Korim et al., 2014). Similarly, activation of hypothalamic orexin neurons by local glucoprivation favours an increase in adrenaline secretion over noradrenaline secretion (Korim et al., 2014). It is also likely that the premotor neurons that control sympathetic vasomotor outflow and adrenal catecholamine secretion are controlled by separate inputs (Figure 2). In the case of the vasomotor neurons, we know that a major input and regulator is the intramedullary baroreflex pathway (Schreihofer and Guyenet, 2002; Schreihofer and Sved, 2011). In contrast, presympathetic neurons that control adrenaline secretion are likely to receive input from central and/or peripheral glucose‐sensing neurons but not from baroreceptors (Verberne and Sartor, 2010; Korim et al., 2014).
Measurement of adrenal sympathetic nerve activity (ASNA) is complicated by the fact that the adrenal nerve supply consists of several fine branches rather than a single discrete nerve trunk. In addition, adrenal nerve discharge consists of activity that is related to sympathetic preganglionic input to chromaffin cells as well as sympathetic postganglionic vasomotor drive to the adrenal vasculature. The relative contributions of preganglionic and postganglionic components to the multiunit recording can be estimated by systemic administration of a short‐acting ganglion blocker, such as trimethaphan (Sapru et al., 1982). Because trimethaphan has become difficult to obtain in recent years, an alternative is to use systemic administration of the ganglion blocker hexamethonium (20 mg·kg−1, i.v.) at the conclusion of the experiment.
Adrenaline and cardiovascular regulation
Adrenaline modulates vascular tone in a fashion that is regionally specific. Adrenaline also increases myocardial contractility, heart rate and cardiac output. High, experimentally‐induced circulating levels of adrenaline can induce hypertension (Majewski et al., 1981; Tung et al., 1981), but adrenaline is not considered an underlying contributor to essential hypertension (see the preceding text). Unlike the release of catecholamine from sympathetic nerve terminals at neurovascular junctions, adrenaline secretion from the isolated adrenal gland is not modulated by presynaptic α‐ or β‐adrenoceptors (Collett and Story, 1982; Collett et al., 1984). Adrenaline can modulate the activity of the baroreflex although probably not through modulation of the activity of the arterial baroreceptors per se (Shoukas, 1982). Inhibitors of PNMT (Figure 1) that do not cross the blood–brain‐barrier do not reduce arterial blood pressure, suggesting that peripheral adrenaline does not contribute significantly to resting arterial blood pressure (Black et al., 1981).
During moderate exercise, plasma levels of adrenaline rise, but there is no significant increase in secretion of adrenaline from the adrenal medulla (Warren et al., 1984). The increase appears to be largely a result of reduced clearance rather than increased adrenal secretion. Infusion of adrenaline to produce plasma levels that are comparable with those found during moderate exercise produces only a modest β2‐adrenoceptor‐mediated vasodilator response (Warren and Dalton, 1983). Infusion of adrenaline into normotensive subjects resulted in increased plasma renin, glucose and free fatty acids but had only minimal effects on the cardiovascular system (Fitzgerald et al., 1980).
Overall, these observations support the view that adrenaline is not of major significance in maintaining cardiovascular homeostasis and is not a major factor in the development of essential hypertension.
Adrenaline and the fight‐or‐flight response
Many previous studies performed on freely moving and anaesthetized animals have shown that electrical or chemical stimulation of the hypothalamic ‘defence’ area elicits fight‐or‐flight responses that involve anterior hypothalamic structures including the PeH, dorsomedial hypothalamic nucleus and the lateral hypothalamic area (Hilton, 1982; Smith et al., 2000). The responses are characterized by reactions such as pupillary dilatation, piloerection, growling, hissing and other aggressive behaviour or flight (escape) behaviour (Fuchs et al., 1985). Connections with caudal brain structures such as the midbrain periaqueductal grey area and the RVLM mediate sympathetic vasomotor adjustments as well secretion of catecholamines (Yardley and Hilton, 1987; Carrive et al., 1988; Carrive et al., 1989; Lovick, 1992; Verberne and Guyenet, 1992). These hindbrain structures mediate regional changes in blood flow produced during defence‐like behaviour. Hindlimb vasodilatation associated with shifting blood away from the splanchnic circulation to active skeletal muscle during defence responses is, at least in part, dependent on adrenaline acting at β‐adrenoceptors on skeletal muscle resistance vessels (Yardley and Hilton, 1987).
Do baroreceptors and chemoceptors modulate adrenaline release?
Presympathetic neurons of the RVLM and SPNs in the IML of the thoracic spinal cord that control adrenaline secretion do not display a prominent cardiac rhythm nor is their activity depressed by activation of the baroreceptor reflex (Natarajan and Morrison, 1999; Morrison and Cao, 2000; Verberne and Sartor, 2010). In contrast, studies of the activity of preganglionic and postganglionic adrenal axons measured during multi‐fibre recordings show similar degrees of baroreflex‐induced inhibition (Carlsson et al., 1992b). The postganglionic activity is barosensitive in nature, which is explained by the fact that these fibres innervate the adrenal vasculature. It is likely that baroreflex‐induced inhibition of the preganglionic activity occurs because the neurons that control the noradrenaline cells of the adrenal medulla are also barosensitive (Morrison and Cao, 2000). In cats but not dogs, baroreceptor unloading results mainly in noradrenaline release, (Critchley et al., 1980).
Presympathetic vasomotor neurons of the RVLM receive an inhibitory GABAergic input from neurons in the caudal ventrolateral medulla (CVLM) that are a critical relay in the baroreflex pathway. Disinhibition of the CVLM neurons results in a marked elevation of arterial pressure, sympathetic nerve activity and plasma noradrenaline but not adrenaline (Natarajan and Morrison, 1999), again supporting the view that the baroreceptor reflex does not influence adrenaline secretion. However, Mundinger and colleagues have demonstrated that haemorrhage in anaesthetized dogs elicits adrenaline secretion (Mundinger et al., 1997). While haemorrhage unloads the arterial baroreceptors, it can also activate cardiopulmonary baroreceptors because the arterial blood pressure drops to very low levels (Morita and Vatner, 1985). Activation of this subset of baroreceptors may cause adrenaline release.
Scislo and colleagues have examined the effect of baroreflex activation on adrenal sympathetic nerve discharge (Scislo et al., 1998). They demonstrated that, when compared with lumbar or renal sympathetic activity, ASNA exhibits prominent barosensitivity characterized by greater maximal sympathoexcitation in response to hypotension. Carlsson and colleagues examined the response of the renal sympathetic nerve to baroreceptor stimulation and haemorrhage and compared these responses with recordings of preganglionic and postganglionic nerve activity of the adrenal sympathetic nerve (Carlsson et al., 1992b), which could be distinguished using trimethaphan. Baroreflex response curves for renal sympathetic activity and preganglionic and postganglionic ASNA were similar although maximal baroreflex‐induced inhibition of preganglionic ASNA was less (adrenal reduced to 87% versus renal reduced to 68% of control) than that of the renal sympathetic nerve. This result is somewhat unexpected because the majority of the adrenal preganglionic input probably targets the adrenaline‐producing cells of the adrenal medulla and this input is largely baroinsensitive (Morrison and Cao, 2000). In contrast, adrenal SPNs that drive noradrenaline‐synthesizing chromaffin cells exhibit pronounced barosensitivity (Morrison and Cao, 2000). Chemoreceptor activation also activates ASNA and increases the plasma adrenaline concentration in rats, cats and dogs (Critchley et al., 1980; Biesold et al., 1989). This effect is abolished by spinal transection, treatment with hexamethonium or adrenal denervation, indicating that the response is dependent on a central pathway (Lee et al., 1987) and the sympathetic preganglionic input to the adrenal gland (Seidler and Slotkin, 1986).
In summary, hypoxia activates adrenaline secretion, while baroreceptor unloading activates mainly noradrenaline secretion from the adrenal medulla.
Current therapeutic uses of adrenaline
Adrenaline is used as an emergency treatment for acute ventricular fibrillation and cardiac arrest, acute anaphylactic shock and angio‐oedema and as an emergency treatment of acute airways obstruction in asthma (‘Epi‐Pen’) (Rang et al., 2012). These therapeutic interventions depend on the positive inotropic and bronchodilator effects of adrenaline mediated by activation of β1‐ and β2‐adrenoceptor respectively. Adrenaline is also often combined with local anaesthetics to retard absorption of the anaesthetic agent by promoting local vasoconstriction induced by activation of α1‐adrenoceptors.
Neuroglucoprivation and adrenaline secretion
Adrenaline is a hormone that, along with the pancreatic hormone glucagon, participates in the counter‐regulatory response to hypoglycaemia (Cryer, 1981). Thus, an additional and important action of adrenaline is to increase plasma glucose by promoting glycogenolysis in the liver and skeletal muscle, liver gluconeogenesis and reduction of glucose uptake by tissues such as skeletal muscle via activation of α1‐ and β2‐adrenoceptors (Moratinos et al., 1986). In both liver and skeletal muscle, glycogenolysis occurs as a result of β1‐adrenoceptor‐mediated activation of glycogen phosphorylase. In type 1 diabetes and advanced type 2 diabetes, adrenaline is of primary importance for the response to hypoglycaemia because the ability to secrete glucagon is lost or impaired (Cryer, 2012). Indeed, the glucagon response to hypoglycaemia is lost within 5 years of diagnosis of type 1 diabetes (Amiel, 2005; McCrimmon, 2009). Repeated bouts of hypoglycaemia can also lead to reduced adrenaline secretion and ‘hypoglycaemia unawareness’, in which the symptoms of hypoglycaemia are no longer perceived, creating a vicious cycle of defective glucose homeostasis (Cryer, 2005).
Depriving the brain of glucose (neuroglucoprivation) activates the glucose counter‐regulatory response to restore normal levels of blood glucose (normoglycaemia). In humans, the glucose counter‐regulatory response consists of release into the circulation of the rapid‐acting hormones: glucagon from the pancreatic α‐cells and adrenaline from the adrenal medulla (Bolli and Fanelli, 1999). Growth hormone and cortisol, referred to as slow‐acting hormones, are also released during prolonged hypoglycaemia, but their counter‐regulatory effects do not become evident for some hours. Glucagon acts exclusively by stimulating glucose production in the liver, whereas adrenaline acts by suppressing endogenous insulin secretion, stimulation of hepatic glucose production, stimulation of lipolysis (β3‐adrenoceptor‐mediated activation of lipase in adipose tissue) and reduction of glucose utilization (Bolli and Fanelli, 1999). Neuroglucoprivation can be produced by induction of insulin‐induced hypoglycaemia or by central or peripheral administration of glucose analogues, such as 2‐DG or 5‐thioglucose. 2‐DG is metabolized to 2‐deoxyglucose‐6‐phosphate, which cannot be metabolized further and as a result blocks glucose utilization (Himsworth, 1970; Weindruch et al., 2001; Pelicano et al., 2006; Hersey et al., 2009). The lateral hypothalamus is a critical area mediating the adrenaline response to neuroglucoprivation. Himsworth showed that the hyperglycaemic response to 3‐O‐methylglucose as a glucoprivic agent was abolished by microinjection of the local anaesthetic agent lignocaine bilaterally into the lateral hypothalamus of rats (Himsworth, 1970). Although lignocaine is not selective for neuronal cell bodies, the importance of the lateral hypothalamus has subsequently been verified with local microinjections into the PeH of the GABAA agonist muscimol, which does not inhibit axons of passage. This treatment abolishes the increase in adrenal nerve discharge produced by systemic 2‐DG (Korim et al., 2014). Figure 4 shows a proposed model of the circuitry that mediates adrenaline secretion during hypoglycaemia or glucoprivation.
Figure 4.
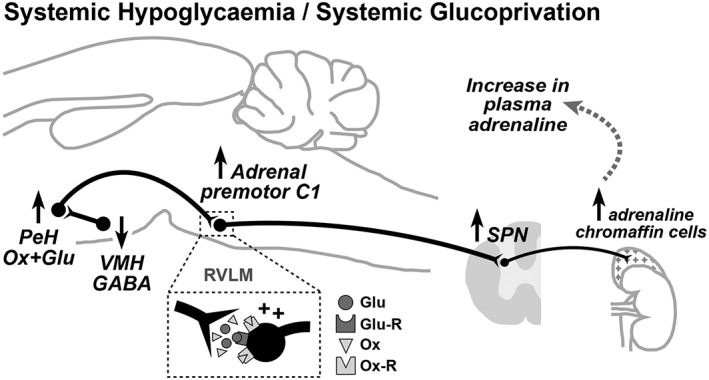
Central neurocircuitry that controls hypoglycaemia‐induced adrenaline release from the adrenal gland. Perifornical hypothalamic (PeH) orexin neurons are activated by falling brain concentrations of glucose (Glu) that are probably detected by GABAergic ventromedial hypothalamic (VMH) neurons. VMH glucose‐sensitive neurons are ‘glucose excited’, and so, the declining brain glucose concentration results in disinhibition of the orexin neurons. Orexin (Ox) release onto ‘adrenal’ sympathetic premotor neurons in the rostral ventrolateral medulla (RVLM) activates sympathetic drive to adrenal chromaffin cells, which release adrenaline thereby raising blood levels of this metabolic hormone.
Orexin neurons in the PeH are activated in response to declining brain glucose concentration (Korim et al., 2016). One mechanism by which this may occur is through disinhibition of PeH orexin neurons as a result of the withdrawal of GABAergic drive from adjacent glucose‐sensing (glucose‐excited) neurons in the ventromedial hypothalamic nucleus (Chan et al., 2006). Most importantly, activation of ASNA during insulin‐induced hypoglycaemia is abolished by blockade of orexin receptors in the RVLM (Korim et al., 2014), where adrenal premotor sympathoexcitatory neurons are located (Verberne and Sartor, 2010). In addition, insulin‐induced activation of ASNA is markedly reduced by excitatory amino acid receptor blockade in the RVLM (Sabetghadam, Korim and Verberne, unpublished observations). This observation is consistent with the finding that a proportion of PeH orexin neurons are glutamatergic (Rosin et al., 2003).
While 2‐DG and insulin‐induced hypoglycaemia can both activate the glucose counter‐regulatory response, there are differences in the mechanism of action. As described above, 2‐DG competes with normal glucose for utilization by brain neurons. The replacement of glucose by non‐metabolizable 2‐DG is perceived as a reduction in the brain glucose concentration and results in an elevation in the plasma levels of glucagon and adrenaline, resulting in hyperglycaemia (Sanders and Ritter, 2000). In contrast, insulin‐induced hypoglycaemia results in elevated plasma levels of glucagon and adrenaline, but glucose levels remain low because of the presence of insulin. Hyperinsulinaemia associated with insulin‐induced hypoglycaemia can be a confounding factor but can be controlled for experimentally by comparing the effects of hyperinsulinaemic hypoglycaemia with hyperinsulinaemic normoglycaemia in which the plasma glucose concentration is held constant by simultaneous glucose infusion (a ‘glucose clamp’) (Ao et al., 2005; Bardgett et al., 2010).
Systemic administration of insulin or 2‐DG results in increased preganglionic ASNA (Carlsson et al., 1992a; Korim et al., 2014). The central circuitry mediating this response is still relatively poorly understood but almost certainly involves central and/or peripheral glucose‐sensing neurons as well as RVLM presympathetic neurons and adrenal SPNs that are activated by 2‐DG‐induced glucoprivation (Morrison and Cao, 2000; Verberne and Sartor, 2010). While the central mechanisms associated with insulin‐induced adrenaline secretion are difficult to study in humans, several important observations have been made. Tetraplegic patients with complete spinal transection between C4 and C8 have lower resting plasma levels of adrenaline and noradrenaline (Mathias et al., 1979). In addition, their plasma levels of adrenaline and noradrenaline do not rise in response to insulin‐induced hypoglycaemia nor do they exhibit the sympathetic symptoms of hypoglycaemia. These observations strongly support the view that a spinal pathway that passes through spinal segments C4–C8 mediates activation of ASNA during hypoglycaemia.
Diabetes and the glucose counter‐regulatory response
The previous section indicates how depriving the brain of glucose activates glucagon and adrenaline secretion. Severe neuroglucoprivation can occur as a result of insulin‐induced hypoglycaemia during the treatment of diabetes (Gabriely and Shamoon, 2004). Indeed, patients with type 1 diabetes frequently report hypoglycaemia particularly when therapy is intensive (Diabetes Control and Complications Trial Research Group, 1993). Hypoglycaemia is feared by patients with type 1 diabetes because of its serious and potentially life‐threatening consequences (Böhme et al., 2013). For reasons that are still incompletely understood, patients with type 1 diabetes soon lose their ability to secrete glucagon during hypoglycaemia (Gerich et al., 1973) despite normal responses to other glucagon‐releasing stimuli such as arginine or the muscarinic agonist carbachol (Fukuda et al., 1988). Loss of the glucagon response to hypoglycaemia also occurs in the alloxan‐treated mouse model of diabetes wherein the glucagon response to insulin and 2‐DG administration is lost despite a normal secretory response to the muscarinic agonist carbachol (Ahren et al., 2002). Similarly, in the streptozotocin rat model of type 1 diabetes, glucagon secretion evoked by electrical stimulation of the cervical vagus was markedly reduced 12 weeks after onset of diabetes despite a normal response to arginine (Hertelendy et al., 1992). These observations suggest (1) that the cellular mechanisms associated with α‐cell glucagon secretion are largely intact in diabetes and (2) possibly that diabetes‐induced autonomic neuropathy may lead to loss of the glucagon response. Ganglionic blockade reduces the glucagon response to insulin‐induced hypoglycaemia by 75–90%, suggesting a pivotal role for the autonomic nervous system in the glucagon response to hypoglycaemia (Havel and Ahren, 1997). This result suggests that diabetes induces a ‘disconnect’ between the regions of the brain that sense declining extracellular levels of glucose and the autonomic output to the pancreas. Despite this recent progress, several important questions remain about the central nerve pathways that control the counter‐regulatory response. First, where is hypoglycaemia sensed (Verberne and Gilbey, 2012; Verberne et al., 2014)? Glucose sensing has been attributed to peripheral neural mechanisms, including vagal and sympathetic afferent glucose sensors (Fujita and Donovan, 2005; Fujita et al., 2007), the carotid body (Koyama et al., 2000), and to several hindbrain structures including the NTS, ventrolateral medulla and several regions of the hypothalamus (Ritter et al., 2000; Routh, 2002; Burdakov and Gonzalez, 2009; Routh, 2010). Donovan and colleagues have proposed that peripheral glucose sensors are important when there is slow and progressive decline in blood glucose concentration while central glucose sensors respond to a rapid decline in brain glucose concentrations (Donovan and Watts, 2014). Second, why are there so many glucose‐sensing regions? Perhaps, this redundancy occurs because glucose homeostasis is not only vital to bodily function but also critical for the functioning of the nervous system. Alternatively, different glucose‐sensing circuits may have differing responsibilities, for example, control of energy balance or feeding or glucose homeostasis.
Conclusion
For over a century, pharmacologists have focussed on the substantial circulatory effects of exogenously administered adrenaline and on the role of adrenaline in the fight‐or‐flight response. However, recent evidence summarized here suggests that endogenous adrenaline is, in fact, primarily a metabolic hormone (Cryer, 1993), which is of critical importance in the glucose counter‐regulatory response evoked by hypoglycaemia. In fact, adrenaline is now acknowledged as the primary counter‐regulatory hormone in people with type 1 diabetes. In these patients, repeated bouts of hypoglycaemia can lead to hypoglycaemia unawareness. Hypoglycaemia unawareness is characterized by marked loss of the sympathetic responses mediating adrenaline secretion and, if left unchecked, can lead to coma or even death. Understanding how hypoglycaemia activates central neurons to cause adrenaline release from chromaffin cells will be important for identifying new therapeutic targets to combat this potentially life‐threatening condition.
Author contributions
All authors contributed equally to the concept, design and preparation of the manuscript.
Conflict of interest
The authors declare no conflicts of interest.
Acknowledgements
This work was supported by grants from the National Health and Medical Research Council of Australia to A.J.M.V and I.J.L‐S, the Austin Medical Research Foundation to A.J.M.V., W.S.K. and A.S. and a University of Melbourne International Postgraduate student award to A.S.
Verberne, A. J. M. , Korim, W. S. , Sabetghadam, A. , and Llewellyn‐Smith, I. J. (2016) Adrenaline: insights into its metabolic roles in hypoglycaemia and diabetes. British Journal of Pharmacology, 173: 1425–1437. doi: 10.1111/bph.13458.
References
- Abel JJ (1898). On epinephrin, the active constituent of the suprarenal capsule and its compounds. Proc Am Physiol Soc 3‐4: iii–iiv. [Google Scholar]
- Addison T (1855). On the constitutional and local effects of disease of the suprarenal capsules. 1st edn Samuel Highley: London. [Google Scholar]
- Ahlquist RP (1948). A study of the adrenotropic receptors. Am J Physiol 153: 586–600. [DOI] [PubMed] [Google Scholar]
- Ahren B, Taborsky GJ Jr, Havel PJ (2002). Differential impairment of glucagon responses to hypoglycemia, neuroglycopenia, arginine, and carbachol in alloxan‐diabetic mice. Metabolism 51: 12–19. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE, et al. (2015a). The Concise Guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE, et al. (2015b). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amiel SA (2005). Iatrogenic hypoglycemia In: Kahn CR, Weir GC, King GL, Jacobson AM, Moses AC, Smith RJ. (eds). Joslin's Diabetes Mellitus, 14th edn. Lippincott Williams & Wilkins: Philadelphia, pp. 671–686. [Google Scholar]
- Ao Y, Wu S, Go VL, Toy N, Yang H (2005). Maintaining euglycemia prevents insulin‐induced Fos expression in brain autonomic regulatory circuits. Pancreas 31: 142–147. [DOI] [PubMed] [Google Scholar]
- Axelrod J (1966). Methylation reactions in the formation and metabolism of catecholamines and other biogenic amines. Pharmacol Rev 18: 95–113. [PubMed] [Google Scholar]
- Bardgett ME, McCarthy JJ, Stocker SD (2010). Glutamatergic receptor activation in the rostral ventrolateral medulla mediates the sympathoexcitatory response to hyperinsulinemia. Hypertension 55: 284–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biesold D, Kurosawa M, Sato A, Trzebski A (1989). Hypoxia and hypercapnia increase the sympathoadrenal medullary functions in anesthetized, artificially ventilated rats. Jpn J Physiol 39: 511–522. [DOI] [PubMed] [Google Scholar]
- Biollaz B, Biollaz J, Kohlman O Jr, Bresnahan M, Gavras I, Gavras H (1984). Acute cardiovascular effects of two central phenylethanolamine‐N‐methyl‐transferase inhibitors in unanesthetized desoxycorticosterone‐salt hypertensive rats. Eur J Pharmacol 102: 515–519. [DOI] [PubMed] [Google Scholar]
- Black J, Waeber B, Bresnahan MR, Gavras I, Gavras H (1981). Blood pressure response to central and/or peripheral inhibition of phenylethanolamine N‐methyltransferase in normotensive and hypertensive rats. Circ Res 49: 518–524. [DOI] [PubMed] [Google Scholar]
- Böhme P, Bertin E, Cosson E, Chevalier N (2013). Fear of hypoglycaemia in patients with type 1 diabetes: do patients and diabetologists feel the same way? Diabetes Metab 39: 63–70. [DOI] [PubMed] [Google Scholar]
- Bolli GB, Fanelli CG (1999). Physiology of glucose counterregulation to hypoglycemia. Endocrinol Metab Clin North Am 28: 467–493. [DOI] [PubMed] [Google Scholar]
- Burdakov D, Gonzalez JA (2009). Physiological functions of glucose‐inhibited neurones. Acta Physiol (Oxf) 195: 71–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon WB, Rosenblueth A (1935). A comparative study of sympathin and adrenine. Am J Physiol 112: 268–276. [Google Scholar]
- Card JP, Sved JC, Craig B, Raizada M, Vazquez J, Sved AF (2006). Efferent projections of rat rostroventrolateral medulla C1 catecholamine neurons: implications for the central control of cardiovascular regulation. J Comp Neurol 499: 840–859. [DOI] [PubMed] [Google Scholar]
- Carlsson S, Jonsdottir IH, Skarphedinsson JO, Thoren P (1993). Evidence for an adrenergic innervation of the adrenal cortical blood vessels in rats. Acta Physiol Scand 149: 23–30. [DOI] [PubMed] [Google Scholar]
- Carlsson S, Skarphedinsson JO, Delle M, Hoffman P, Thoren P (1992a). Differential responses in post‐ and pre‐ganglionic adrenal sympathetic nerve activity and renal sympathetic nerve activity after injection of 2‐deoxy‐d‐glucose and insulin in rats. Acta Physiol Scand 145: 169–175. [DOI] [PubMed] [Google Scholar]
- Carlsson S, Skarphedinsson JO, Delle M, Hoffman P, Thoren P (1992b). Reflex changes in post‐ and preganglionic sympathetic adrenal nerve activity and postganglionic sympathetic renal nerve activity upon arterial baroreceptor activation and during severe haemorrhage in the rat. Acta Physiol Scand 144: 317–323. [DOI] [PubMed] [Google Scholar]
- Carrive P, Bandler R, Dampney RA (1988). Anatomical evidence that hypertension associated with the defence reaction in the cat is mediated by a direct projection from a restricted portion of the midbrain periaqueductal grey to the subretrofacial nucleus of the medulla. Brain Res 460: 339–345. [DOI] [PubMed] [Google Scholar]
- Carrive P, Bandler R, Dampney RA (1989). Viscerotopic control of regional vascular beds by discrete groups of neurons within the midbrain periaqueductal gray. Brain Res 493: 385–390. [DOI] [PubMed] [Google Scholar]
- Celler BG, Schramm LP (1981). Pre‐ and postganglionic sympathetic activity in splanchnic nerves of rats. Am J Physiol 241: R55–R61. [DOI] [PubMed] [Google Scholar]
- Chan O, Zhu W, Ding Y, McCrimmon RJ, Sherwin RS (2006). Blockade of GABA(A) receptors in the ventromedial hypothalamus further stimulates glucagon and sympathoadrenal but not the hypothalamo–pituitary–adrenal response to hypoglycemia. Diabetes 55: 1080–1087. [DOI] [PubMed] [Google Scholar]
- Collett AR, Rand MJ, Story DF (1984). Catecholamine secretion from the rabbit adrenal gland is not modulated by a mechanism involving beta‐adrenoceptors. Arch Int Pharmacodyn Ther 269: 63–69. [PubMed] [Google Scholar]
- Collett AR, Story DF (1982). Release of 3H‐adrenaline from an isolated intact preparation of the rabbit adrenal gland: no evidence for release modulatory alpha‐adrenoreceptors. J Auton Pharmacol 2: 25–34. [DOI] [PubMed] [Google Scholar]
- Coupland RE (1956). The development and fate of the abdominal chromaffin tissue in the rabbit. J Anat 90: 527–537. [PMC free article] [PubMed] [Google Scholar]
- Coupland RE (1953). On the morphology and adrenaline‐nor‐adrenaline content of chromaffin tissue. J Endocrinol 9: 194–203. [DOI] [PubMed] [Google Scholar]
- Coupland RE, Parker TL, Kesse WK, Mohamed AA (1989). The innervation of the adrenal gland. III. Vagal innervation. J Anat 163: 173–181. [PMC free article] [PubMed] [Google Scholar]
- Critchley JAJH, Ellis P, Ungar A (1980). The reflex release of adrenaline and noradrenaline from the adrenal gland of cats and dogs. J Physiol (Lond) 298: 71–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cryer PE (1993). Adrenaline: a physiological metabolic regulatory hormone in humans? Int J Obes Relat Metab Disord 17 (Suppl 3): S43–S46. [PubMed] [Google Scholar]
- Cryer PE (1981). Glucose counterregulation in man. Diabetes 30: 261–264. [DOI] [PubMed] [Google Scholar]
- Cryer PE (2005). Mechanisms of hypoglycemia‐associated autonomic failure and its component syndromes in diabetes. Diabetes 54: 3592–3601. [DOI] [PubMed] [Google Scholar]
- Cryer PE (2012). Minireview: glucagon in the pathogenesis of hypoglycemia and hyperglycemia in diabetes. Endocrinology 153: 1039–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davenport HW (1982). Epinephrin(e). Physiologist 25: 76–82. [PubMed] [Google Scholar]
- Diabetes Control and Complications Trial Research Group (1993). The effect of intensive treatment of diabetes on the development and progression of long‐term complications in insulin‐dependent diabetes mellitus. The Diabetes Control and Complications Trial Research Group. N Engl J Med 329: 977‐986. [DOI] [PubMed] [Google Scholar]
- Ding ZQ, Li YW, Wesselingh SL, Blessing WW (1993). Transneuronal labelling of neurons in rabbit brain after injection of herpes simplex virus type 1 into the renal nerve. J Auton Nerv Syst 42: 23–31. [DOI] [PubMed] [Google Scholar]
- Donovan CM, Watts AG (2014). Peripheral and central glucose sensing in hypoglycemic detection. Physiology (Bethesda) 29: 314–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards AV (1982). Adrenal catecholamine output in response to stimulation of the splanchnic nerve in bursts in the conscious calf. J Physiol (Lond) 327: 409–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards SL, Anderson CR, Southwell BR, McAllen RM (1996). Distinct preganglionic neurons innervate noradrenaline and adrenaline cells in the cat adrenal medulla. Neuroscience 70: 825–832. [DOI] [PubMed] [Google Scholar]
- Esler MD (1993). Catecholamines and essential hypertension. Baillieres Clin Endocrinol Metab 7: 415–438. [DOI] [PubMed] [Google Scholar]
- Fenwick NM, Martin CL, Llewellyn‐Smith IJ (2006). Immunoreactivity for cocaine‐ and amphetamine‐regulated transcript in rat sympathetic preganglionic neurons projecting to sympathetic ganglia and the adrenal medulla. J Comp Neurol 495: 422–433. [DOI] [PubMed] [Google Scholar]
- Fitzgerald GA, Barnes P, Hamilton CA, Dollery CT (1980). Circulating adrenaline and blood pressure: the metabolic effects and kinetics of infused adrenaline in man. Eur J Clin Invest 10: 401–406. [DOI] [PubMed] [Google Scholar]
- Folkow B, Von Euler US (1954). Selective activation of noradrenaline and adrenaline producing cells in the cat's adrenal gland by hypothalamic stimulation. Circ Res 2: 191–195. [DOI] [PubMed] [Google Scholar]
- Fuchs SA, Edinger HM, Siegel A (1985). The organization of the hypothalamic pathways mediating affective defense behavior in the cat. Brain Res 330: 77–92. [DOI] [PubMed] [Google Scholar]
- Fujita S, Bohland MA, Sanchez‐Watts G, Watts AG, Donovan CM (2007). Hypoglycemic detection at the portal vein is mediated by capsaicin‐sensitive primary sensory neurons. Am J Physiol 293: E96–E101. [DOI] [PubMed] [Google Scholar]
- Fujita S, Donovan CM (2005). Celiac‐superior mesenteric ganglionectomy, but not vagotomy, suppresses the sympathoadrenal response to insulin‐induced hypoglycemia. Diabetes 54: 3258–3264. [DOI] [PubMed] [Google Scholar]
- Fukuda M, Tanaka A, Tahara Y, Ikegami H, Yamamoto Y, Kumahara Y, et al. (1988). Correlation between minimal secretory capacity of pancreatic beta‐cells and stability of diabetic control. Diabetes 37: 81–88. [DOI] [PubMed] [Google Scholar]
- Gabriely I, Shamoon H (2004). Hypoglycemia in diabetes: common, often unrecognized. Cleve Clin J Med 71: 335–342. [DOI] [PubMed] [Google Scholar]
- Geerling JC, Mettenleiter TC, Loewy AD (2003). Orexin neurons project to diverse sympathetic outflow systems. Neuroscience 122: 541–550. [DOI] [PubMed] [Google Scholar]
- Gerich JE, Langlois M, Noacco C, Karam JH, Forsham PH (1973). Lack of glucagon response to hypoglycemia in diabetes: evidence for an intrinsic pancreatic alpha cell defect. Science 182: 171–173. [DOI] [PubMed] [Google Scholar]
- Goldstein DS (1983). Plasma catecholamines and essential hypertension. An analytical review. Hypertension 5: 86–99. [DOI] [PubMed] [Google Scholar]
- Goldstein DS (1999). Clinical pharmacology of the autonomic nervous system In: Appenzeller O. (ed). Handbook of Clinical Neurology, The Autonomic Nervous System Part I, edn, Vol. 74 Elsevier: Amsterdam, pp. 135–179. [Google Scholar]
- Gonsalvez DG, Kerman IA, McAllen RM, Anderson CR (2010). Chemical coding for cardiovascular sympathetic preganglionic neurons in rats. J Neurosci 30: 11781–11791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Havel PJ, Ahren B (1997). Activation of autonomic nerves and the adrenal medulla contributes to increased glucagon secretion during moderate insulin‐induced hypoglycemia in women. Diabetes 46: 801–807. [DOI] [PubMed] [Google Scholar]
- Hersey P, Watts RN, Zhang XD, Hackett J (2009). Metabolic approaches to treatment of melanoma. Clin Cancer Res 15: 6490–6494. [DOI] [PubMed] [Google Scholar]
- Hertelendy ZI, Patel DG, Skau KA (1992). Progressive and concurrent deterioration of vagus‐stimulated and hypoglycemia‐induced glucagon secretion in streptozotocin‐diabetic rats. Acta Endocrinol 126: 80–84. [DOI] [PubMed] [Google Scholar]
- Hilton SM (1982). The defence‐arousal system and its relevance for circulatory and respiratory control. J Exp Biol 100: 159–174. [DOI] [PubMed] [Google Scholar]
- Himsworth RL (1970). Hypothalamic control of adrenaline secretion in response to insufficient glucose. J Physiol (Lond) 206: 411–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holgert H, Aman K, Cozzari C, Hartman BK, Brimijoin S, Emson P, et al. (1995). The cholinergic innervation of the adrenal gland and its relation to enkephalin and nitric oxide synthase. Neuroreport 6: 2576–2580. [DOI] [PubMed] [Google Scholar]
- Jensen I, Llewellyn‐Smith IJ, Pilowsky P, Minson JB, Chalmers J (1995). Serotonin inputs to rabbit sympathetic preganglionic neurons projecting to the superior cervical ganglion or adrenal medulla. J Comp Neurol 353: 427–438. [DOI] [PubMed] [Google Scholar]
- Jensen I, Pilowsky P, Llewellyn‐Smith IJ, Minson J, Chalmers J (1992). Sympathetic preganglionic neurons projecting to the adrenal medulla and aorticorenal ganglion in the rabbit. Brain Res 586: 125–129. [DOI] [PubMed] [Google Scholar]
- Kerman IA, Bernard R, Rosenthal D, Beals J, Akil H, Watson SJ (2007). Distinct populations of presympathetic‐premotor neurons express orexin or melanin‐concentrating hormone in the rat lateral hypothalamus. J Comp Neurol 505: 586–601. [DOI] [PubMed] [Google Scholar]
- Kesse WK, Parker TL, Coupland RE (1988). The innervation of the adrenal gland. I. The source of pre‐ and postganglionic nerve fibres to the rat adrenal gland. J Anat 157: 33–41. [PMC free article] [PubMed] [Google Scholar]
- Korim WS, Bou‐Farah L, McMullan S, Verberne AJ (2014). Orexinergic activation of medullary premotor neurons modulates the adrenal sympathoexcitation to hypothalamic glucoprivation. Diabetes 63: 1895–1906. [DOI] [PubMed] [Google Scholar]
- Korim WS, Llewellyn‐Smith IJ, Verberne AJM (2016). Activation of medulla‐projecting perifornical neurons modulates the adrenal sympathetic response to hypoglycemia: involvement of orexin type 2 (OX2‐R) receptors. Endocrinology 157: 810–819. [DOI] [PubMed] [Google Scholar]
- Koyama Y, Coker RH, Stone EE, Lacy DB, Jabbour K, Williams PE, et al. (2000). Evidence that carotid bodies play an important role in glucoregulation in vivo . Diabetes 49: 1434–1442. [DOI] [PubMed] [Google Scholar]
- Kuhar MJ, Couceyro P, Lambert PD (1999). Biosynthesis of catecholamines In: Siegel GJ, Agranoff BW, Albers RW, Fisher SK, Uhler MD. (eds). Basic Neurochemistry: Molecular, Cellular and Medical Aspects, 6th edn. Lippincott‐Raven: Philadelphia. [Google Scholar]
- Lee K, Miwa S, Fujiwara M, Magaribuchi T (1987). Differential effects of hypoxia on the turnover of norepinephrine and epinephrine in the heart, adrenal gland, submaxillary gland and stomach. J Pharmacol Exp Ther 240: 954–958. [PubMed] [Google Scholar]
- Li YW, Wesselingh SL, Blessing WW (1992). Projections from rabbit caudal medulla to C1 and A5 sympathetic premotor neurons, demonstrated with phaseolus leucoagglutinin and herpes simplex virus. J Comp Neurol 317: 379–395. [DOI] [PubMed] [Google Scholar]
- Lovick TA (1992). Midbrain influences on ventrolateral medullo‐spinal neurones in the rat. Exp Brain Res 90: 147–152. [DOI] [PubMed] [Google Scholar]
- Majewski H, Tung LH, Rand MJ (1981). Adrenaline‐induced hypertension in rats. J Cardiovasc Pharmacol 3: 179–185. [DOI] [PubMed] [Google Scholar]
- Mathias CJ, Frankel HL, Turner RC, Christensen NJ (1979). Physiological responses to insulin hypoglycaemia in spinal man. Paraplegia 17: 319–326. [DOI] [PubMed] [Google Scholar]
- McCrimmon R (2009). Glucose sensing during hypoglycemia: lessons from the lab. Diabetes Care 32: 1357–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIsaac RJ, Millerschoen NR (1966). A comparison of the effects of mecamylamine and hexamethonium on transmission in the superior cervical ganglion of the cat. J Pharmacol Exp Ther 139: 18–24. [Google Scholar]
- Menuet C, Sevigny CP, Connelly AA, Bassi JK, Jancovski N, Williams DA, et al. (2014). Catecholaminergic C3 neurons are sympathoexcitatory and involved in glucose homeostasis. J Neurosci 34: 15110–15122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merck I (1968). The Merck Index. 8th edn Merck & Co. Inc.: Rahway NJ. [Google Scholar]
- Mohamed AA, Parker TL, Coupland RE (1988). The innervation of the adrenal gland. II. The source of spinal afferent nerve fibres to the guinea‐pig adrenal gland. J Anat 160: 51–58. [PMC free article] [PubMed] [Google Scholar]
- Moratinos J, Olmedilla B, de Pablos I, Vigueras MD (1986). Alpha‐adrenoceptor involvement in catecholamine‐induced hyperglycaemia in conscious fasted rabbits. Br J Pharmacol 89: 55–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita H, Vatner SF (1985). Effects of hemorrhage on renal nerve activity in conscious dogs. Circ Res 57: 788–793. [DOI] [PubMed] [Google Scholar]
- Morrison SF, Cao WH (2000). Different adrenal sympathetic preganglionic neurons regulate epinephrine and norepinephrine secretion. Am J Physiol 279: R1763–R1775. [DOI] [PubMed] [Google Scholar]
- Mundinger TO, Boyle MR, Taborsky GJ Jr (1997). Activation of hepatic sympathetic nerves during hypoxic, hypotensive and glucopenic stress. J Auton Nerv Syst 63: 153–160. [DOI] [PubMed] [Google Scholar]
- Natarajan M, Morrison SF (1999). Adrenal epinephrine secretion is not regulated by sympathoinhibitory neurons in the caudal ventrolateral medulla. Brain Res 827: 169–175. [DOI] [PubMed] [Google Scholar]
- Niijima A (1992). Electrophysiological study on the vagal innervation of the adrenal gland in the rat. J Auton Nerv Syst 41: 87–92. [DOI] [PubMed] [Google Scholar]
- Oliver G, Schafer EA (1895). The physiological effects of extracts of the suprarenal capsules. J Physiol (Lond) 18: 230–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker TL, Mohamed AA, Coupland RE (1990). The innervation of the adrenal gland. IV. The source of pre‐ and postganglionic nerve fibres to the guinea‐pig adrenal gland. J Anat 172: 17–24. [PMC free article] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. (2014). The IUPHAR/BPS guide to PHARMACOLOGY: an expert‐driven knowledge base of drug targets and their ligands. Nucleic Acids Res 42: D1098–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelicano H, Martin DS, Xu RH, Huang P (2006). Glycolysis inhibition for anticancer treatment. Oncogene 25: 4633–4646. [DOI] [PubMed] [Google Scholar]
- Rang HP, Dale MM, Ritter JM, Flower RJ, Henderson G (2012). Rang and Dale's Pharmacology. 7th edn Elsevier Churchill Livingstone: Edinburgh. [Google Scholar]
- Rea RF, Biaggioni I, Robertson RM, Haile V, Robertson D (1990). Reflex control of sympathetic nerve activity in dopamine beta‐hydroxylase deficiency. Hypertension 15: 107–112. [DOI] [PubMed] [Google Scholar]
- Reed JD, Sanders DJ, Thorpe V (1971). The effect of splanchnic nerve stimulation on gastric acid secretion and mucosal blood flow in the anaesthetized cat. J Physiol (Lond) 214: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritter S, Bugarith K, Dinh TT (2001). Immunotoxic destruction of distinct catecholamine subgroups produces selective impairment of glucoregulatory responses and neuronal activation. J Comp Neurol 432: 197–216. [DOI] [PubMed] [Google Scholar]
- Ritter S, Dinh TT, Zhang Y (2000). Localization of hindbrain glucoreceptive sites controlling food intake and blood glucose. Brain Res 856: 37–47. [DOI] [PubMed] [Google Scholar]
- Ritter S, Llewellyn‐Smith IJ, Dinh TT (1998). Subgroups of hindbrain catecholamine neurons are selectively activated by 2‐deoxy‐d‐glucose induced metabolic challenge. Brain Res 805: 41–54. [DOI] [PubMed] [Google Scholar]
- Rogers PF, Head GA, Lungershausen YK, Howe PR (1991). Effects of depleting central and peripheral adrenaline stores on blood pressure in stroke‐prone spontaneously hypertensive rats. J Auton Nerv Syst 34: 9–16. [DOI] [PubMed] [Google Scholar]
- Rosin DL, Weston MC, Sevigny CP, Stornetta RL, Guyenet PG (2003). Hypothalamic orexin (hypocretin) neurons express vesicular glutamate transporters VGLUT1 or VGLUT2. J Comp Neurol 465: 593–603. [DOI] [PubMed] [Google Scholar]
- Routh VH (2002). Glucose‐sensing neurons: are they physiologically relevant? Physiol Behav 76: 403–413. [DOI] [PubMed] [Google Scholar]
- Routh VH (2010). Glucose sensing neurons in the ventromedial hypothalamus. Sensors (Basel) 10: 9002–9025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders NM, Ritter S (2000). Repeated 2‐deoxy‐d‐glucose‐induced glucoprivation attenuates Fos expression and glucoregulatory responses during subsequent glucoprivation. Diabetes 49: 1865–1874. [DOI] [PubMed] [Google Scholar]
- Sapru HN, Gonzalez ER, Krieger AJ (1982). Greater splanchnic nerve activity in the rat. Brain Res Bull 8: 267–272. [DOI] [PubMed] [Google Scholar]
- Schadt JC, Gaddis RR (1988). Role of adrenal medulla in hemodynamic response to hemorrhage and naloxone. Am J Physiol 254: R559–R565. [DOI] [PubMed] [Google Scholar]
- Schramm LP, Strack AM, Platt KB, Loewy AD (1993). Peripheral and central pathways regulating the kidney: a study using pseudorabies virus. Brain Res 616: 251–262. [DOI] [PubMed] [Google Scholar]
- Schreihofer AM, Guyenet PG (2002). The baroreflex and beyond: control of sympathetic vasomotor tone by GABAergic neurons in the ventrolateral medulla. Clin Exp Pharmacol Physiol 29: 514–521. [DOI] [PubMed] [Google Scholar]
- Schreihofer AM, Sved AF (2011). The ventrolateral medulla and sympathetic regulation of arterial pressure In: Llewellyn‐Smith IJ, Verberne AJ. (eds). Central Regulation of Autonomic Functions, 2nd edn. Oxford University Press: New York, pp. 78–97. [Google Scholar]
- Scislo TJ, Augustyniak RA, Oleary DS (1998). Differential arterial baroreflex regulation of renal, lumbar, and adrenal sympathetic nerve activity in the rat. Am J Physiol 44: R995–R1002. [DOI] [PubMed] [Google Scholar]
- Seidler FJ, Slotkin TA (1986). Ontogeny of adrenomedullary responses to hypoxia and hypoglycemia: role of splanchnic innervation. Brain Res Bull 16: 11–14. [DOI] [PubMed] [Google Scholar]
- Shoukas AA (1982). Carotid sinus baroreceptor reflex control and epinephrine. Influence on capacitive and resistive properties of the total pulmonary vascular bed of the dog. Circ Res 51: 95–101. [DOI] [PubMed] [Google Scholar]
- Sly DJ, Colvill L, McKinley MJ, Oldfield BJ (1999). Identification of neural projections from the forebrain to the kidney, using the virus pseudorabies. J Auton Nerv Syst 77: 73–82. [PubMed] [Google Scholar]
- Smith OA, Astley CA, Spelman FA, Golanov EV, Bowden DM, Chesney MA, et al. (2000). Cardiovascular responses in anticipation of changes in posture and locomotion. Brain Res Bull 53: 69–76. [DOI] [PubMed] [Google Scholar]
- Standish A, Enquist LW, Schwaber JS (1994). Innervation of the heart and its central medullary origin defined by viral tracing. Science 263: 232–235. [DOI] [PubMed] [Google Scholar]
- Strack AM, Sawyer WB, Hughes JH, Platt KB, Loewy AD (1989a). A general pattern of CNS innervation of the sympathetic outflow demonstrated by transneuronal pseudorabies viral infections. Brain Res 491: 156–162. [DOI] [PubMed] [Google Scholar]
- Strack AM, Sawyer WB, Marubio LM, Loewy AD (1988). Spinal origin of sympathetic preganglionic neurons in the rat. Brain Res 455: 187–191. [DOI] [PubMed] [Google Scholar]
- Strack AM, Sawyer WB, Platt KB, Loewy AD (1989b). CNS cell groups regulating the sympathetic outflow to adrenal gland as revealed by transneuronal cell body labeling with pseudorabies virus. Brain Res 491: 274–296. [DOI] [PubMed] [Google Scholar]
- Takamine J (1902). The blood‐pressure raising principle of the suprarenal gland. JAMA 38: 153–155. [Google Scholar]
- Thompson JM, O'Callaghan CJ, Kingwell BA, Lambert GW, Jennings GL, Esler MD (1995). Total norepinephrine spillover, muscle sympathetic nerve activity and heart‐rate spectral analysis in a patient with dopamine beta‐hydroxylase deficiency. J Auton Nerv Syst 55: 198–206. [DOI] [PubMed] [Google Scholar]
- Toth IE, Banczerowski P, Boldogkoi Z, Toth JS, Szabo A, Halasz B, et al. (2008a). Cerebral neurons involved in the innervation of both the adrenal gland and the ovary: a double viral tracing study. Brain Res Bull 77: 306–311. [DOI] [PubMed] [Google Scholar]
- Toth IE, Vizi ES, Hinson JP, Vinson GP (1997). Innervation of the adrenal cortex, its physiological relevance, with primary focus on the noradrenergic transmission. Microsc Res Tech 36: 534–545. [DOI] [PubMed] [Google Scholar]
- Toth IE, Wiesel O, Toth DE, Boldogkoi Z, Halasz B, Gerendai I (2008b). Transneuronal retrograde viral labeling in the brain stem and hypothalamus is more intense from the left than from the right adrenal gland. Microsc Res Tech 71: 503–509. [DOI] [PubMed] [Google Scholar]
- Tsuchimochi H, Nakamoto T, Matsukawa K (2010). Centrally evoked increase in adrenal sympathetic outflow elicits immediate secretion of adrenaline in anaesthetized rats. Exp Physiol 95: 93–106. [DOI] [PubMed] [Google Scholar]
- Tung LH, Rand MJ, Majewski H (1981). Adrenaline‐induced hypertension in rats. Clin Sci (Lond) 61 (Suppl 7): 191s–193s. [DOI] [PubMed] [Google Scholar]
- Verberne AJ, Gilbey MP (2012). Highlights in basic autonomic neurosciences: autonomic control of the counter‐regulatory response and glucose homeostasis. Auton Neurosci Basic Clin 169: 1–3. [DOI] [PubMed] [Google Scholar]
- Verberne AJ, Sabetghadam A, Korim WS (2014). Neural pathways that control the glucose counterregulatory response. Front Autonom Neurosci 8: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verberne AJ, Sartor DM (2010). Rostroventrolateral medulla neurons modulate glucose homeostasis in the rat. Am J Physiol 299: E802–E807. [DOI] [PubMed] [Google Scholar]
- Verberne AJM, Guyenet PG (1992). Midbrain central gray: influence on medullary sympathoexcitatory neurons and the baroreflex in rats. Am J Physiol 263: R24–R33. [DOI] [PubMed] [Google Scholar]
- Verberne AJM, Stornetta RL, Guyenet PG (1999). Properties of C1 and other ventrolateral medullary neurones with hypothalamic projections in the rat. J Physiol (Lond) 517: 477–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhofstad AA, Coupland RE, Parker TR, Goldstein M (1985). Immunohistochemical and biochemical study on the development of the noradrenaline‐ and adrenaline‐storing cells of the adrenal medulla of the rat. Cell Tissue Res 242: 233–243. [DOI] [PubMed] [Google Scholar]
- Vollmer RR, Baruchin A, Kolibal‐Pegher SS, Corey SP, Stricker EM, Kaplan BB (1992). Selective activation of norepinephrine‐ and epinephrine‐secreting chromaffin cells in rat adrenal medulla. Am J Physiol 263: R716–R721. [DOI] [PubMed] [Google Scholar]
- von Euler US (1946). A specific sympathomimetic ergone in adrenergic nerve fibres (sympathin) and its relations to adrenaline and nor‐adrenaline. Acta Physiol Scand 12: 73–96. [Google Scholar]
- von Euler US (1966). Twenty years of noradrenaline. Pharmacol Rev 18: 29–38. [PubMed] [Google Scholar]
- Wakade AR (1981). Studies on secretion of catecholamines evoked by acetylcholine or transmural stimulation of the rat adrenal gland. J Physiol (Lond) 313: 463–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakade AR, Wakade TD (1983). Contribution of nicotinic and muscarinic receptors in the secretion of catecholamines evoked by endogenous and exogenous acetylcholine. Neuroscience 10: 973–978. [DOI] [PubMed] [Google Scholar]
- Warren JB, Dalton N (1983). A comparison of the bronchodilator and vasopressor effects of exercise levels of adrenaline in man. Clin Sci (Lond) 64: 475–479. [DOI] [PubMed] [Google Scholar]
- Warren JB, Dalton N, Turner C, Clark TJ, Toseland PA (1984). Adrenaline secretion during exercise. Clin Sci (Lond) 66: 87–90. [DOI] [PubMed] [Google Scholar]
- Weindruch R, Keenan KP, Carney JM, Fernandes G, Feuers RJ, Floyd RA, et al. (2001). Caloric restriction mimetics: metabolic interventions. J Gerontol A Biol Sci Med Sci 56 (Spec No 1): 20–33. [DOI] [PubMed] [Google Scholar]
- Wesselingh SL, Li YW, Blessing WW (1989). PNMT‐containing neurons in the rostral medulla oblongata (C1, C3 groups) are transneuronally labelled after injection of herpes simplex virus type 1 into the adrenal gland. Neurosci Lett 106: 99–104. [DOI] [PubMed] [Google Scholar]
- Westerhaus MJ, Loewy AD (2001). Central representation of the sympathetic nervous system in the cerebral cortex. Brain Res 903: 117–127. [DOI] [PubMed] [Google Scholar]
- Westerhaus MJ, Loewy AD (1999). Sympathetic‐related neurons in the preoptic region of the rat identified by viral transneuronal labeling. J Comp Neurol 414: 361–378. [PubMed] [Google Scholar]
- Westfall TC, Westfall DP (2011). Adrenergic agonists and antagonists In: Brunton LL. (ed). Goodman & Gilman's The Pharmacological Basis of Therapeutics, 12th edn. McGraw‐Hill: New York, pp. 277–333. [Google Scholar]
- Wurtman RJ, Axelrod J (1965). Adrenaline synthesis: control by the pituitary gland and adrenal glucocorticoids. Science 150: 1464–1465. [DOI] [PubMed] [Google Scholar]
- Wurtman RJ, Axelrod J (1966). Control of enzymatic synthesis of adrenaline in the adrenal medulla by adrenal cortical steroids. J Biol Chem 241: 2301–2305. [PubMed] [Google Scholar]
- Wurtman RJ, Pohorecky LA, Baliga BS (1972). Adrenocortical control of the biosynthesis of epinephrine and proteins in the adrenal medulla. Pharmacol Rev 24: 411–426. [PubMed] [Google Scholar]
- Yardley CP, Hilton SM (1987). Vasodilatation in hind‐limb skeletal muscle evoked as part of the defence reaction in the rat. J Auton Nerv Syst 19: 127–136. [DOI] [PubMed] [Google Scholar]