

Abstract
Air pollution is a major challenge to public health. Ambient fine particulate matter (PM) is the key component for air pollution, and associated with significant mortality. The majority of the mortality following PM exposure is related to cardiovascular diseases. However, the mechanisms for the adverse effects of PM exposure on cardiovascular system remain largely unknown and under active investigation. Endothelial dysfunction or injury is considered one of the major factors that contribute to the development of cardiovascular diseases such as atherosclerosis and coronary heart disease. Endothelial progenitor cells (EPCs) play a critical role in maintaining the structural and functional integrity of vasculature. Particulate matter exposure significantly suppressed the number and function of EPCs in animals and humans. However, the mechanisms for the detrimental effects of PM on EPCs remain to be fully defined. One of the important mechanisms might be related to increased level of reactive oxygen species (ROS) and inflammation. Bone marrow (BM) is a major source of EPCs. Thus, the number and function of EPCs could be intimately associated with the population and functional status of stem cells (SCs) in the BM. Bone marrow stem cells and other SCs have the potential for cardiovascular regeneration and repair. The present review is focused on summarizing the detrimental effects of PM exposure on EPCs and SCs, and potential mechanisms including ROS formation as well as clinical implications.
Keywords: air pollution, PM, cardiovascular disease, stem cell, endothelial progenitor cell
Introduction
Air pollution is a major challenge to public health. Ambient particulate matter (PM) is the key component for air pollution. A recent Global Burden of Disease Study showed that PM exposure is responsible for 3.2 million deaths per year and 76 million years of healthy life lost 1. Particulate matter exposure could be especially a major health problem in the developing countries as the fine PM levels in some developing countries are reported to be 10 times higher than that in the developed countries 2. The majority of mortality following fine PM exposure has been related to cardiovascular diseases 1.
Particulate matter is a mixture of various particles including metals, crustal material and bio‐aerosols 3, 4. The particles with a median aerodynamic diameter of <2.5 μm (PM2.5) and <10 μm (PM10) are of serious global health concerns because of their close association (especially PM2.5) with the detrimental effects of air pollution on our health 5, 6. The PM2.5 exposure has been reported to produce a variety of deleterious effects on cardiovascular system including (but not limited to) vascular dysfunction, reduced heart rate variability and enhanced risk for thrombosis 3, 7. Long‐term exposure to PM2.5 has been demonstrated to accelerate the process of atherosclerosis and vascular inflammation in apolipoprotein E−/− mice with high‐fat diet 8, and increase blood pressure in human and animal models 9, while short‐term exposure to PM2.5 could also induce hypertension 10.
Endothelial dysfunction or injury is considered one of the major factors that contribute to the development of atherosclerosis and coronary heart disease 11, 12. Endothelial progenitor cells (EPCs) play a critical role in vascular re‐endothelialization, angiogenesis and prevention of neointima formation after vascular injury 13, 14, 15, 16. The number and function of stem cells (SCs) and EPCs are significantly decreased in the animals exposed to PM2.5‐10. However, the mechanism(s) for PM2.5‐10 exposure‐induced impairment of EPCs is not fully understood. Current data strongly support the concept that the effect of PM‐exposure on EPCs might be related to increased level of reactive oxygen species (ROS) and inflammation 17, 18, 19, 20. Accumulating pre‐clinical and clinical data suggest that bone marrow stem cells (BMSCs) and other SCs could significantly contribute to cardiovascular regeneration and repair after damage like myocardial infarction and myocarditis. Exposure to ambient PM related to air pollution is a constant lifelong hazard for all of us. It is therefore important to summarize the adverse consequence of PM exposure on human participants. The present review is focused on the impact of PM exposure on cardiovascular system with special efforts on progenitor cells and SCs as well as related mechanisms especially ROS formation.
PM and cardiovascular diseases
It has been demonstrated that PM2.5 exposure could induce various cardiovascular diseases including atherosclerosis, hypertension, stroke and Type 2 diabetes mellitus (DM) 21. American Heart Association and the Environmental Protection Agency have officially acknowledged the detrimental effects of PM2.5 on cardiovascular system and related morbidity outcome 2. The Harvard Six Cities study showed that the cardiopulmonary mortality was increased up to 37% in the population exposed to high levels of ambient PM2.5 over a period of 14–16 years 22. The analysis from a population of 50 million living in the major U.S. cities (The National Morbidity, Mortality and Air Pollution Study) indicated that an increase of 10 μg/m3 in PM10 was related to an increase in 0.68% in cardiopulmonary mortality 23, 24, 25. Every 10 μg/m3 increase in PM2.5 exposure was also associated with an increase in 4.5% in coronary artery disease (CAD) 26. Conversely, it was estimated that each 10 μg/m3 decrease in PM2.5 was associated with an increase in 0.61 years in mean life expectancy in the United States 27. In addition, there was a close relationship between NO2 and PM2.5 and the risk of acute myocardial infarction and hospitalization in the U.S 28, 29. Similarly, the Air Pollution and Health European Approach study analysed a population of 43 million in 29 large European cities, and showed that PM10 was closely related to cardiovascular diseases 30.
Particulate matter pollution was also correlated with a significant increase in blood pressure. It has been reported that there was a 2.8 mmHg increase in systolic blood pressure and 2.7 mmHg increase in diastolic blood pressure in patients in Boston over 5 days for every 10.5 μg/m3 increase in PM2.5 levels 31. Similarly, studies have shown that increased PM2.5 levels were associated with mean increases in systolic blood pressure of 3.2 mmHg in Detroit, Michigan, USA 32. In addition, it was observed that a significant rise in diastolic blood pressure (6 mmHg) in 23 normotensive patients after a 2‐hr exposure to PM2.5 and O3 compared with the patients without exposure 33. These observations support the conclusion that there is indeed a close association between increased blood pressure and PM2.5 exposure in human participants 34.
Air pollution has been shown to increase the risk for obesity, hypertension, chronic pulmonary disease and cardiovascular disease in the elderly 35, 36. Long‐term exposure to PM2.5 could induce insulin resistance and mitochondrial alteration in adipose tissue 37, thus further causing or exaggerating DM 38, 39. These studies provide additional evidence that PM exposure is directly associated with cardiovascular diseases, and also closely related with conditions like DM directly associated with increased cardiovascular morbidity and mortality.
Progenitor cells and cardiovascular diseases
Endothelial dysfunction or injury is considered one of the major factors that contribute to the development of cardiovascular diseases like atherosclerosis 11, coronary heart disease 12, congestive heart failure 40, 41, 42 and periphery artery disease 43. Bone marrow‐derived EPCs play a critical role in vascular re‐endothelialization, angiogenesis and prevention of neointima formation after vascular injury 13, 15, 44. There is an obvious inverse relation between the level of circulating EPCs and the risk of cardiovascular events in the patients with angiographically documented CAD 45. Similarly, impaired function of EPCs such as deficiency in migratory response and poor angiogenic capability has a negative correlation with the severity of CAD 46. The important role of EPCs in maintaining the structural and functional integrity of the blood vessels has been well‐established and extensively discussed in many excellent reviews 47, 48. Thus, the level of circulating EPCs has been an important and independent predictor for cardiovascular outcome in CAD patients 45, and it is crucial to preserve the number and function of EPCs at a healthy level for the normal functionality of vasculature in patients with cardiovascular diseases. A variety of factors are critically involved in the regulation of the in vivo dynamics of EPC number and function, including (but not limited to) cytokines and growth factors like granulocyte‐stimulating colony stimulating factor and VEGF 40, 41, 42, nitric oxide, pharmacological agents like statins 49 and environmental factors like air pollution 19, 50. Some disease states like hyperlipidaemia, DM, inflammation, oxidative stress, ischaemia and chronic heart failure are also important for the dynamic changes of EPCs in vivo 19, 51, 52, 53, 54.
It is important to point out that the identification and characterization of EPCs have been very challenging and complex, and even controversial as excellently summarized in a few recent review articles 55, 56, 57. There are currently no unified criteria to define EPCs as yet. Therefore, the terminology ‘EPC’ was adopted from the original papers without modification to preserve the originality. The obvious limitation or confusion was that ‘EPCs’ from different studies might not be the same cell populations with different with cell markers in the literature. There are also multiple sources for circulating EPCs, including BM and non‐BM origins such as liver and spleen 58, 59. The number and function of circulating EPCs could be delicately determined by the combined outcome of EPC mobilization, differentiation, proliferation and apoptosis at sites of different sources. Accumulating evidence from pre‐clinical and clinical studies suggests that cell‐based therapy with progenitor cells (such as EPCs, CD34+ cells, c‐kit+ cells and adipose tissue progenitor cells, APCs) and SCs (including BMSCs) remains an attractive option for tissue regeneration and repair after significant damages like myocardial infarction and myocarditis or stroke 60, 61, 62. To achieve the optimal outcome for cell‐based therapy, the quality of the cells needs to be preserved both in the donors and in the recipients before and after the in vivo delivery. It is well known that only a small fraction of cells could survive after in vivo delivery (both locally and systematically) 63, 64. However, very little is known on how the quality (including the number and function as well as differentiation potential) of the progenitor cells and SCs could be affected by the potential factors in vivo.
PM and progenitor cells
Epidemiological and experimental studies have shown that there is an obvious relationship between exposure to airborne pollutants and poor cardiovascular health 50. Although very limited data are available on the mechanisms for air pollution‐related cardiovascular diseases, induction of endothelial dysfunction by PM2.5 (not able to be filtered by the respiratory tract) is believed to be one of the mechanisms for the adverse effects of air pollution on cardiovascular system in a population‐based study with children and adolescents 65.
It is well known that EPCs play a critical role in vascular repair, angiogenesis and maintaining normal endothelial function 13, 14, 15, 16. Particulate matter exposure has been reported to significantly decrease the number and function of EPCs, and thus increase the risk of cardiovascular diseases and adverse cardiovascular events. In 2010, O'Toole et al. recruited 16 healthy college students from Provo, UT, in the United States to participate in their study 18. The city of Provo is located in a valley and the temperature inversion in the valley could lead to a temporary increase in the concentration of PM2.5 in the atmosphere. In this study, the investigators demonstrated that episodic exposure to PM2.5 induced reversible vascular injury, decreased circulating EPC (CD34+/CD31+/CD45+/CD133+) levels, enhanced platelet activation, and increased plasma level of nonalbumin protein in vivo. In the same year, Liberda et al. reported that inhalation of nickel nanoparticles could result in a decrease in tube formation and chemotaxis function of EPCs (CD34+/VEGF‐R2+/CD11b−) in vitro as well as a reduction in EPC number in murine BM in vivo 66. A study performed in China in 2013 also showed that PM2.5 exposure decreased the number of EPCs (CD34+/KDR+, CD34+/KDR+/CD45− or CD34+/KDR+/CD133+) in circulation 67. This Chinese study was conducted in two large adjacent communities in Jinchang and Zhangye with comparable ambient concentrations of PM2.5. Jinchang was identified as a heavily Nickel‐polluted area because of its proximity to the second largest Nickel refinery in the world. Zhangye, 250 miles northwest and upwind from Jinchang, was selected to serve as a control community. A total of 60 healthy non‐smoking adult women residents were recruited in the study. It was observed that the circulating EPCs were significantly lower in the participants from Jinchang than those from Zhangye. Diesel exhaust particles were reported to reduce the number and function of EPCs with impaired stromal cell‐derived factor (SDF)‐1‐induced migratory capacity and neoangiogenesis both in vivo and in vitro in a murine model. Consistent with above observation, we recently reported that PM treatment significantly decreased murine‐circulating EPC population, promoted apoptosis of murine EPCs (CD34+/CD133+) in association with increased ROS production and serum TNF‐α and IL‐1β levels in vivo (Fig. 1) 19.
Figure 1.
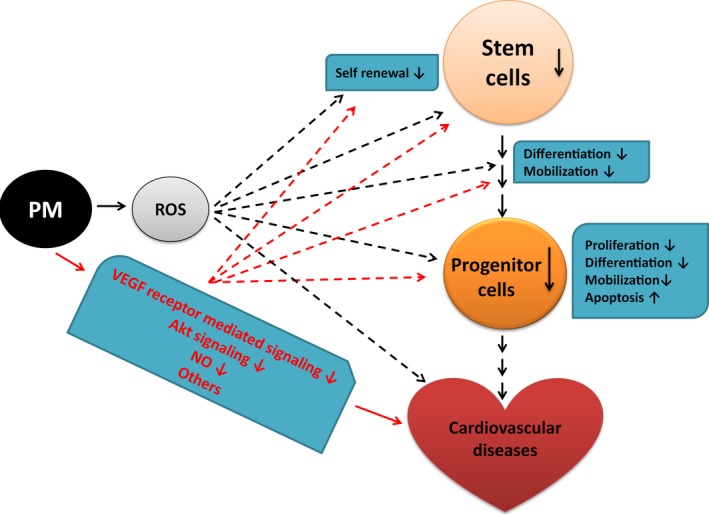
Illustration of potential mechanisms for the effect of PM exposure on cardiovascular system and progenitor/stem cells. PM exposure resulted in ROS formation that in turn could lead to the detrimental effects on cardiovascular system and impaired number and function of progenitor/stem cells. The mechanisms for decreased number and function of progenitor/stem cells following PM exposure might be because of the inhibition of self‐renewal, proliferation, survival (enhanced apoptosis), homing, mobilization, adhesion to extracellular matrices or differentiation. Blocking PM‐induced ROS formation might be an effective treatment option to attenuate or prevent the adverse effect of PM exposure on the progenitor/stem cells and cardiovascular system. Other mechanisms like reduction in VEGF receptor‐mediated signalling, decreased Akt signalling and nitric oxide level could be also important for the effect of PM exposure. PM: particulate matter; ROS: reactive oxygen species. ‐: diminish; ↓: decrease.
However, some studies demonstrated that circulating EPCs could be increased after PM2.5‐10 exposure. Brook et al. recruited 32 healthy non‐smoking adults (18–50 years old) in Dexter, a town in Michigan, United States, an area with coarse PM2.5‐10 exposures 68. Dexter is 410 km from major freeways and 460 km west of the Detroit metropolitan area. The study showed that increased number of EPCs (CD34+/CD133+/CD3−/CD79b−/CD56−) in vivo persisted for at least 20 hrs following brief inhalation of coarse PM2.5‐10. The mechanism was believed to be related to a systemic reaction to an acute ‘endothelial injury’ and/or a circulating EPCs response to sympathetic nervous system activation. Haberzettl et al. delineated that exposure to PM2.5 could increase murine EPC (Sca‐1+/Flk‐1+) levels in the BM by preventing their mobilization to the peripheral blood via inhibition of signalling events triggered by VEGF‐receptor stimulation based on in vivo and ex vivo experiment 17. This might also explain the decreased circulating EPCs as a result of the decreased mobilization of BM EPCs into the circulation.
PM2.5 exposure might also have significant impact on APCs as well. Adipose tissue progenitor cells (Lin−/CD34+/CD29+/Sca‐1+/CD24+) in brown adipose tissue are closely correlated with the normal functionality of brown adipose tissue and reduction in obesity 69. It has been shown that high level exposure to PM2.5 in murine early life is associated with decreased number of murine APCs and increased risk factor for the development of insulin resistance, adiposity and inflammation in association with ROS generation by NADPH oxidase in vivo 70. The effect of PM exposure on different progenitor cells and their function in both human and animals was summarized in Tables 1 and 2.
Table 1.
The effect of particulate air pollution on human progenitor/stem cells
| Authors | Key findings | Participants | Exposure time | Location |
|---|---|---|---|---|
| O'Toole et al. | PM2.5 exposure decreases circulating EPC level | 18–25 years adults | 3 months | Utah, US |
| Brook et al. | Brief PM inhalation could increase EPC number for at least 20 hrs | 18–50 years adults | 2 hrs | Michigan US |
| Niu et al. | Specific metals in PM2.5 may be responsible for decreased circulating EPC level | 60–65 years women | 12 months | China |
| Lin et al. | CS inhibits ESCs growth | ESCs | 6–24 hrs | Lab |
| Talbot et al. | CS could lead to poor adhesion to extracellular matrices, diminished survival and proliferation and increased apoptosis of ESCs | ESCs | 6–24 hrs | Lab |
| Liszewski et al. | Tobacco smoking impairs foetal development | ESCs | 8–21 days | Lab |
| Zhou et al. | Smoking inhibits BMSC recruitment and differentiation | MSCs | 1 month | Lab |
PM: particulate matter; SCs: stem cell; ESCs: embryonic stem cells; MSCs: mesenchymal stem cells; HSC: haematopoietic stem cells; BM: bone marrow; BMSCs: bone marrow stem cells; CSC: cigarette smoke condensate.
Table 2.
The effect of particulate air pollution on animal progenitor/stem cells
| Authors | Key findings | Participants | Exposure time |
|---|---|---|---|
| Xu et al. | PM2.5 exposure induces oxidative stress | Mouse APCs | 10 weeks |
| Liberda et al. | Ni nanoparticles result in reduced number and function of EPCs in bone marrow | Mouse EPCs | 2–5 days |
| Haberzettl et al. | PM2.5 exposure increases EPC levels in the bone marrow by preventing mobilization via inhibition of VEGF‐receptor signalling | Mouse EPCs | 18 months |
| Poss et al. | Diesel exhaust particles impair EPC number and function in vivo and in vitro | Mouse EPCs | 3–6 weeks |
| Cui et al. | PM exposure significantly decreased circulating EPCs population due to increased apoptosis via ROS formation | Mouse EPCs | 1 month |
| Cui et al. | PM suppresses BMSC in vivo proliferation via ROS formation | Mouse SCs | 1 month |
| Huang et al. | CS induces oxidative stress, telomere shortening, and apoptosis | Mouse EPCs | 1 month |
| Yauk et al. | CS leads to mutations of spermatogonial SCs | Mouse spermatogonial SCs | 6–12 weeks |
| Huang et al. | Acute CS exposure causes cell death and reduces pluripotency, while chronic CS exposure leads to DNA damage and telomere shortening | Mouse ESCs | 20 hrs–2 weeks |
| Lin et al. | CS impairs ESC function | Mouse ESCs | 6–24 hrs |
| Albrecht et al. | Titanium dioxide in coal dust induces hyperplasia of Clara cells | Rat Clara cell | 126–129 weeks |
| Izzotti et al. | CS could induce recruitment of undifferentiated SC into lung | Mouse MSCs | 1–4 months |
| De Flora et al. | Same as above | Mouse MSCs | 1 week–11 months |
| Khaldoyanidi et al. | Nicotine could impair the function of the haematopoiesis‐supportive stromal microenvironment, and interfere with SCs homing | Mouse HSCs | 1–4 weeks |
| Zhou et al. | Smoking inhibits BMSC recruitment and differentiation | Mouse MSCs | 1 month |
PM: particulate matter; APCs: adipose tissue progenitor cells; SCs: stem cell; ESCs: embryonic stem cells; MSCs: mesenchymal stem cells; HSC: haematopoietic stem cells; BM: bone marrow; BMSCs: bone marrow stem cells; CSC: cigarette smoke condensate.
PM and stem cells
Cell‐based therapy with progenitor cells and SCs appears to be a promising option for the regeneration and/or repair of damaged tissues in cardiovascular system 71, 72, 73, 74. Many types of SCs have been studies as the potential sources for cell‐based therapy including embryonic SCs (ESCs) 75, 76, neural SCs 77, cardiac SCs 78, 79, 80, 81, BM‐derived haematopoietic SCs (HSCs) 82, BM‐derived c‐kit+/Lin− cells 83, 84, mesenchymal SCs (MSCs) 72, 85, 86, adipose‐derived SCs 87 and inducible pluripotent stem cells from somatic cells 88. Progenitor/SCs in circulating blood and in the vascular wall could serve as the endogenous pool of SCs to restore the structural and functional integrity of the vasculature through rapid repair of the endothelial cells and/or formation of new vessels after injuries 89. The progenitor/SCs residing in vascular intima, media and adventitia may participate in vascular repair and the formation of neointimal lesions in severely damaged vessels 90. Recently, a new type of SCs was identified in the murine arterial media, named multipotent vascular SCs, which could differentiate into neural cells and MSC‐like cells and subsequently differentiate into SMCs 91. In addition, abundant progenitor/SCs expressing Sca‐1 have been identified in the adventitia, which may contribute to endothelial regeneration and smooth muscle accumulation in the neointimal lesions 92.
The therapeutic efficacy of cell therapy for cardiovascular diseases is associated with a variety of factors including cell types, myocardial ischemia, cardiac dysfunction or their combination 93. The outcome of cell therapy with stem cells could be also related to the engraftment and survival of the cells transplanted into the target area such as an infarcted myocardial area. It is known that one of the major challenges for cell therapy with BMSCs is the low viability of the implanted cells with the loss of cells occurring mainly in the first few days after in vivo delivery 94. However, the mechanisms for the poor in vivo survival of the cells are complex, and have yet to be defined. It is believed that an acute inflammatory reaction with formation of various inflammatory factors including inducible nitric oxide synthase in the delivery site is a critical factor for the cell death in the first 24–72 hr period 94, 95, 96.
Unfortunately, there is very little data available in the area of PM2.5 and SCs. We recently found that PM exposure significantly decreased murine BMSCs population in vivo, defined as lineage negative/Sca‐1 positive (LS) and lineage negative/CD133 positive (Lin−/CD133+) cells, in association with increased ROS formation, decreased level of Akt phosphorylation and inhibition of in vivo proliferation of murine BMSCs without induction of apoptosis 20. We further demonstrated that PM‐induced ROS production was the major mechanism for decreased in vivo proliferation and population of murine BMSCs. Treating mice with antioxidant N‐acetylcysteine (NAC) or using a triple transgenic mouse line with overexpression of antioxidant enzyme network (AON) composed of superoxide dismutase (SOD)1, SOD3 and glutathione peroxidase‐1 with decreased in vivo ROS production significantly decreased murine BMSCs intracellular ROS level, partially reversed the suppression of p‐Akt expression, effectively reversed the inhibition of BMSCs proliferation rate and restored the BMSCs population in the mice with PM exposure in vivo (Fig. 1).
There are a variety of sources for PM exposure 97. Recent studies showed that the median concentration of PM2.5 in the smoking area (both indoor and outdoor) was significantly higher than in the control area 98, 99, 100. Thus, environmental tobacco smoke‐associated PM could be an important independent health hazard in addition to the well‐known toxic and carcinogenic compounds contained in cigarette smoking (CS). It has been reported that CS could significantly impair the number and function of various SCs including ESCs, spermatogonial SCs (SSCs) and Clara cell (SCs of the bronchiolar epithelium). Cigarette smoking could produce cytotoxic action on human ESCs (hESCs) and mouse ESCs (mESCs), induce oxidative stress, apoptosis and telomere shortening in ESCs in vitro, inhibit cell adhesion and growth in vitro, and compromise embryo development in vivo 101, 102, 103. In addition, CS might also induce mutation of SSCs gene and alterations in hESCs gene expression (especially those characteristic for mesoderm and ectoderm development) in vivo 104. Increased expression of Notch, Wnt or transforming growth factor‐β genes by smoking resulted in retention of the cells in pluripotent state in vivo 105. In addition, acute exposure of mESCs to CS or cadmium could cause immediate cell death, and decrease their pluripotency, while chronic exposure could lead to DNA damage and telomere shortening in vivo 106, 107. Coal dust exposure resulted in the disappearance of proliferating cell nuclear antigen in rat Clara cell in vivo 108. Although CS could recruit SCs into murine lung in vivo 109, 110, negative impact including interfering murine and human MSCs homing by targeting microvascular endothelial cells and differentiation into endometrial cells and blood vessel ex vivo were reported 111, 112. The detrimental effects of PM exposure and CS on SCs were summarized in Tables 1 and 2. However, it is important to differentiate the effect of PM exposure from that of other toxic and carcinogenic compounds in CS on SCs.
Possible mechanisms for the effects of PM exposure on progenitor/stem cells
There is growing evidence that supports an important role of oxidative stress in response to air pollution in different organ systems 113. Reactive oxygen species could function as signalling molecules in PM2.5‐trigged autophagy in human epithelia A549 cells 114. Oxidative stress could be triggered by PM2.5 and result in alterations in mitochondrial gene expression in brown adipose tissue 115. Clinical studies suggested that ROS formation, oxidative stress and inflammation induced by PM2.5 exposure were closely related to paediatric asthma 116. A relationship has been observed between ambient PM10, oxidative burden and carotid intima‐media thickness (a change and indicator for subclinical atherosclerosis) 117. Studies, using a simulated respiratory tract lining fluid model with three major water soluble antioxidants (glutathione, urate and ascorbate) at physiological concentrations that served as the first‐line defence in the airway against the oxidative activity of PM, showed that PM could deplete the antioxidants 118. It was also demonstrated that a close relationship was present for ultrafine particles and NO2/NOx 119.
Reactive oxygen species and oxidative stress are involved in EPCs dysfunction in many disease states including hyperlipidaemia, DM and CAD 13, 15, 44. It was observed that the functional impairment of human EPCs by diesel exhaust particles was associated with an increased superoxide production 120. We also observed that ROS production was significantly increased in the EPCs and BMSCs from the mice exposed to PM. Blockage of ROS formation using pharmacological agent NAC or transgenic model with overexpression of NOA effectively prevented PM‐induced decrease in the numbers of circulating EPCs and BMSCs. These data suggested that ROS formation was an important cause for decreased number of EPCs 19 and BMSCs following PM exposure (Fig. 1).
Particulate matter exposure was shown to suppress VEGF‐induced Akt activation and endothelial nitric oxide synthase (eNOS) phosphorylation in the aorta, and prevented VEGF/AMD3100‐induced mobilization of EPCs into the peripheral circulation without change in the plasma levels of human SDF‐1α and VEGF 17, 66. Second‐hand smoke exposure was also reported to block VEGF‐stimulated nitric oxide production 121. There are extensive and complex interactions between ROS and Akt pathway in both normal and cancer cells. We observed that PM exposure inhibited BMSC proliferation via ROS‐mediated mechanism(s) partially through suppression of Akt signalling. It is certainly possible that other pathways might also be affected by PM exposure. Future studies are needed to define the role of other pathways in the effect of PM exposure on BMSCs and progenitor cells.
Clinical implications for PM‐induced detrimental effects on progenitor/stem cells in cardiovascular system
It is clear that exposure to PM increases the risk of cardiovascular diseases with ROS formation as the predominant mechanism. It could be ideal to avoid inhaling PM physically via wearing masks or using filters. However, fine PM such as PM2.5 is very difficult to be removed or isolated from the air because of their extremely small size, especially in those cities with severe air pollution. Moreover, PM2.5 widely exists in the environment and may carry ROS within gas phase 122 or water phase (aerosol) 123 into the lower respiratory tract to create an increased risk for adverse cardiovascular events.
Antioxidant enzyme and antioxidant supplementation have been examined for its impact on cardio‐respiratory effects of PM2.5 exposure. Animal studies have shown an increase in the levels of antioxidant gene expression in epithelial cells after exposure to diesel exhaust particles 124. It was reported that omega‐3 polyunsaturated fatty acid could attenuate the adverse effect of PM2.5 on heart rate variability 125. Antioxidant supplementation such as vitamin C and E was shown to have beneficial effects against human lung damage by air pollution 126. Antioxidant probucol could reduce CS‐induced impairment of neovascularization associated with improved function of EPCs 127. Inhibition of ROS accumulation/production or oxidative stress with pharmacological agents like NAC and SOD‐mimics, or overexpression of antioxidant enzymes like Hsp20 and SOD could reduce ROS accumulation in human MSCs, and attenuate oxidative cell damage in BMSCs in vitro 128, protect stem cells against ROS‐induced apoptosis in vitro 129, protect MSCs against cell death triggered by oxidative stress in vitro in association with enhanced Akt activation and increased secretion of growth factors (such as VEGF, fibroblast growth factor‐2, and insulin‐like growth factor 1) 130, increase the differentiation of EPCs into endothelial cells 131, inhibit cell senescence in HSCs in the BM 132 and restore the impaired self‐renewal potential and functional activity of HSCs with high ROS level 133. N‐acetylcysteine treatment also protected BMSCs against the toxic effect of low concentration ox‐LDL, and restored their endothelial differentiation potential impaired by ox‐LDL 134. Recently, we observed that after PM≤4 exposure, NAC or overexpression of AON could completely block intracellular ROS production in BMSCs, partially restore p‐Akt level, decrease serum TNF‐α and IL‐1β level, reduce EPCs apoptotic rate, effectively reversed the decreased proliferation rate of BMSCs and increased the BMSCs and EPCs number to normal level 19. Thus, inhibition of ROS production and oxidative stress might be an effective option to ameliorate PM‐induced detrimental effects on progenitor cells and SCs as well as cardiovascular system.
Other considerations
There is no question that PM exposure has significant impact on the number and function of progenitor cells and stem cells. However, studies are needed to address a variety of important issues in this area including (but not limited to): (i) determining the size and active components of PM that are responsible for detrimental effect of PM exposure on progenitor cells and SCs, as well as cardiovascular diseases and related mechanism(s) since the size and components are critical to the action of PM 135; and (ii) defining the mode of actions (direct or indirect) for PM exposure on the progenitor cells and SCs.
The number and function of progenitor cells and SCs are associated with other factors and cells like monocytes and platelets through a wide range of cytokines and growth factors 136, 137, 138, 139. Both monocytes and platelets are important to cardiovascular physiology (like angiogenesis and haemostasis), and closely related to cardiovascular diseases like CAD. It is known that monocytes display certain plasticity and could function as pluripotent stem cells with regenerative capability, and produce a variety of cytokines and inflammatory factors 140, 141. Particulate matter exposure could exhibit its effects on progenitor cells and SCs through functional and/or structural modifications of monocytes and platelets. Indeed, PM exposure is able to significantly alter the function and responses of platelets both in vitro and in vivo including induction of Ca(2+) release, dense granule secretion and surface expression of platelet activation markers like P‐selectin expression, as well as aggregation, and change in the mean platelet volume 142, 143, 144, 145, 146. Particulate matter exposure/treatment has been shown to modify the function of monocytes significantly including inflammatory response and cytokine production, recruitment and mobilization, and transcriptional and translational modulations of gene expressions in monocytes 135, 136, 147, 148, 149, 150, 151, 152. Further studies are needed to determine the complex relations between PM exposure, monocyte and platelet function, and progenitor cells/SCs.
Conclusion
In this review, we discussed the adverse effects of PM exposure on cardiovascular diseases with specific effort on PM‐induced detrimental impact on progenitor/SCs. Indeed, PM exposure correlated with the reduction in life expectancy primarily via cardiovascular diseases, and the resultant abnormality in the number and function of progenitor/SCs might play an important role in cardiovascular diseases related to PM exposure (Fig. 1). However, there are lots of questions that need to be addressed on PM‐induced structural and functional impairment on progenitor/SCs. For example, does PM affect the differentiation potential of BMSCs and how? Does PM affect other SCs and how? All these questions require further studies. Although prevention of ROS formation and oxidative stress might be an effective way to attenuate PM‐induced deleterious effects on progenitor/SCs, we believe that other mechanisms may be also important for the effect of PM exposure, which merit further investigations.
Conflicts of interest
No conflict of interest.
Acknowledgements
This work was supported by US NIH grants (R01HL094650 and RO1HL124122 to Z.L., and RO1ES018900 to Q.S.). Y.C., Q.S. and Z.L. wrote and revised the article.
References
- 1. Lim SS, Vos T, Flaxman AD, et al A comparative risk assessment of burden of disease and injury attributable to 67 risk factors and risk factor clusters in 21 regions, 1990‐2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet. 2012; 380: 2224–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lee BJ, Kim B, Lee K. Air pollution exposure and cardiovascular disease. Toxicol Res. 2014; 30: 71–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Brook RD, Rajagopalan S, Pope CA 3rd, et al American Heart Association Council on E, Prevention CotKiCD, Council on Nutrition PA, Metabolism. Particulate matter air pollution and cardiovascular disease: an update to the scientific statement from the American Heart Association. Circulation. 2010; 121: 2331–78. [DOI] [PubMed] [Google Scholar]
- 4. Brunekreef B, Forsberg B. Epidemiological evidence of effects of coarse airborne particles on health. Eur Respir J. 2005; 26: 309–18. [DOI] [PubMed] [Google Scholar]
- 5. WHO's global air‐quality guidelines. Lancet. 2006; 368: 1302. [DOI] [PubMed] [Google Scholar]
- 6. Marks GB. A critical‐appraisal of the evidence for adverse respiratory effects due to exposure to environmental ozone and particulate pollution ‐ relevance to air‐quality guidelines. Aust Nz J Med. 1994; 24: 202–13. [DOI] [PubMed] [Google Scholar]
- 7. Brook RD, Urch B, Dvonch JT, et al Insights into the mechanisms and mediators of the effects of air pollution exposure on blood pressure and vascular function in healthy humans. Hypertension. 2009; 54: 659–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sun QH, Wang AX, Jin XM, et al Long‐term air pollution exposure and acceleration of atherosclerosis and vascular inflammation in an animal model. J Am Med Assoc. 2005; 294: 3003–10. [DOI] [PubMed] [Google Scholar]
- 9. Ying ZK, Xu XH, Bai YT, et al Long‐term exposure to concentrated ambient PM2.5 increases mouse blood pressure through abnormal activation of the sympathetic nervous system: a role for hypothalamic inflammation. Environ Health Perspect. 2014; 122: 79–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sun QH, Yue PB, Ying ZK, et al Air pollution exposure potentiates hypertension through reactive oxygen species‐mediated activation of Rho/ROCK. Arterioscl Throm Vas. 2008; 28: 1760–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Davignon J, Ganz P. Role of endothelial dysfunction in atherosclerosis. Circulation. 2004; 109: III27–32. [DOI] [PubMed] [Google Scholar]
- 12. Heitzer T, Schlinzig T, Krohn K, et al Endothelial dysfunction, oxidative stress, and risk of cardiovascular events in patients with coronary artery disease. Circulation. 2001; 104: 2673–8. [DOI] [PubMed] [Google Scholar]
- 13. Rauscher FM, Goldschmidt‐Clermont PJ, Davis BH, et al Aging, progenitor cell exhaustion, and atherosclerosis. Circulation. 2003; 108: 457–63. [DOI] [PubMed] [Google Scholar]
- 14. Strehlow K, Werner N, Berweiler J, et al Estrogen increases bone marrow‐derived endothelial progenitor cell production and diminishes neointima formation. Circulation. 2003; 107: 3059–65. [DOI] [PubMed] [Google Scholar]
- 15. Urbich C, Dimmeler S. Endothelial progenitor cells: characterization and role in vascular biology. Circ Res. 2004; 95: 343–53. [DOI] [PubMed] [Google Scholar]
- 16. Werner N, Junk S, Laufs U, et al Intravenous transfusion of endothelial progenitor cells reduces neointima formation after vascular injury. Circ Res. 2003; 93: E17–24. [DOI] [PubMed] [Google Scholar]
- 17. Haberzettl P, Lee J, Duggineni D, et al Exposure to ambient air fine particulate matter prevents VEGF‐induced mobilization of endothelial progenitor cells from the bone marrow. Environ Health Perspect. 2012; 120: 848–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. O'Toole TE, Hellmann J, Wheat L, et al Episodic exposure to fine particulate air pollution decreases circulating levels of endothelial progenitor cells. Circ Res. 2010; 107: 200–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cui Y, Xie X, Jia F, et al Ambient fine particulate matter induces apoptosis of endothelial progenitor cells through reactive oxygen species formation. Cell Physiol Biochem. 2015; 35: 353–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cui Y, Jia F, He J, et al Ambient fine particulate matter suppresses in vivo proliferation of bone marrow stem cells through reactive oxygen species formation. PLoS ONE. 2015; 10: e0127309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sun Q, Hong X, Wold LE. Cardiovascular effects of ambient particulate air pollution exposure. Circulation. 2010; 121: 2755–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dockery DW, Pope CA 3rd, Xu X, et al An association between air pollution and mortality in six U.S. cities. N Engl J Med. 1993; 329: 1753–9. [DOI] [PubMed] [Google Scholar]
- 23. Sarnat JA, Schwartz J, Suh HH. Fine particulate air pollution and mortality in 20 U.S. cities. N Engl J Med. 2001; 344: 1253–4. [DOI] [PubMed] [Google Scholar]
- 24. Brook RD. Cardiovascular effects of air pollution. Clin Sci. 2008; 115: 175–87. [DOI] [PubMed] [Google Scholar]
- 25. Brook RD, Brook JR, Rajagopalan S. Air pollution: the “Heart” of the problem. Curr Hypertens Rep. 2003; 5: 32–9. [DOI] [PubMed] [Google Scholar]
- 26. Pope CA 3rd, Muhlestein JB, May HT, et al Ischemic heart disease events triggered by short‐term exposure to fine particulate air pollution. Circulation. 2006; 114: 2443–8. [DOI] [PubMed] [Google Scholar]
- 27. Pope CA 3rd, Ezzati M, Dockery DW. Fine‐particulate air pollution and life expectancy in the United States. N Engl J Med. 2009; 360: 376–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wellenius GA, Schwartz J, Mittleman MA. Particulate air pollution and hospital admissions for congestive heart failure in seven United States cities. Am J Cardiol. 2006; 97: 404–8. [DOI] [PubMed] [Google Scholar]
- 29. Zanobetti A, Schwartz J. The effect of particulate air pollution on emergency admissions for myocardial infarction: a multicity case‐crossover analysis. Environ Health Perspect. 2005; 113: 978–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Katsouyanni K, Samet JM, Anderson HR, et al Committee HEIHR. Air pollution and health: a European and North American approach (APHENA). Res Rep. 2009; 142: 5–90. [PubMed] [Google Scholar]
- 31. Zanobetti A, Canner MJ, Stone PH, et al Ambient pollution and blood pressure in cardiac rehabilitation patients. Circulation. 2004; 110: 2184–9. [DOI] [PubMed] [Google Scholar]
- 32. Dvonch JT, Kannan S, Schulz AJ, et al Acute effects of ambient particulate matter on blood pressure: differential effects across urban communities. Hypertension. 2009; 53: 853–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Urch B, Silverman F, Corey P, et al Acute blood pressure responses in healthy adults during controlled air pollution exposures. Environ Health Perspect. 2005; 113: 1052–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Brook RD. Why physicians who treat hypertension should know more about air pollution. J Clin Hypertens. 2007; 9: 629–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dubowsky SD, Suh H, Schwartz J, et al Diabetes, obesity, and hypertension may enhance associations between air pollution and markers of systemic inflammation. Environ Health Perspect. 2006; 114: 992–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Peel JL, Metzger KB, Klein M, et al Ambient air pollution and cardiovascular emergency department visits in potentially sensitive groups. Am J Epidemiol. 2007; 165: 625–33. [DOI] [PubMed] [Google Scholar]
- 37. Xu X, Liu C, Xu Z, et al Long‐term exposure to ambient fine particulate pollution induces insulin resistance and mitochondrial alteration in adipose tissue. Toxicol Sci. 2011; 124: 88–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Liu C, Bai Y, Xu X, et al Exaggerated effects of particulate matter air pollution in genetic type II diabetes mellitus. Part Fibre Toxicol. 2014; 11: 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Liu C, Fonken LK, Wang A, et al Central IKK beta inhibition prevents air pollution mediated peripheral inflammation and exaggeration of type II diabetes. Part Fibre Toxicol. 2014; 11: 53–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chong AY, Blann AD, Patel J, et al Endothelial dysfunction and damage in congestive heart failure: relation of flow‐mediated dilation to circulating endothelial cells, plasma indexes of endothelial damage, and brain natriuretic peptide. Circulation. 2004; 110: 1794–8. [DOI] [PubMed] [Google Scholar]
- 41. Fischer D, Rossa S, Landmesser U, et al Endothelial dysfunction in patients with chronic heart failure is independently associated with increased incidence of hospitalization, cardiac transplantation, or death. Eur Heart J. 2005; 26: 65–9. [DOI] [PubMed] [Google Scholar]
- 42. Marti CN, Gheorghiade M, Kalogeropoulos AP, et al Endothelial dysfunction, arterial stiffness, and heart failure. J Am Coll Cardiol. 2012; 60: 1455–69. [DOI] [PubMed] [Google Scholar]
- 43. Fadini GP, Sartore S, Albiero M, et al Number and function of endothelial progenitor cells as a marker of severity for diabetic vasculopathy. Arterioscl Throm Vas. 2006; 26: 2140–6. [DOI] [PubMed] [Google Scholar]
- 44. Werner N, Kosiol S, Schiegl T, et al Circulating endothelial progenitor cells and cardiovascular outcomes. N Engl J Med. 2005; 353: 999–1007. [DOI] [PubMed] [Google Scholar]
- 45. Werner N, Wassmann S, Ahlers P, et al Endothelial progenitor cells correlate with endothelial function in patients with coronary artery disease. Basic Res Cardiol. 2007; 102: 565–71. [DOI] [PubMed] [Google Scholar]
- 46. Vasa M, Fichtlscherer S, Aicher A, et al Number and migratory activity of circulating endothelial progenitor cells inversely correlate with risk factors for coronary artery disease. Circ Res. 2001; 89: E1–7. [DOI] [PubMed] [Google Scholar]
- 47. Lee PS, Poh KK. Endothelial progenitor cells in cardiovascular diseases. World J Stem Cells. 2014; 6: 355–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mak A, Kow NY. Imbalance between endothelial damage and repair: a gateway to cardiovascular disease in systemic lupus erythematosus. Biomed Res Int. 2014; 2014: 178721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Liguori A, Fiorito C, Balestrieri ML, et al Functional impairment of hematopoietic progenitor cells in patients with coronary heart disease. Eur J Haematol. 2008; 80: 258–64. [DOI] [PubMed] [Google Scholar]
- 50. King A. Air pollution reduces EPC levels. Nat Rev Cardiol. 2010; 7: 540. [DOI] [PubMed] [Google Scholar]
- 51. Li TB, Zhang JJ, Liu B, et al Involvement of NADPH oxidases and non‐muscle myosin light chain in senescence of endothelial progenitor cells in hyperlipidemia. Naunyn Schmiedebergs Arch Pharmacol. 2016; 389: 289–302. [DOI] [PubMed] [Google Scholar]
- 52. Sukmawati D, Tanaka R, Ito‐Hirano R, et al The role of Notch signaling in diabetic endothelial progenitor cells dysfunction. J Diabetes Complications. 2016; 30: 12–20. [DOI] [PubMed] [Google Scholar]
- 53. Lam YT. Critical roles of reactive oxygen species in age‐related impairment in ischemia‐induced neovascularization by regulating stem and progenitor cell function. Oxid Med Cell Longev. 2015; 2015: 7095901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Berezin AE, Kremzer AA. [Content of circulating endothelial progenitor cells in patients with chronic ischemic heart failure with preserved left ventricular ejection fraction]. Kardiologiia. 2015; 55: 14–22. [DOI] [PubMed] [Google Scholar]
- 55. Timmermans F, Plum J, Yoder MC, et al Endothelial progenitor cells: identity defined? J Cell Mol Med. 2009; 13: 87–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Fadini GP, Losordo D, Dimmeler S. Critical reevaluation of endothelial progenitor cell phenotypes for therapeutic and diagnostic use. Circ Res. 2012; 110: 624–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Favre J, Terborg N, Horrevoets AJ. The diverse identity of angiogenic monocytes. Eur J Clin Invest. 2013; 43: 100–7. [DOI] [PubMed] [Google Scholar]
- 58. King TF, McDermott JH. Endothelial progenitor cells and cardiovascular disease. J Stem Cells. 2014; 9: 93–106. [PubMed] [Google Scholar]
- 59. Saito H, Yamamoto Y, Yamamoto H. Diabetes alters subsets of endothelial progenitor cells that reside in blood, bone marrow, and spleen. Am J Physiol Cell Physiol. 2012; 302: C892–901. [DOI] [PubMed] [Google Scholar]
- 60. Werner L, Deutsch V, Barshack I, et al Transfer of endothelial progenitor cells improves myocardial performance in rats with dilated cardiomyopathy induced following experimental myocarditis. J Mol Cell Cardiol. 2005; 39: 691–7. [DOI] [PubMed] [Google Scholar]
- 61. Cheng M, Huang K, Zhou J, et al A critical role of Src family kinase in SDF‐1/CXCR4‐mediated bone‐marrow progenitor cell recruitment to the ischemic heart. J Mol Cell Cardiol. 2015; 81: 49–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Nucci LP, Silva HR, Giampaoli V, et al Stem cells labeled with superparamagnetic iron oxide nanoparticles in a preclinical model of cerebral ischemia: a systematic review with meta‐analysis. Stem Cell Res Ther. 2015; 6: 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Henning RJ. Stem cells in cardiac repair. Future Cardiol. 2011; 7: 99–117. [DOI] [PubMed] [Google Scholar]
- 64. Stamm C, Liebold A, Steinhoff G, et al Stem cell therapy for ischemic heart disease: beginning or end of the road? Cell Transplant. 2006; 15: S47–56. [DOI] [PubMed] [Google Scholar]
- 65. Poursafa P, Kelishadi R, Haghjooy‐Javanmard S, et al Synergistic effects of genetic polymorphism and air pollution on markers of endothelial dysfunction in children. J Res Med Sci. 2012; 17: 718–23. [PMC free article] [PubMed] [Google Scholar]
- 66. Liberda EN, Cuevas AK, Gillespie PA, et al Exposure to inhaled nickel nanoparticles causes a reduction in number and function of bone marrow endothelial progenitor cells. Inhalation Toxicol. 2010; 22: 95–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Niu J, Liberda EN, Qu S, et al The role of metal components in the cardiovascular effects of PM2.5. PLoS ONE. 2013; 8: e83782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Brook RD, Bard RL, Kaplan MJ, et al The effect of acute exposure to coarse particulate matter air pollution in a rural location on circulating endothelial progenitor cells: results from a randomized controlled study. Inhalation Toxicol. 2013; 25: 587–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Xu X, Ying Z, Cai M, et al Exercise ameliorates high‐fat diet‐induced metabolic and vascular dysfunction, and increases adipocyte progenitor cell population in brown adipose tissue. Am J Physiol Regul Integr Comp Physiol. 2011; 300: R1115–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Xu X, Yavar Z, Verdin M, et al Effect of early particulate air pollution exposure on obesity in mice: role of p47phox. Arterioscler Thromb Vasc Biol. 2010; 30: 2518–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Campagnolo P, Wong MM, Xu Q. Progenitor cells in arteriosclerosis: good or bad guys? Antioxid Redox Signal. 2011; 15: 1013–27. [DOI] [PubMed] [Google Scholar]
- 72. Mazhari R, Hare JM. Mechanisms of action of mesenchymal stem cells in cardiac repair: potential influences on the cardiac stem cell niche. Nat Clin Pract Cardiovasc Med. 2007; 4: S21–6. [DOI] [PubMed] [Google Scholar]
- 73. Chou SH, Lin SZ, Kuo WW, et al Mesenchymal stem cell insights: prospects in cardiovascular therapy. Cell Transplant. 2014; 23: 513–29. [DOI] [PubMed] [Google Scholar]
- 74. Kane NM, Xiao QZ, Baker AH, et al Pluripotent stem cell differentiation into vascular cells: a novel technology with promises for vascular re(generation). Pharmacol Therapeut. 2011; 129: 29–49. [DOI] [PubMed] [Google Scholar]
- 75. Doetschman TC, Eistetter H, Katz M, et al The in vitro development of blastocyst‐derived embryonic stem cell lines: formation of visceral yolk sac, blood islands and myocardium. J Embryol Exp Morphol. 1985; 87: 27–45. [PubMed] [Google Scholar]
- 76. Kehat I, Kenyagin‐Karsenti D, Snir M, et al Human embryonic stem cells can differentiate into myocytes with structural and functional properties of cardiomyocytes. J Clin Invest. 2001; 108: 407–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Katare R, Stroemer P, Hicks C, et al Clinical‐grade human neural stem cells promote reparative neovascularization in mouse models of hindlimb ischemia. Arterioscl Throm Vas. 2014; 34: 408–18. [DOI] [PubMed] [Google Scholar]
- 78. Beltrami AP, Barlucchi L, Torella D, et al Adult cardiac stem cells are multipotent and support myocardial regeneration. Cell. 2003; 114: 763–76. [DOI] [PubMed] [Google Scholar]
- 79. Laugwitz KL, Moretti A, Lam J, et al Postnatal isl1+cardioblasts enter fully differentiated cardiomyocyte lineages. Nature. 2005; 433: 647–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Oh H, Bradfute SB, Gallardo TD, et al Cardiac progenitor cells from adult myocardium: homing, differentiation, and fusion after infarction. Proc Natl Acad Sci USA. 2003; 100: 12313–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Winter EM, Grauss RW, Hogers B, et al Preservation of left ventricular function and attenuation of remodeling after transplantation of human epicardium‐derived cells into the infarcted mouse heart. Circulation. 2007; 116: 917–27. [DOI] [PubMed] [Google Scholar]
- 82. Yeh ETH, Zhang S, Wu HD, et al Transdifferentiation of human peripheral blood CD34(+)‐enriched cell population into cardiomyocytes, endothelial cells, and smooth muscle cells in vivo . Circulation. 2003; 108: 2070–3. [DOI] [PubMed] [Google Scholar]
- 83. Orlic D, Kajstura J, Chimenti S, et al Bone marrow cells regenerate infarcted myocardium. Nature. 2001; 410: 701–5. [DOI] [PubMed] [Google Scholar]
- 84. Orlic D, Kajstura J, Chimenti S, et al Mobilized bone marrow cells repair the infarcted heart, improving function and survival. Proc Natl Acad Sci USA. 2001; 98: 10344–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Chen SL, Fang WW, Ye F, et al Effect on left ventricular function of intracoronary transplantation of autologous bone marrow mesenchymal stem cell in patients with acute myocardial infarction. Am J Cardiol. 2004; 94: 92–5. [DOI] [PubMed] [Google Scholar]
- 86. Hare JM, Traverse JH, Henry TD, et al A randomized, double‐blind, placebo‐controlled, dose‐escalation study of intravenous adult human mesenchymal stem cells (prochymal) after acute myocardial infarction. J Am Coll Cardiol. 2009; 54: 2277–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Freitag J, Ford J, Bates D, et al Adipose derived mesenchymal stem cell therapy in the treatment of isolated knee chondral lesions: design of a randomised controlled pilot study comparing arthroscopic microfracture versus arthroscopic microfracture combined with postoperative mesenchymal stem cell injections. BMJ Open. 2015; 5: e009332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Choi HW, Kim JS, Choi S, et al Neural stem cells differentiated from iPS cells spontaneously regain pluripotency. Stem Cells. 2014; 32: 2596–604. [DOI] [PubMed] [Google Scholar]
- 89. Xu Q. The impact of progenitor cells in atherosclerosis. Nat Clin Pract Cardiovasc Med. 2006; 3: 94–101. [DOI] [PubMed] [Google Scholar]
- 90. Torsney E, Xu Q. Resident vascular progenitor cells. J Mol Cell Cardiol. 2011; 50: 304–11. [DOI] [PubMed] [Google Scholar]
- 91. Tang Z, Wang A, Yuan F, et al Differentiation of multipotent vascular stem cells contributes to vascular diseases. Nat Commun. 2012; 3: 875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Hu Y, Zhang Z, Torsney E, et al Abundant progenitor cells in the adventitia contribute to atherosclerosis of vein grafts in ApoE‐deficient mice. J Clin Invest. 2004; 113: 1258–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Jain M, DerSimonian H, Brenner DA, et al Cell therapy attenuates deleterious ventricular remodeling and improves cardiac performance after myocardial infarction. Circulation. 2001; 103: 1920–7. [DOI] [PubMed] [Google Scholar]
- 94. Yasuda T, Weisel RD, Kiani C, et al Quantitative analysis of survival of transplanted smooth muscle cells with real‐time polymerase chain reaction. J Thorac Cardiovasc Surg. 2005; 129: 904–11. [DOI] [PubMed] [Google Scholar]
- 95. Suzuki K, Murtuza B, Beauchamp JR, et al Role of interleukin‐1beta in acute inflammation and graft death after cell transplantation to the heart. Circulation. 2004; 110: II219–24. [DOI] [PubMed] [Google Scholar]
- 96. Li HM, Liu L, Mei X, et al Overexpression of inducible nitric oxide synthase impairs the survival of bone marrow stem cells transplanted into rat infarcted myocardium. Life Sci. 2014; 106: 50–7. [DOI] [PubMed] [Google Scholar]
- 97. Orozco D, Delgado R, Wesloh D, et al Aerosol particulate matter in the Baltimore metropolitan area: temporal variation over a six‐year period. J Air Waste Manag Assoc. 2015; 65: 1050–61. [DOI] [PubMed] [Google Scholar]
- 98. Mueller D, Schulze J, Ackermann H, et al Particulate matter (PM) 2.5 levels in ETS emissions of a Marlboro Red cigarette in comparison to the 3R4F reference cigarette under open‐ and closed‐door condition. J Occup Med Toxicol. 2012; 7: 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Gerber A, Hofen‐Hohloch AV, Schulze J, et al Tobacco smoke particles and indoor air quality (ToPIQ‐II) ‐ a modified study protocol and first results. J Occup Med Toxicol. 2015; 10: 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. da Silveira Fleck A, Carneiro MF, Barbosa F Jr, et al Monitoring an outdoor smoking area by means of PM measurement and vegetal biomonitoring. Environ Sci Pollut Res Int. 2015; DOI: 10.1007/s11356‐015‐5878‐4. [DOI] [PubMed] [Google Scholar]
- 101. Huang J, Okuka M, McLean M, et al Effects of cigarette smoke on fertilization and embryo development in vivo . Fertil Steril. 2009; 92: 1456–65. [DOI] [PubMed] [Google Scholar]
- 102. Talbot P, Lin S. The effect of cigarette smoke on fertilization and pre‐implantation development: assessment using animal models, clinical data, and stem cells. Biol Res. 2011; 44: 189–94. [PubMed] [Google Scholar]
- 103. Lin S, Fonteno S, Weng JH, et al Comparison of the toxicity of smoke from conventional and harm reduction cigarettes using human embryonic stem cells. Toxicol Sci. 2010; 118: 202–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Yauk CL, Berndt ML, Williams A, et al Mainstream tobacco smoke causes paternal germ‐line DNA mutation. Cancer Res. 2007; 67: 5103–6. [DOI] [PubMed] [Google Scholar]
- 105. Liszewski W, Ritner C, Aurigui J, et al Developmental effects of tobacco smoke exposure during human embryonic stem cell differentiation are mediated through the transforming growth factor‐beta superfamily member. Nodal Differentiation. 2012; 83: 169–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Huang J, Okuka M, Lu W, et al Telomere shortening and DNA damage of embryonic stem cells induced by cigarette smoke. Reprod Toxicol. 2013; 35: 89–95. [DOI] [PubMed] [Google Scholar]
- 107. Lin S, Tran V, Talbot P. Comparison of toxicity of smoke from traditional and harm‐reduction cigarettes using mouse embryonic stem cells as a novel model for preimplantation development. Hum Reprod. 2009; 24: 386–97. [DOI] [PubMed] [Google Scholar]
- 108. Albrecht C, Adolf B, Weishaupt C, et al Clara‐cell hyperplasia after quartz and coal‐dust instillation in rat lung. Inhalation Toxicol. 2001; 13: 191–205. [DOI] [PubMed] [Google Scholar]
- 109. Izzotti A, Larghero P, Balansky R, et al Interplay between histopathological alterations, cigarette smoke and chemopreventive agents in defining microRNA profiles in mouse lung. Mutat Res. 2011; 717: 17–24. [DOI] [PubMed] [Google Scholar]
- 110. De Flora S, Balansky R, D'Agostini F, et al Smoke‐induced microRNA and related proteome alterations. Modulation by chemopreventive agents. Int J Cancer. 2012; 131: 2763–73. [DOI] [PubMed] [Google Scholar]
- 111. Zhou Y, Gan Y, Taylor HS. Cigarette smoke inhibits recruitment of bone‐marrow‐derived stem cells to the uterus. Reprod Toxicol. 2011; 31: 123–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Khaldoyanidi S, Sikora L, Orlovskaya I, et al Correlation between nicotine‐induced inhibition of hematopoiesis and decreased CD44 expression on bone marrow stromal cells. Blood. 2001; 98: 303–12. [DOI] [PubMed] [Google Scholar]
- 113. Xiao GG, Wang MY, Li N, et al Use of proteomics to demonstrate a hierarchical oxidative stress response to diesel exhaust particle chemicals in a macrophage cell line. J Biol Chem. 2003; 278: 50781–90. [DOI] [PubMed] [Google Scholar]
- 114. Deng X, Zhang F, Wang L, et al Airborne fine particulate matter induces multiple cell death pathways in human lung epithelial cells. Apoptosis. 2014; 19: 1099–112. [DOI] [PubMed] [Google Scholar]
- 115. Xu Z, Xu X, Zhong M, et al Ambient particulate air pollution induces oxidative stress and alterations of mitochondria and gene expression in brown and white adipose tissues. Part Fibre Toxicol. 2011; 8: 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Delfino RJ, Staimer N, Tjoa T, et al Airway inflammation and oxidative potential of air pollutant particles in a pediatric asthma panel. J Eposure Sci Environ Epidemiol. 2013; 23: 466–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Tonne C, Yanosky JD, Beevers S, et al PM mass concentration and PM oxidative potential in relation to carotid intima‐media thickness. Epidemiology. 2012; 23: 486–94. [DOI] [PubMed] [Google Scholar]
- 118. Godri KJ, Green DC, Fuller GW, et al Particulate oxidative burden associated with firework activity. Environ Sci Technol. 2010; 44: 8295–301. [DOI] [PubMed] [Google Scholar]
- 119. Strak M, Janssen NAH, Godri KJ, et al Respiratory health effects of airborne particulate matter: the role of particle size, composition, and oxidative potential‐the RAPTES project. Environ Health Persp. 2012; 120: 1183–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Poss J, Lorenz D, Werner C, et al Diesel exhaust particles impair endothelial progenitor cells, compromise endothelial integrity, reduce neoangiogenesis, and increase atherogenesis in mice. Cardiovasc Toxicol. 2013; 13: 290–300. [DOI] [PubMed] [Google Scholar]
- 121. Heiss C, Amabile N, Lee AC, et al Brief secondhand smoke exposure depresses endothelial progenitor cells activity and endothelial function: sustained vascular injury and blunted nitric oxide production. J Am Coll Cardiol. 2008; 51: 1760–71. [DOI] [PubMed] [Google Scholar]
- 122. Khurshid SS, Siegel JA, Kinney KA. Indoor particulate reactive oxygen species concentrations. Environ Res. 2014; 132: 46–53. [DOI] [PubMed] [Google Scholar]
- 123. Shafer MM, Perkins DA, Antkiewicz DS, et al Reactive oxygen species activity and chemical speciation of size‐fractionated atmospheric particulate matter from Lahore, Pakistan: an important role for transition metals. J Environ Monit. 2010; 12: 704–15. [DOI] [PubMed] [Google Scholar]
- 124. Koike E, Hirano S, Furuyama A, et al cDNA microarray analysis of rat alveolar epithelial cells following exposure to organic extract of diesel exhaust particles. Toxicol Appl Pharm. 2004; 201: 178–85. [DOI] [PubMed] [Google Scholar]
- 125. Romieu I, Tellez‐Rojo MM, Lazo M, et al Omega‐3 fatty acid prevents heart rate variability reductions associated with particulate matter. Am J Respir Crit Care Med. 2005; 172: 1534–40. [DOI] [PubMed] [Google Scholar]
- 126. Tashakkor AY, Chow KS, Carlsten C. Modification by antioxidant supplementation of changes in human lung function associated with air pollutant exposure: a systematic review. Bmc Public Health. 2011; 11: 532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Turgeon J, Dussault S, Haddad P, et al Probucol and antioxidant vitamins rescue ischemia‐induced neovascularization in mice exposed to cigarette smoke: potential role of endothelial progenitor cells. Atherosclerosis. 2010; 208: 342–9. [DOI] [PubMed] [Google Scholar]
- 128. Ebert R, Ulmer M, Zeck S, et al Selenium supplementation restores the antioxidative capacity and prevents cell damage in bone marrow stromal cells in vitro . Stem Cells. 2006; 24: 1226–35. [DOI] [PubMed] [Google Scholar]
- 129. Chang J, Li Y, Huang Y, et al Adiponectin prevents diabetic premature senescence of endothelial progenitor cells and promotes endothelial repair by suppressing the p38 MAP kinase/p16INK4A signaling pathway. Diabetes. 2010; 59: 2949–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Wang X, Zhao T, Huang W, et al Hsp20‐engineered mesenchymal stem cells are resistant to oxidative stress via enhanced activation of Akt and increased secretion of growth factors. Stem Cells. 2009; 27: 3021–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Ohshima M, Li TS, Kubo M, et al Antioxidant therapy attenuates diabetes‐related impairment of bone marrow stem cells. Circ J. 2009; 73: 162–6. [DOI] [PubMed] [Google Scholar]
- 132. Li HL, Wang Y, Pazhanisarny SK, et al Mn(III) meso‐tetrakis‐(N‐ethylpyridinium‐2‐yl) porphyrin mitigates total body irradiation‐induced long‐term bone marrow suppression. Free Radical Bio Med. 2011; 51: 30–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Pervaiz S, Taneja R, Ghaffari S. Oxidative stress regulation of stem and progenitor cells. Antioxid Redox Signal. 2009; 11: 2777–89. [DOI] [PubMed] [Google Scholar]
- 134. Lu T, Parthasarathy S, Hao H, et al Reactive oxygen species mediate oxidized low‐density lipoprotein‐induced inhibition of oct‐4 expression and endothelial differentiation of bone marrow stem cells. Antioxid Redox Signal. 2010; 13: 1845–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Steenhof M, Gosens I, Strak M, et al In vitro toxicity of particulate matter (PM) collected at different sites in the Netherlands is associated with PM composition, size fraction and oxidative potential–the RAPTES project. Part Fibre Toxicol. 2011; 8: 26 Doi:10.1186/1743‐8977‐8‐26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Wu W, Muller R, Berhane K, et al Inflammatory response of monocytes to ambient particles varies by highway proximity. Am J Respir Cell Mol Biol. 2014; 51: 802–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Xu X, Jiang SY, Wang TY, et al Inflammatory response to fine particulate air pollution exposure: neutrophil versus monocyte. PLoS ONE. 2013; 8: e71414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Massberg S, Konrad I, Schürzinger K, et al Platelets secrete stromal cell‐derived factor 1alpha and recruit bone marrow‐derived progenitor cells to arterial thrombi in vivo . J Exp Med. 2006; 203: 1221–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Stellos K, Gawaz M. Platelet interaction with progenitor cells: potential implications for regenerative medicine. Thromb Haemost. 2007; 98: 922–9. [DOI] [PubMed] [Google Scholar]
- 140. Zhao Y, Glesne D, Huberman E. A human peripheral blood monocyte‐derived subset acts as pluripotent stem cells. Proc Natl Acad Sci USA. 2003; 100: 2426–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Soltan M, Rohrer MD, Prasad HS. Monocytes: super cells for bone regeneration. Implant Dent. 2012; 21: 13–20. [DOI] [PubMed] [Google Scholar]
- 142. Van Winkle LS, Bein K, Anderson D, et al Biological dose response to PM2.5: effect of particle extraction method on platelet and lung responses. Toxicol Sci. 2015; 143: 349–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Solomon A, Smyth E, Mitha N, et al Induction of platelet aggregation after a direct physical interaction with diesel exhaust particles. J Thromb Haemost. 2013; 11: 325–34. [DOI] [PubMed] [Google Scholar]
- 144. Kim H, Oh SJ, Kwak HC, et al The impact of intratracheally instilled carbon black on the cardiovascular system of rats: elevation of bloodhomocysteine and hyperactivity of platelets. J Toxicol Environ Health A. 2012; 75: 1471–83. [DOI] [PubMed] [Google Scholar]
- 145. Yarlioglues M, Ardic I, Dogdu O, et al The acute effects of passive smoking on mean platelet volume in healthy volunteers. Angiology. 2012; 63: 353–7. [DOI] [PubMed] [Google Scholar]
- 146. Poursafa P, Kelishadi R. Air pollution, platelet activation and atherosclerosis. Inflamm Allergy Drug Targets. 2010; 9: 387–92. [DOI] [PubMed] [Google Scholar]
- 147. Bastonini E, Verdone L, Morrone S, et al Transcriptional modulation of a human monocytic cell line exposed to PM(10) from an urban area. Environ Res. 2011; 111: 765–74. [DOI] [PubMed] [Google Scholar]
- 148. Schneider A, Alexis NE, Diaz‐Sanchez D, et al Ambient PM2.5 exposure up‐regulates the expression of costimulatory receptors on circulating monocytes in diabetic individuals. Environ Health Perspect. 2011; 119: 778–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Yatera K, Hsieh J, Hogg JC, et al Particulate matter air pollution exposure promotes recruitment of monocytes into atherosclerotic plaques. Am J Physiol Heart Circ Physiol. 2008; 294: H944–53. [DOI] [PubMed] [Google Scholar]
- 150. Goto Y, Ishii H, Hogg JC, et al Particulate matter air pollution stimulates monocyte release from the bone marrow. Am J Respir Crit Care Med. 2004; 170: 891–7. [DOI] [PubMed] [Google Scholar]
- 151. van Eeden SF, Hogg JC. Systemic inflammatory response induced by particulate matter air pollution: the importance of bone‐marrow stimulation. J Toxicol Environ Health A. 2002; 65: 1597–613. [DOI] [PubMed] [Google Scholar]
- 152. Goto Y, Hogg JC, Shih CH, et al Exposure to ambient particles accelerates monocyte release from bone marrow in atherosclerotic rabbits. Am J Physiol Lung Cell Mol Physiol. 2004; 287: L79–85. [DOI] [PubMed] [Google Scholar]