

Key Clinical Message
Nicolaides–Baraitser syndrome (NCBRS) is a rare genetic condition associated with SMARCA2 gene mutations. Clinical diagnosis is challenging as its features evolve with time. The 20 years follow‐up of our NCBRS patient, with a previously unreported SMARCA2 mutation, illustrates the syndrome's natural history and its clinical variability, especially in a milder form.
Keywords: follow‐up studies, intellectual disability, natural history, Nicolaides–Baraitser syndrome, SMARCA2, sparse hair
Introduction
Nicolaides–Baraitser syndrome (NCBRS, OMIM #601358) was first reported as a consolidated syndrome in 2003 after being initially described in 1993 1, 2. It is defined by developmental delay, sparse hair, seizures, short stature, dysmorphic facies, and prominent interphalangeal joints. Since 2011, nontruncating mutations in the SMARCA2 gene have been found in 80% of affected individuals in the original cohort. SMARCA2 is part of the SWI/SNF‐related ATP‐dependent chromatin remodeling complexes and plays an important role in gene expression and neural development 3. Given that the median patient age in the literature is 10 years, little data are available on the long‐term evolution of NCBRS 4, 5. The original reported patient died at 33 years from status epilepticus with history of worsening motor and language function 5. This course is divergent from that seen in our presented patient. By reviewing the 20‐year follow‐up of our patient, we were able to gain insight into the possible varying progression of development and symptoms in NCBRS.
Clinical Report
The patient was born at term to a 28‐year‐old primigravida mother and a 29‐year‐old father. Parents were nonconsanguineous, of English descent, with no dysmorphic features or intellectual disability (ID). Pregnancy was complicated by a urinary tract infection in first trimester requiring antibiotics, placental abruption at 30 weeks, and preeclampsia at 39 weeks. There was no history of teratogen exposure. His birth weight was 3.4 kg (50th percentile), length was 48 cm (10th percentile), and occipitofrontal circumference (OFC) was 34 cm (50th percentile). Apgars were 9 and 9 at 1 and 5 min.
He was first assessed at 6 weeks for failure to thrive with weight 3.57 kg (<3rd percentile), and OFC 34.5 cm (−3 SD). Length is not available. Clinical examination revealed left‐sided torticollis, cryptorchidism, hypospadias, and umbilical hernia. He had sparse hair, droopy eyelids, curly eyelashes, prominent nasal root, bulbous nose, malar hypoplasia, and thin upper vermilion. Karyotype was 46, XY and basic metabolic work‐up, brain magnetic resonance imaging (MRI), abdominal ultrasound, and electroencephalogram (EEG) were normal. By 4 months, his feeding difficulties necessitated gastrojejunostomy (GJ) tube insertion. Occupational therapy and speech language pathology were initiated before 1 year of age.
At his 19 months review, examination revealed weight of 8.12 kg (<3rd percentile), length 76 cm (<3rd percentile), and OFC 45 cm (−3 SD). He had increased prominence of the lower vermilion border with emergence of mild scoliosis. He had no words; comprehension was normal. He had walked at 18 months with appropriate fine motor skills. His first generalized tonic–clonic seizure was noted at 21 months, for which he was started on carbamazepine. EEG was normal.
At 3 years of age, facial features and examination remained the same. His delays persisted, particularly with regard to language. He could not balance on one foot, had 5–10 words, and was unable to put two words together. Behaviorally, he had a short attention span with tactile sensitivities. Seizures ceased at 4 years of age.
On examination at 6.5 years of age, his weight was 18.1 kg (3rd–10th percentile), height was 106 cm (<3rd percentile), and OFC was 49.5 cm (−2 SD). Emerging features such as distal phalangeal broadening in the upper and lower limbs and prognathism were noted (Fig. 1). Hand, foot, and spine radiographs revealed no soft tissue or bony abnormality (Fig. 2).
Figure 1.

Hands and feet in Nicolaides–Baraitser syndrome. Patient's hands (A) display characteristic prominence of the phalangeal joints with distal phalangeal broadening and feet (B) also show broadening of the distal first and second toes with prominent interphalangeal joints.
Figure 2.
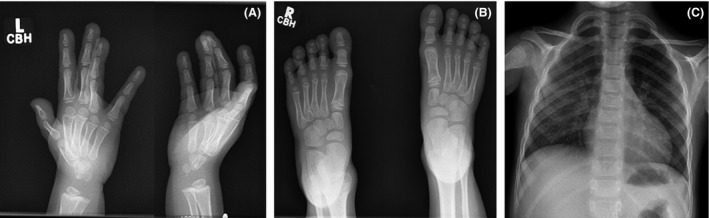
Radiographs of patient’ hands (A), feet (B), and thoracic spine (C) at 6 years of age. There were no bony or soft tissue abnormalities on hands and feet AP radiographs (A, B). AP chest radiograph shows mild downward sloping ribs with no skeletal abnormalities (C).
Developmentally, there was substantial improvement in language skills with the ability to speak in complete sentences. He was able to ride a bicycle. He was enrolled in mainstream grade 1 with one‐to‐one assistance with difficulties in long‐term recall and counting. Psychoeducational testing at 6 years 1 month indicated his function was at a 3 year 6 month level. He showed improvement in attention span but had developed poor impulse control. He had no comprehension of danger. His next available Wechsler Individual Intelligence Scale–Fourth Edition (WISC‐IV) result at age 11 years placed him in the moderate ID category.
His GJ tube was removed at age 12 years and he was orally fed with preference for puree and soft textures. A heightened choke and gag reflex persisted. Puberty occurred at 15–16 years of age and was accompanied by increased self‐aggression.
The patient was reassessed at age 20 years, after delayed follow‐up. His weight was 59.9 kg (10–25th percentile), height was 163.4 cm (5th percentile), and OFC was 52.5 cm (−2 SD). Over the years, his mild dysmorphism became more prominent with coarsening of facial features especially following puberty (Fig. 3). He had sparse hair, droopy eyelids, deep‐set eyes, malar hypoplasia, prominent nose, overhanging columella, full lips, and prognathism. Skin was normal apart from cystic acne on back. He remained seizure free. On development assessment, he was independent, with balance sufficient to skateboard. He had normal language with a mild speech impediment. He was able to prepare meals for himself. Parental encouragement was required for hygiene, and behavioral concerns were unchanged. He gained comprehension of danger. He continued to show gains in specialized education classes, with the ability to read and write basic English. He is registered to attend the special stream in a local college.
Figure 3.
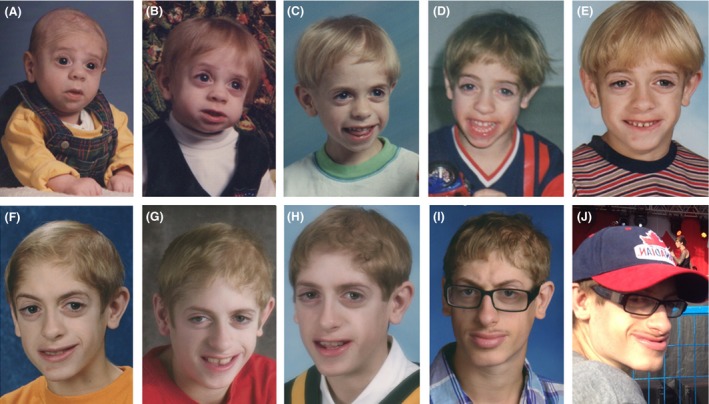
Evolving features of the patient at age 3 months (A), 1 year (B), 3 years (C), 5 years (D), 8 years (E), 11 years (F), 13 years (G), 15 years (H), 18 years (I), and 20 years (J). Note the progressive coarsening of facial features, sparse hair (A, B), eversion and filling of lower vermilion (F–J), and prognathism (I, J). Triangular face, downslanting palpebral fissures, malar hypoplasia, and thick alae nasi are seen throughout.
Genetic Testing
Nicolaides–Baraitser syndrome was clinically diagnosed at the 20 years reassessment. Targeted DNA sequence analysis of the SMARCA2 gene at age 20 years, completed using Illumina technology and confirmed by Sanger sequencing, showed a heterozygous mutation, specifically c.3493C>A (p.Gln1165Lys). This is a previously unreported mutation which was predicted to be deleterious by SIFT, PolyPhen2, and MutationTaster in silico pathogenicity prediction tools. It occurs in exon 25, which is part of domain VI helicase, in a highly conserved ATPase functional domain of the SMARCA2 gene. Parental testing for the above mutation in the SMARCA2 gene was negative, confirming a de novo event. Of the sixty‐one mutations observed in individuals with NCBRS, two are multiexon deletions and 59 are missense mutations, 58 of which occur in the ATPase domain (Fig. 4) 5. The proposed effect is that the missense mutations interfere with the ability to hydrolyze ATP, resulting in a functionally inactive protein due to a dominant negative effect 6.
Figure 4.

Schematic diagram of SMARCA2 gene and location of 59 known missense germline mutations (adopted from Sousa et al. 5). The arrow indicates the mutation identified in our patient.
Discussion
Through assessing the gradual development of the NCBRS features in our patient and reviewing previously reported cases, we are able to broaden the phenotype hitherto described in the literature. Our patient's facial features are consistent with NCBRS. Approximately 50% of 60 reported patients have either absent speech or speech decline whereas our patient showed remarkable gains in language acquisition 5. Whether this is related to degree and persistence of seizures requires further study in larger cohorts. Our patient is also unique in having feeding difficulties necessitating GJ tube feeds for 12 years. While feeding difficulties and poor weight gain have been documented in other patients, our report highlights that this can be a long‐term concern for some families. Other less frequently occurring anomalies include hypospadias, noted in only one patient 5, and scoliosis, noted in 9 of the 22 patients 5. With relatively advanced speech, chronic feeding concerns, and milder spectrum ID, our patient expands the variable phenotypic features described in NCBRS thus far.
Previous genotype–phenotype correlations explored by Sousa and Hennekam 5 in 11 patients suggested that mutations in exon 25 of domain VI are implicated in more severe ID and seizures. There are six reported cases with missense mutations affecting arginine at position 1159, described with severe ID and epilepsy 5. Patient 7 reported by Mari et al. 4 also has a domain VI mutation p.Ala1156Pro and has absent speech with seizures. Our patient has a mutation in the same region with milder ID and resolution of seizures. Thus, genotype–phenotype correlations remain difficult to be made with certainty in NCBRS.
Given that features of NCBRS take time to fully manifest, assessment of mildly affected patients in childhood can be particularly challenging. Some patients initially tentatively diagnosed with NCBRS have been later reclassified to Coffin–Siris syndrome (CSS) due to similarities in gestalt in infancy 7. Retrospective review of our patient's development supports this challenge. The characteristic facial features and prominent interphalangeal joints became progressively evident after 6 years of age. The overlap of features with CSS, and the possible lack of fully developed NCBRS features at an initial assessment, makes it likely for cases of NCBRS to be underrecognized or misclassified.
Moreover, our long‐term NCBRS follow‐up underscores the challenges of prognosis given phenotypic variability, as well as what anticipatory guidance could be provided. In the case of the original patient, poorly controlled seizures may have been a prognosticator for neurological complications and decline in function 8. After 6 years of age, our patient was medically and developmentally stable although his ID and behavioral concerns still impaired his activities of daily living. Behavioral concerns as an emerging feature were also noted 9 Thus, in less severely affected patients, one might need to focus on behavioral modifications as these could have the greatest impact on the patient's family unit.
Conclusion
Our patient broadens the NCBRS phenotype, as his mild developmental delay with preserved speech has been rarely described previously in NCBRS. He also differs from the previously reported genotype–phenotype correlation between mutations in SMARCA2 domain VI mutations and a severe phenotype. The 20‐year follow‐up of our patient highlights the changes in physical findings over the years, including the coarse facial features and interphalangeal prominence which may not be as evident in infancy. In addition, it suggests that behavior tendencies may become a source of distress in some families, as uncontrolled seizures or language development may be in others. In our patient we noted no evidence of regression. On the contrary, there was progressive improvement in milestones and academic skills as well as stature. Thus, early recognition of NCBRS is challenging and requires long‐term follow‐up.
Acknowledgments
We thank our patient and his family for their cooperation and participation in this follow‐up. The authors have no conflict of interest to declare.
Conflict of Interest
None declared.
Clinical Case Reports 2016; 4(4): 351–355
References
- 1. Nicolaides, P. , and Baraitser M.. 2003. An unusual syndrome with mental retardation and sparse hair. Clin. Dysmorphol. 2:232–236. [PubMed] [Google Scholar]
- 2. Morin, G. , Villemain L., Baumann C., Mathieu M., Blanc N., and Verloes A.. 2003. Nicolaides‐Baraitser syndrome: confirmatory report of a syndrome with sparse hair, mental retardation, and short stature and metacarpals. Clin. Dysmorphol. 12:237–240. [DOI] [PubMed] [Google Scholar]
- 3. Yoo, S. , and Crabtree G. R.. 2009. ATP‐dependent chromatin remodeling in neural development. Curr. Opin. Neurobiol. 19:120–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mari, F. , Marozza A., Mencarelli M. A., Lo Rizzo C., Fallerini C., Dosa L., et al. 2014. Coffin‐Siris and Nicolaides‐Baraitser syndromes are a common well recognizable cause of intellectual disability. Brain Dev. Brain & Development 37:527–536. [DOI] [PubMed] [Google Scholar]
- 5. Sousa, S. B. , Hennekam, R. C. , the Nicolaides‐Baraitser Syndrome International Consortium . 2014. Phenotype and genotype in Nicolaides‐Baraitser syndrome. Am. J. Med. Genet. Part C 166C:302–314. [DOI] [PubMed] [Google Scholar]
- 6. Van Houdt, J. K. J. , Nowakowska B. A., Sousa S. B., van Schaik B. D. C., Seuntjens E., Avonce N., et al. 2012. Heterozygous missense mutations in SMARCA2 cause Nicolaides‐Baraitser syndrome. Nat. Genet. 44:445–450. [DOI] [PubMed] [Google Scholar]
- 7. Wieczorek, D. , Bögershausen N., Beleggia F., Steiner‐Haldenstätt S., Pohl E., Li Y., et al. 2013. A comprehensive molecular study on Coffin‐Siris and Nicolaides‐Baraitser syndromes identifies a broad molecular and clinical spectrum converging on altered chromatin remodeling. Hum. Mol. Genet. 22:5121–5135. [DOI] [PubMed] [Google Scholar]
- 8. Sousa, S. B. , Abdul‐Rahman O. A., Bottani A., Cormier‐Daire V., Fryer A., Gillessen‐Kaesback G., et al. 2009. Nicolaides‐Baraitser syndrome: delineation of the phenotype. Am. J. Med. Genet. Part A 149A:1628–1640. [DOI] [PubMed] [Google Scholar]
- 9. Wolff, D. , Endele S., Azzarello‐Burri S., Hoyer J., Zweier M., Schanze I., et al. 2012. In‐frame deletion and missense mutations of the C‐terminal helicase domain of SMARCA2 in three patients with Nicolaides‐Baraitser syndrome. Mol. Syndromol. 2:237–244. [DOI] [PMC free article] [PubMed] [Google Scholar]