

Abstract
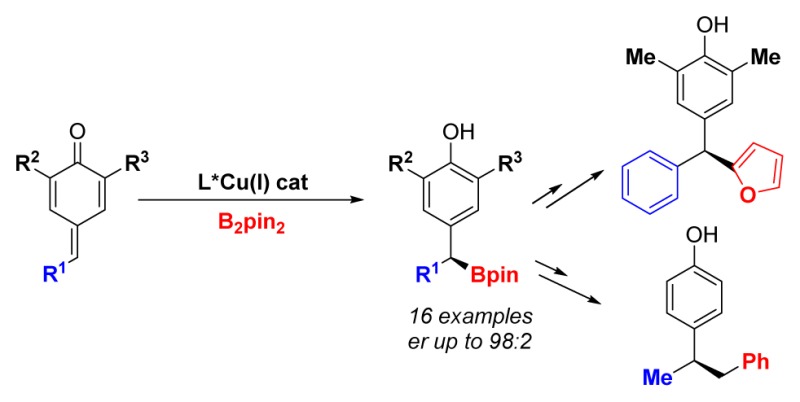
In this report, we establish that DM-Segphos copper(I) complexes are efficient catalysts for the enantioselective borylation of para-quinone methides. This method provides straightforward access to chiral monobenzylic and dibenzylic boronic esters, with enantiomeric ratios up to 96:4, using a commercially available chiral phosphine. Standard manipulations of the C–B bond afford a variety of chiral diaryl derivatives.
Keywords: asymmetric catalysis, boron, copper, synthetic methods, asymmetric synthesis
Chiral secondary boronic esters are important intermediates in organic synthesis, because they are precursors of chiral alcohols, chiral amines, and tertiary stereocenters.1 Among them, dibenzylic boronates such as B are especially interesting, because they can provide a variety of enantiomerically enriched diaryl derivatives (see Scheme 1). The diarylmethane framework represents a privileged structural motif widely found in pharmaceuticals.2 Most of these biologically active compounds present a chiral center at the benzylic position with a stereodefined C–O, C–N, or C–C bond. We envisioned that functionalization of the C–B bond in B could offer a unified strategy for the preparation of these compounds, from a common intermediate. However, the enantioselective synthesis of dibenzylic boronic esters is still a difficult challenge in chemical synthesis.
Scheme 1. Chiral Dibenzylic Boronic Esters.
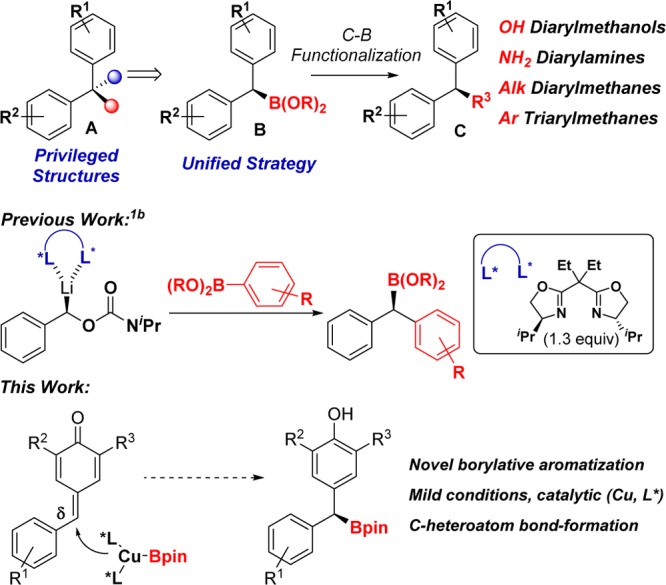
At the outset of this project, the only method available for the synthesis of boronates such as B involved the use of chiral lithiated carbamates and aryl boronic esters (Scheme 1).1b Despite the undoubted significance of this approach, the yields were moderate, and a stoichiometric amount of a chiral ligand was required. As part of our interest in unconventional C–B bond formation,3 we envisioned a new approach toward the synthesis of dibenzylic boronates through the enantioselective 1,6-addition of a chiral copper-(I) boryl complex to a p-quinone methide (Scheme 1).4,5 Formally, p-quinone methides are neutral entities with a zwitterionic resonance structure that enhances the electrophilic character at the δ-position. Surprisingly, while ortho-quinone methides have been broadly used in asymmetric synthesis,6 only two catalytic enantioselective additions to para-quinone methides have been reported.7,8 Both methods use carbon-based nucleophiles and an organocatalyst to control the enantioselectivity. Therefore, we became intrigued in exploring these compounds for several reasons:
-
(1)
the use of asymmetric metal catalysis to functionalize p-quinone methides remained unexplored;
-
(2)
the introduction of a boronic ester unit in ortho- or para-quinone methides had not been reported; and
-
(3)
the stereoselective addition of heteroatomic nucleophiles to p-quinone methides had not been studied to date.
Herein, we describe the synthesis of dibenzylic boronates through the borylative aromatization of p-quinone methides with good yields and high enantiomeric ratio (er) values, under mild reaction conditions and using a commercially available chiral phosphine.9,10
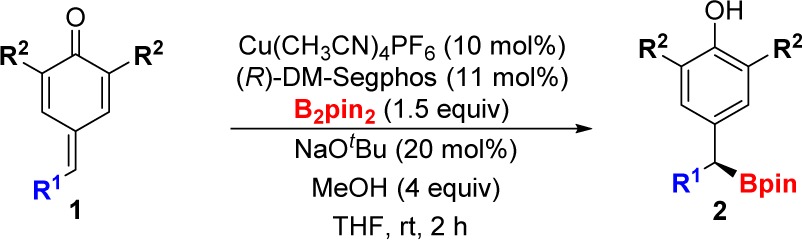
While unsubstituted p-quinone methides (R2, R3 = H) are too reactive to be isolated, 2,6-disubstituted derivatives are easy to handle. We began our study with p-quinone methide 1a, which contains removable t-Bu groups at the α-positions (Table 1).11 When 1a was treated in THF with Cu(CH3CN)4PF6 (10 mol %), B2pin2 (1.5 equiv), NaOt-Bu (0.2 equiv), and methanol (MeOH) (4 equiv) in the absence of ligand, we observed the formation of product 2a with moderate yield (Table 1, entry 1). This background reaction showed the feasibility of the transformation but also revealed a serious handicap for the development of an asymmetric version. We soon realized that the yields and stereoselectivities were highly dependent on the ligand (Table 1, entries 2–7).12 Commercially available (R)-DM-Segphos was superior to other chiral ligands affording the desired dibenzylic boronate 2a in good yield and high enantiomeric ratio at room temperature (Table 1, entry 7, er = 96:4). The reaction can be carried out with 5 mol % copper salt and 5.5 mol % of chiral phosphine (Table 1, entry 8) without affecting the enantioselectivity, although the yield observed under these conditions was slightly lower. Interestingly, in the absence of MeOH, we observed product formation but moderate er values (Table 1, entry 9).
Table 1. Effect of the Chiral Ligand in the Borylative Aromatization of p-Quinone Methides[a].
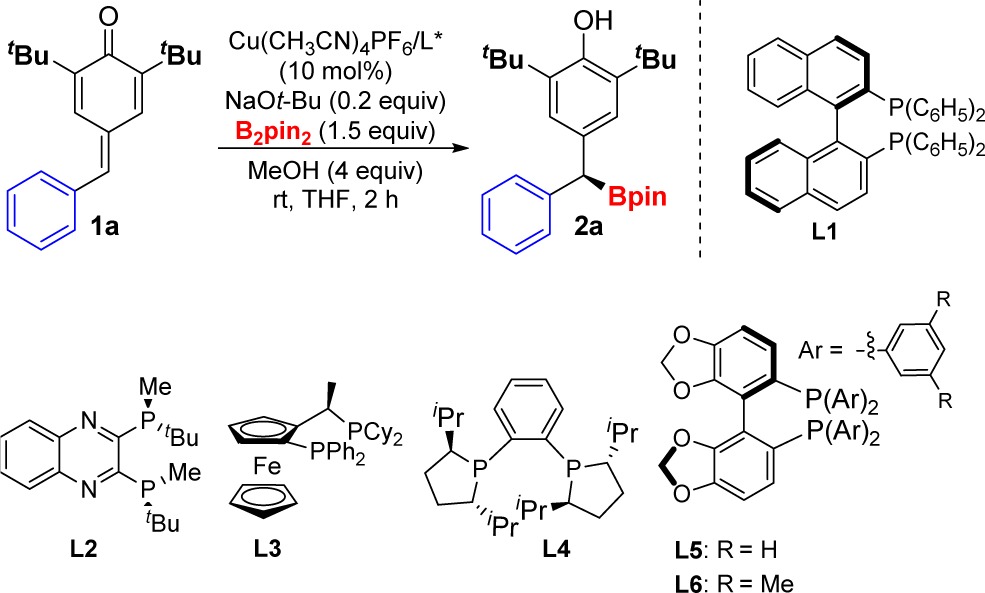
| entry | L* | enantiomeric ratio, er[b] | yield (%)[c] |
|---|---|---|---|
| 1[a] | 35 | ||
| 2[a] | L1 | 54.5:45.5 | 47 |
| 3[a] | L2 | 65:35 | 52 |
| 4[a] | L3 | 77:23 | 76 |
| 5[a] | L4 | 83:17 | 68 |
| 6[a] | L5 | 66.5:33.5 | 41 |
| 7[a] | L6 | 96:4 | 95 |
| 8[d] | L6 | 94:6 | 79 |
| 9e | L6 | 74:26 | 54 |
Reaction conditions: 1 (0.2 mmol), B2pin2 (0.30 mmol), NaOt-Bu (20 mol %), Cu(CH3CN)4PF6 (10 mol %), L* (11 mol %), MeOH (0.8 mmol), THF (0.2 M).
er determined by chiral SFC previous oxidation of the C–B bond.
Yield of isolated 2.
Reaction conditions: 1 (0.2 mmol), B2pin2 (0.30 mmol), NaOt-Bu (20 mol %), Cu(CH3CN)4PF6 (5 mol %), L* (5.5 mol %), MeOH (0.8 mmol), THF (0.2 M).
The reaction was carried out in the absence of MeOH.
With the optimal conditions in hand, we studied the scope of the borylative aromatization. The catalytic system was robust for p-quinone methides with different aromatic R1 substituents at the δ-carbon (Table 2, structures 2a–2g). Dibenzylic boronic esters with a larger naphthyl group (2b) or with electron-rich aromatic substituents (2c and 2d) were prepared in similarly good yields and high er values.
Table 2. Substrate Scope[a],[b].
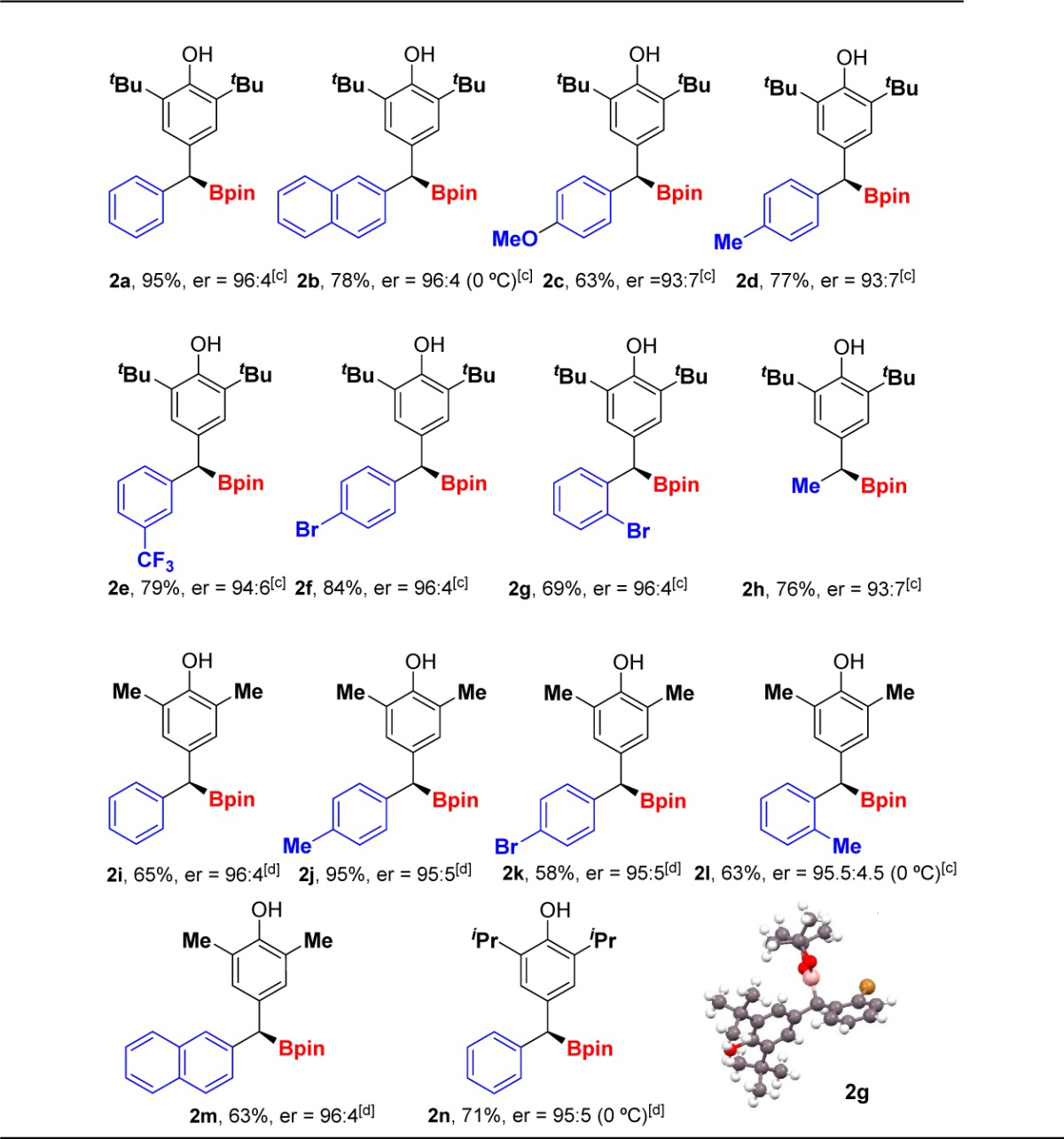
Reaction conditions: 1 (0.2 mmol), B2pin2 (0.30 mmol), NaOt-Bu (20 mol %), Cu(CH3CN)4PF6 (10 mol %), (R)-DM-Segphos (11 mol %), MeOH (0.8 mmol), THF (0.2 M).
Yield of isolated 2.
er value as determined by chiral SFC or HPLC previous oxidation of the C–B bond.
er value as determined by chiral SFC.
Diaryl derivatives with electron-withdrawing groups at the meta- (2e), para- (2f), or ortho- position (2g) were also synthesized, with excellent stereocontrol. In addition, the absolute configuration of 2g was determined by single-crystal X-ray crystallography.13 Importantly, a p-quinone methide bearing an alkyl group at the δ-carbon afforded the monobenzylic boronic ester 2h with good yield and high er value. Alkyl groups with different steric hindrance (methyl (Me) and isopropyl (i-Pr)) can also be introduced at the α-position (R2) without affecting the yields and enantioselectivities (2i–2n).
To further explore the scope of this reaction, we studied the borylative aromatization reaction with more challenging nonsymmetric p-quinone methides (1o and 1p; see Scheme 2). These substrates were synthesized as E/Z mixtures of the exocyclic double bond. Surprisingly, E/Zp-quinone methides 1o and 1p afforded dibenzylic boronic esters 2o and 2p with high enantioselectivities. This result is striking and significantly increases the potential structural scope of the method. These experiments indicate that the stereodiscrimination of the prochiral Si-face is not dependent on the geometry of the exocyclic double bond in the p-quinone methide.14,15
Scheme 2. Borylative Aromatization of Nonsymmetric p-Quinone Methides.
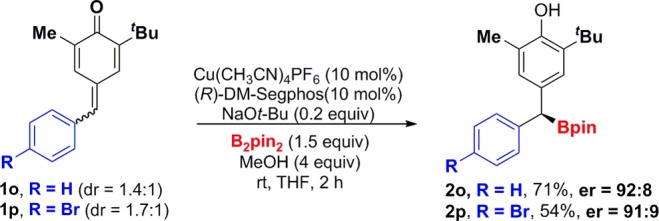
Overall, commercially available DM-Segphos consistently provides high er values and overcomes some of the structural limitations found with the use of chiral sulfoxide-phosphine ligands (prepared in four steps), recently reported by Liao.10 In the latter case, p-quinone methides with ortho- substitution on the aromatic ring (similar to 1g) and groups with less steric hindrance α to the carbonyl (similar to 1i–1n) afforded only moderate enantioselectivities (er = 84:16/75:25). Our catalyst system provides similar compounds with higher stereocontrol (compounds 2g, 2i–2n, er ≥ 95:5). More significantly, we have expanded the scope of the reaction to p-quinone methides bearing alkyl substituents at the R1 position (1h) and to the challenging nonsymmetric p-quinone methides (1o and 1p), which previously have not been studied.
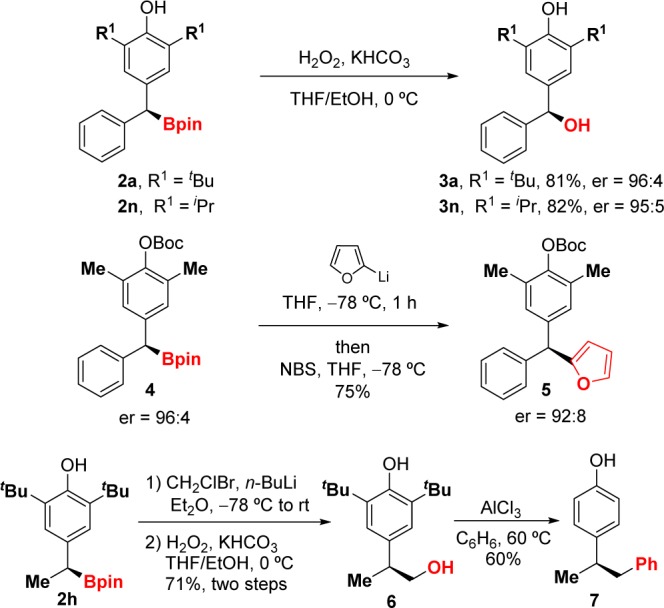
Functionalization of the C–B bond provided several monobenzylic, dibenzylic, and triaryl derivatives (Scheme 3). Oxidation of the C–B bond allows for the synthesis of enantiomerically enriched diarylmethanols in high yields. In addition, we have prepared triarylmethane 5 from boronate 4 with excellent stereoretention (97% specificity).1c To the best of our knowledge, this transition-metal-free C(sp3)–C(sp2) coupling has not been used before with secondary dibenzylic boronates. Homologation and oxidation of monobenzylic boronate 2h gave alcohol 6 in good overall yield. Finally, treatment of 6 with AlCl3 in refluxing benzene resulted in removal of the tert-butyl groups, followed by Friedel–Crafts reaction to afford phenol 7.
Scheme 3. C–B Bond Functionalization.
In order to gain insight into the reaction mechanism and to ascertain the reasons for the observed enantioselectivity, we performed quantum calculations at the DFT level. For this study, we used (R)-Segphos-Cu(I) complexes and p-quinone methide 1i (R2 = Me, R1 = Ph) as models. Noteworthy, no simplification of the ligand structure was used in order to properly consider the steric effects around the metal center. As shown in Scheme 4, the boryl cupration is a highly exoergic and, therefore, irreversible process. Since the absolute configuration of the new stereogenic carbon is fixed in this step, enantioselectivity is kinetically controlled. Transition states for the boryl cupration (TSR and TSS) were located and allowed the calculation of the corresponding activation energies for the formation of both enantiomers. The free energy of TSS is 0.9 kcal mol–1 lower than that corresponding to TSR, because of better substrate accommodation within the complex pocket for the former. This value is in accord with the observed enantioselectivity using 1a and (R)-Segphos (Table 1, entry 6). Intrinsic reaction coordinate studies connect these TS with long distance association adducts formed prior to the boryl cupration step (IS and IR; see Scheme 4).16 Calculated energies suggest the boryl cupration reaction as the rate-limiting step, as well as the enantioselective step.
Scheme 4. Calculated Reaction Profile for Both Diastereomeric Approaches (Re and Si) of the Boryl Cupration at the B3LYP/6-31G(d) (C,H,B,O,P) LANL2DZ (Cu) Level.
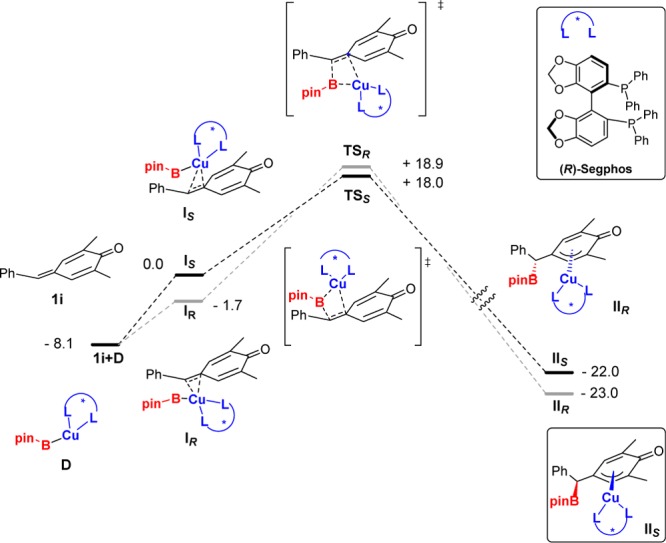
ΔG values are represented in kcal mol–1, considering IS as 0.0.
The Cu complexes formed after the alkene insertion show a long Cu–C distance with the C atom involved in the reaction (IIS and IIR; see Scheme 4). In fact, the structure is reminiscent of a (π-allyl)Cu complex. These complexes would become protonated in a subsequent step. We have also calculated the energy for isomer IIIs corresponding to the slipping of the borylated substrate to afford a copper-phenoxide complex (Figure 1). This process is highly exoergic, and for that reason, we propose that protonation most likely takes place at the Cu–O bond.17
Figure 1.
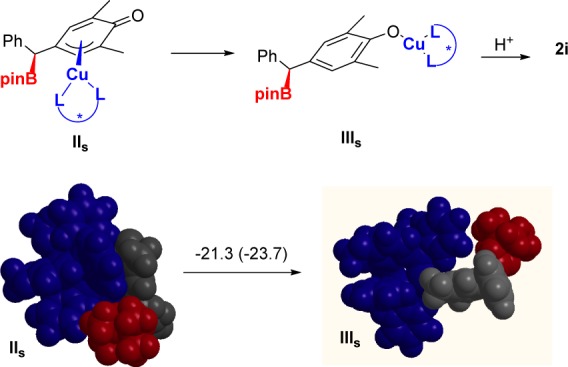
Calculated energy difference and equilibrium geometries for the formation of the copper phenoxide complex at B3LYP/STO-3G (C,H,B,O,P) LANL2DZ (Cu) level. Δ(E +ZPE) and ΔG values (given in brackets) in kcal mol–1.
In summary, we have developed a new method for the asymmetric synthesis of useful monobenzylic and dibenzylic boronic esters via a novel copper-catalyzed borylation of p-quinone methides. For the first time, consistently high enantioselectivities are observed using a commercially available phosphine ligand. The products are versatile intermediates for the enantioselective synthesis of monoaryl, diaryl, and triaryl derivatives. Calculations at the density functional theory (DFT) level fully agree with experimental observations and provide insight for the development of new asymmetric transformations.
Acknowledgments
This paper is dedicated to José Luis García Ruano on occasion of his retirement. We thank the European Research Council (No. ERC-337776) and MINECO (Nos. CTQ2012-35957 and CTQ2013-42806-R) for financial support. M.T. and A.P. thank MICINN for RyC and JdC contracts. D.C.-S. thanks MECD for a FPU fellowship. F.C. thanks Fundación Manuel Morales for a postdoctoral fellowship. We acknowledge Dr. Josefina Perles for X-ray structure analysis. We acknowledge the generous allocation of computer time at the Centro de Computación Científica at the Universidad Autónoma de Madrid (CCC-UAM).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscatal.5b02742.
The authors declare no competing financial interests.
Supplementary Material
References
- a Hall D. In Boronic Acids: Preparation and Applications in Organic Synthesis and Medicine; Hall D., Ed.; Wiley–VCH: Weinheim, Germany, 2005. [Google Scholar]; For recent applications of chiral secondary boronic esters, see:; b Matthew S. C.; Glasspoole B. W.; Eisenberger P.; Crudden C. M. J. Am. Chem. Soc. 2014, 136, 5828. 10.1021/ja412159g. [DOI] [PubMed] [Google Scholar]; c Bonet A.; Odachowski M.; Leonori D.; Essafi S.; Aggarwal V. K. Nat. Chem. 2014, 6, 584. 10.1038/nchem.1971. [DOI] [PubMed] [Google Scholar]; d Burns M.; Essafi S.; Bame J. R.; Bull S. P.; Webster M. P.; Balieu S.; Dale J. W.; Butts C. P.; Harvey J. N.; Aggarwal V. K. Nature 2014, 513, 183. 10.1038/nature13711. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Rasappan R.; Aggarwal V. K. Nat. Chem. 2014, 6, 810. 10.1038/nchem.2010. [DOI] [PubMed] [Google Scholar]; f Mlynarski S. N.; Karns A. S.; Morken J. P. J. Am. Chem. Soc. 2012, 134, 16449. 10.1021/ja305448w. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Larouche-Gauthier R.; Elford T. G.; Aggarwal V. K. J. Am. Chem. Soc. 2011, 133, 16794. 10.1021/ja2077813. [DOI] [PubMed] [Google Scholar]
- For biologically active diaryl derivatives, see the following. For diarylmethanols:; a Miki T.; Kori M.; Mabuchi H.; Tozawa R.; Nishimoto T.; Sugiyama Y.; Teshima K.; Yukimasa H. J. Med. Chem. 2002, 45, 4571. 10.1021/jm020234o. [DOI] [PubMed] [Google Scholar]; For diarylamines: ; b Spencer C. M.; Faulds D.; Peters D. H. Drugs 1993, 46, 1055. 10.2165/00003495-199346060-00008. [DOI] [PubMed] [Google Scholar]; For diarylmethanes: ; c Guay D.; Hamel P.; Blouin M.; Brideau C.; Chan C. C.; Chauret N.; Ducharme Y.; Huang Z.; Girard M.; Jones T. R.; Laliberté F.; Masson P.; McAuliffe M.; Piechuta H.; Silva J.; Young R.; Girard Y. Bioorg. Med. Chem. Lett. 2002, 12, 1457. 10.1016/S0960-894X(02)00190-7. [DOI] [PubMed] [Google Scholar]; For triarylmethanes: ; d Parai M. K.; Panda G.; Chaturvedi V.; Manju Y. K.; Sinha S. Bioorg. Med. Chem. Lett. 2008, 18, 289. 10.1016/j.bmcl.2007.10.083. [DOI] [PubMed] [Google Scholar]
- a Tortosa M. Angew. Chem., Int. Ed. 2011, 50, 3950. 10.1002/anie.201100613. [DOI] [PubMed] [Google Scholar]; b Alfaro R.; Parra A.; Alemán J.; García Ruano J. L.; Tortosa M. J. Am. Chem. Soc. 2012, 134, 15165. 10.1021/ja307670k. [DOI] [PubMed] [Google Scholar]; c Parra A.; Amenós L.; Guisán-Ceinos M.; López A.; Garcia Ruano J. L.; Tortosa M. J. Am. Chem. Soc. 2014, 136, 15833. 10.1021/ja510419z. [DOI] [PubMed] [Google Scholar]; d Parra A.; Lopez A.; Diaz-Tendero S.; Amenos L.; Garcia-Ruano J. L.; Tortosa M. Synlett 2015, 26, 494. 10.1055/s-0034-1379882. [DOI] [Google Scholar]
- For the only previous example of enantioselective 1,6-copper-catalyzed borylation of acyclic α,β,δ,γ-unsaturated esters, see:Luo Y.; Roy I. D.; Madec A. G. E.; Lam H. W. Angew. Chem., Int. Ed. Engl. 2014, 53, 4186. 10.1002/anie.201310380. [DOI] [PubMed] [Google Scholar]
- For seminal publications on 1,4-copper-catalyzed borylations, see:; a Takahashi K.; Ishiyama T.; Miyaura N. Chem. Lett. 2000, 982. 10.1246/cl.2000.982. [DOI] [Google Scholar]; b Takahashi K.; Ishiyama T.; Miyaura N. J. Organomet. Chem. 2001, 625, 47. 10.1016/S0022-328X(00)00826-3. [DOI] [Google Scholar]; c Ito H.; Yamanaka H.; Tateiwa J.; Hosomi A. Tetrahedron Lett. 2000, 41, 6821. 10.1016/S0040-4039(00)01161-8. [DOI] [Google Scholar]; For representative examples of enantioselective 1,4-copper-catalyzed borylations of electron-poor alkenes, see:; d Hornillos V.; Vila C.; Otten E.; Feringa B. L. Angew. Chem., Int. Ed. 2015, 54, 7867. 10.1002/anie.201502987. [DOI] [PubMed] [Google Scholar]; e Kobayashi S.; Xu P.; Endo T.; Ueno M.; Kitanosono T. Angew. Chem., Int. Ed. 2012, 51, 12763. 10.1002/anie.201207343. [DOI] [PubMed] [Google Scholar]; f Hartmann E.; Oestreich M. Org. Lett. 2012, 14, 2406. 10.1021/ol300832f. [DOI] [PubMed] [Google Scholar]; g Burns A. R.; Solana Gonzalez J. S.; Lam H. W. Angew. Chem., Int. Ed. 2012, 51, 10827. 10.1002/anie.201205899. [DOI] [PubMed] [Google Scholar]; h Moure A. L.; Gomez Arrayas R.; Carretero J. C. Chem. Commun. 2011, 47, 6701. 10.1039/c1cc11949d. [DOI] [PubMed] [Google Scholar]; i Lee J. C.; McDonald R.; Hall D. G. Nat. Chem. 2011, 3, 894. 10.1038/nchem.1150. [DOI] [PubMed] [Google Scholar]; j Fernandez E.; Gulyas H.; Sole C.; Mata J. A.; Tatla A.; Whiting A. Chem. - Eur. J. 2011, 17, 14248. 10.1002/chem.201102081. [DOI] [PubMed] [Google Scholar]; k O’Brien J. M.; Lee K.-S.; Hoveyda A. H. J. Am. Chem. Soc. 2010, 132, 10630. 10.1021/ja104777u. [DOI] [PMC free article] [PubMed] [Google Scholar]; l Chen I.-H.; Yin L.; Itano W.; Kanai M.; Shibasaki M. J. Am. Chem. Soc. 2009, 131, 11664. 10.1021/ja9045839. [DOI] [PubMed] [Google Scholar]; m Sim H.-S.; Feng X.; Yun J. Chem.—Eur. J. 2009, 15, 1939. 10.1002/chem.200802150. [DOI] [PubMed] [Google Scholar]; n Lillo V.; Prieto A.; Bonet A.; Diaz-Requejo M. M.; Ramirez J.; Perez P. J.; Fernandez E. Organometallics 2009, 28, 659. 10.1021/om800946k. [DOI] [Google Scholar]; o Lee E.; Yun J. Angew. Chem., Int. Ed. 2008, 47, 145. 10.1002/anie.200703699. [DOI] [PubMed] [Google Scholar]
- For recent reviews, see:; a Bai W.-J.; David J. G.; Feng Z.-G.; Weaver M. G.; Wu K.-L.; Pettus T. R. R. Acc. Chem. Res. 2014, 47, 3655. 10.1021/ar500330x. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Pathak T. P.; Sigman M. S. J. Org. Chem. 2011, 76, 9210. 10.1021/jo201789k. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Caruana L.; Fochi M.; Bernardi L. Molecules 2015, 20, 11733. 10.3390/molecules200711733. [DOI] [PMC free article] [PubMed] [Google Scholar]; For recent examples, see:; d Hsiao C.-C.; Raja S.; Liao H.-H.; Atodiresei I.; Rueping M. Angew. Chem., Int. Ed. 2015, 54, 5762. 10.1002/anie.201409850. [DOI] [PubMed] [Google Scholar]; e Zhao J.-J.; Sun S.-B.; He S.-H.; Wu Q.; Shi F. Angew. Chem., Int. Ed. 2015, 54, 5460. 10.1002/anie.201500215. [DOI] [PubMed] [Google Scholar]; f Hsiao C.-C.; Liao H.-H.; Rueping M. Angew. Chem., Int. Ed. 2014, 53, 13258. 10.1002/anie.201406587. [DOI] [PubMed] [Google Scholar]; g Lv H.; Jia W.-Q.; Sun L.-H.; Ye S. Angew. Chem., Int. Ed. 2013, 52, 8607. 10.1002/anie.201303903. [DOI] [PubMed] [Google Scholar]; h Izquierdo J.; Orue A.; Scheidt K. A. J. Am. Chem. Soc. 2013, 135, 10634. 10.1021/ja405833m. [DOI] [PMC free article] [PubMed] [Google Scholar]; I Luan Y.; Schaus S. E. J. Am. Chem. Soc. 2012, 134, 19965. 10.1021/ja309076g. [DOI] [PMC free article] [PubMed] [Google Scholar]; j Jensen K. H.; Webb J. D.; Sigman M. S. J. Am. Chem. Soc. 2010, 132, 17471–17482. 10.1021/ja108106h. [DOI] [PMC free article] [PubMed] [Google Scholar]; k Jensen K. H.; Pathak T. P.; Zhang Y.; Sigman M. S. J. Am. Chem. Soc. 2009, 131, 17074. 10.1021/ja909030c. [DOI] [PMC free article] [PubMed] [Google Scholar]; l Zhang Y.; Sigman M. S. J. Am. Chem. Soc. 2007, 129, 3076. 10.1021/ja070263u. [DOI] [PubMed] [Google Scholar]; m Mattson A. E.; Scheidt K. A. J. Am. Chem. Soc. 2007, 129, 4508. 10.1021/ja068189n. [DOI] [PMC free article] [PubMed] [Google Scholar]; n Gligorich K. M.; Schultz M. J.; Sigman M. S. J. Am. Chem. Soc. 2006, 128, 2794. 10.1021/ja0585533. [DOI] [PubMed] [Google Scholar]; o Schultz M. J.; Sigman M. S. J. Am. Chem. Soc. 2006, 128, 1460. 10.1021/ja0579053. [DOI] [PubMed] [Google Scholar]
- a Caruana L.; Kniep F.; Johansen T. K.; Poulsen P. H.; Jørgensen K. A. J. Am. Chem. Soc. 2014, 136, 15929. 10.1021/ja510475n. [DOI] [PubMed] [Google Scholar]; b Chu W.-D.; Zhang L.-F.; Bao X.; Zhao X.-H.; Zeng C.; Du J.-Y.; Zhang G.-B.; Wang F.-X.; Ma X.-Y.; Fan C.-A. Angew. Chem., Int. Ed. 2013, 52, 9229. 10.1002/anie.201303928. [DOI] [PubMed] [Google Scholar]; For an asymmetric addition of indoles using chiral Brønsted acid catalysis which appeared during the preparation of this manuscript, see:; c Wang Z.; Wong Y. F.; Sun J. Angew. Chem., Int. Ed. 2015, 54, 13711. 10.1002/anie.201506701. [DOI] [PubMed] [Google Scholar]; For examples of 1,6-additions in the context of asymmetric polymerization, see:; d Nakagaki N.; Uno T.; Kubo M.; Itoh T. J. J. Polym. Sci., Part A: Polym. Chem. 2009, 47, 5923. 10.1002/pola.23654. [DOI] [Google Scholar]; e Iizuka S.; Nakagaki N.; Uno T.; Kubo M.; Itoh T. Macromolecules 2010, 43, 6962. 10.1021/ma100866n. [DOI] [Google Scholar]; f Nagai T.; Uno T.; Kubo M.; Itoh T. J. J. Polym. Sci., Part A: Polym. Chem. 2012, 50, 466. 10.1002/pola.25054. [DOI] [Google Scholar]
- For a recent highlight, see:Parra A.; Tortosa M. ChemCatChem 2015, 7, 1524. 10.1002/cctc.201500176. [DOI] [Google Scholar]
- a Parra A.; Jarava C.; López A.; Cruz F.; Tortosa M. Asymmetric synthesis of Versatil Benzylic Boronic Esters through a Borylation-Aromation Process. Presented at the 18th IUPAC International Symposium on Organometallic Chemistry Directed Towards Organic Synthesis (OMCOS 18), Sitges–Barcelona, Spain, June 28–July 2, 2015; Paper No. P-065.; b López A.; Jarava C.; Parra A.; Cruz F.; Tortosa M. Asymmetric Synthesis of Versatil Benzylic Boronic Esters through a Borylation-Aromation Process. Presented at the 19th European Symposium of Organic Chemistry (ESOC2015), Lisboa, Spain, July 12–16, 2015; Paper No. P181.; c Jarava C.; López A.; Parra A.; Cruz F.; Tortosa M. Asymmetric Synthesis of Versatil Benzylic Boronic Esters through a Borylation-Aromatization Process . Presented at the XXXV Spanish Biannual Chemistry Congress, A Coruña, Spain, July 19–23, 2015; Paper No. S3-FC-12.
- During the preparation of this manuscript, Liao et al. published a closely related paper using a sulfoxide-phosphine ligand (published online Aug. 28, 2015):Lou Y.; Cao P.; Jia T.; Zhang Y.; Wang M.; Liao J.. Angew. Chem., Int. Ed. 2015, 54, 12134. 10.1002/anie.201505926We have observed similar enantioselectivities using a commercially available ligand and running the reaction at room temperature. In addition, our catalytic system can overcome some of the structural limitations found in the excellent work by Liao. [DOI] [PubMed] [Google Scholar]
- For a review on the use of the t-Bu group as a protecting group in the synthesis of aromatic compounds, see:Saleh S. A.; Tashtoush H. I. Tetrahedron 1998, 54, 14157. 10.1016/S0040-4020(98)00756-X. [DOI] [Google Scholar]
- For a full account on all the ligands used and other parameters, see the Supporting Information.
- CCDC 1405406 contains the supplementary crystallographic data. These data can be obtained free of charge at www.ccdc.cam.ac.uk/conts/retrieving.html. The absolute configuration of the other dibenzylic boronates was assigned by analogy.
- 1H NMR experiments at shorter reaction times did not show olefin isomerization of the starting E/Z mixture.
- We have proposed a stereochemical model based on DFT calculations, which show that steric interactions between the double bond substituent and the ligand play an important role in the stereodiscrimination. See Supporting Information for details.
- Formation of these adducts is endoergic, for entropy reasons, as expected for an associative process. Other higher-energy real coordination complex could be located (see Supporting Information, IVR), but they are not productive, since they are not connected to the transition states, as shown by IRC studies.
- We cannot rule out direct reaction of the copper phenoxide with B2pin2 to generate a borylated phenol and a copper(I)-boryl complex that would start over the catalytic cycle.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.