

Abstract
Enzymes are important and effective biological catalyst proteins participating in almost all active cell processes. Identification of multi-functional enzymes is essential in understanding the function of enzymes. Machine learning methods perform better in protein structure and function prediction than traditional biological wet experiments. Thus, in this study, we explore an efficient and effective machine learning method to categorize enzymes according to their function. Multi-functional enzymes are predicted with a special machine learning strategy, namely, multi-label classifier. Sequence features are extracted from a position-specific scoring matrix with autocross-covariance transformation. Experiment results show that the proposed method obtains an accuracy rate of 94.1% in classifying six main functional classes through five cross-validation tests and outperforms state-of-the-art methods. In addition, 91.25% accuracy is achieved in multi-functional enzyme prediction, which is often ignored in other enzyme function prediction studies. The online prediction server and datasets can be accessed from the link http://server.malab.cn/MEC/.
Introduction
Enzymes play a crucial role in the catalysis of biological and chemical reactions. As effective catalyzers, they are not consumed and do not participate in the reactions. After they are catalyzed, more than 400 types of reactions can be accelerated. The enzyme commission (EC) number, which is based on the chemical reactions catalyzed by enzymes, is utilized to characterize different enzymes as a numerical classification scheme[1]. Enzymes are divided into six main classes, namely, oxidoreductases, transferases, hydrolases, lyases, isomerases, and ligases, and then subdivided into three hierarchical levels. Most studies on enzyme classification focused on monofunctional enzyme prediction. However, identification of the multifunctional enzyme, which is a specific type of enzyme that can catalyze two or more chemical reactions, has not been provided much attention.
Various approaches have been utilized to achieve high accuracy in monofunctional enzyme prediction. Bioinformatics approach has attained considerable achievements by using information on the protein sequence and structure[2]. Huang et al.[3] proposed an adaptive fuzzy k-nearest neighbor method with Am-Pse-AAC feature extraction method, which was first developed by Kou-Chen Chou for enzyme subfamily class prediction, and attained an excellent accuracy of 92.1% for the six main families. EzyPred[4] is a three-layer predictor that is based on PSSM; it considers protein evolutionary information abundant in the profiles. The second layer responsible for predicting the main function class achieves 93.7% accuracy. EFICAz[5] has a high accuracy of 92% in predicting four EC digit levels in a jackknife test on test sequences that are <40% identical to any sequences in the training dataset.
With regard to multifunctional enzyme prediction, Luna De Ferrari et al.[6] and Zou[7] achieved good results. Luna De Ferrari presented EnzyML, a multi-label classification method that employs InterPro signatures. This method can efficiently provide an explanation for proteins with multiple enzymatic functions and achieves over 98% subset accuracy without utilizing any feature extraction algorithms. Zou proposed two feature algorithms to make predictions and obtained 99.54% and 98.73% accuracy by using 20-D and 188-D features, respectively; however, dataset redundancy was not mentioned in the paper.
The enzyme sequence in the present study was obtained from the Swiss-Prot Database (release 2014.9), an authoritative organization that provides high-quality annotated protein sequences. After redundancy removal with cluster database—high identity with tolerance (CD—HIT)[8], the similarity of the sequence is established below 65% to ensure the effectiveness of the experiments. ACC is then applied[9, 10] for feature extraction. This method was first proposed by Dong as a taxonomy-based protein fold recognition approach and has not been utilized in enzyme classification yet. Accuracy of 94.1% in monofunctional enzyme classification is obtained by using the K-nearest neighbor classifier. With regard to multifunctional enzymes, an average precision of 95.54% and 91.25% is obtained after five cross-validation tests on all enzymes and multifunctional enzymes, respectively.
Method
Data preprocessing
The original downloaded dataset consists of 214,375 sequences. However, each enzyme class has duplicate sequences. 207,430 sequences remained after duplicate elimination. To eliminate the negative effect of sequence similarity, CD-HIT, a widely utilized procedure to reduce sequence redundancy and improve the performance of other sequence analyses using clustering (known as high computing speed) was applied to perform redundancy removal in the experiments. A total of 59,763 sequences with similarity below 65% were obtained. The CD-HIT algorithm progresses as follows. First, the http://cn.bing.com/dict/clientsearch?mkt=zh-CN&setLang=zh&form=BDVEHC&ClientVer=BDDTV3.5.0.4311&q=%E9%80%92%E5%87%8F%E6%8E%92%E5%BA%8F sequences are sorted in length-descending order. Second, the first series class is formed from the longest sequence, and subsequent sequences are compared with the representative sequence of the known series class. If the similarity is above the threshold set beforehand, the sequence is added in this class; otherwise, a new series class is formed. Third, the longest sequence is extracted from each class to form the final dataset. In the experiments, the threshold is set to 0.65, and the word length to compare is 5. Table 1 shows the situation before and after redundancy removal.
Table 1. Distribution of six enzyme classes before and after CD-HIT(0.65).
| Dataset | EC 1 | EC 2 | EC 3 | EC 4 | EC 5 | EC 6 | Total |
|---|---|---|---|---|---|---|---|
| original data | 32958 | 82735 | 38611 | 22754 | 14096 | 23221 | 214375 |
| after duplicate-elimination | 32016 | 79144 | 36862 | 22421 | 13872 | 23115 | 207430 |
| after CD-HIT | 8781 | 23716 | 11994 | 5331 | 4037 | 5904 | 59763 |
Notably, the multifunctional enzymes in the six classes have not been removed yet. Table 2 shows the distribution of multifunctional enzymes in the six classes.
Table 2. Distribution of multifunctional enzymes before and after CD-HIT(0.65).
| Multifunctional enzymes | EC 1 | EC 2 | EC 3 | EC 4 | EC 5 | EC 6 | Total |
|---|---|---|---|---|---|---|---|
| before redundancy | 1534 | 1924 | 2657 | 1698 | 616 | 179 | 4076 |
| after CD-HIT | 386 | 503 | 689 | 473 | 137 | 52 | 1085 |
Feature extraction algorithm
Position-specific scoring matrix
For convenience of discussion, we denote a protein sequence as S, which is expressed as
| (1) |
where L represents the length of S and si(1 ≤ i ≤ L) represents one item of the amino acid alphabet, which is expressed as {A, C, D, E, F, G, H, I, K, L, M, N, P, Q, R, S, T, V, W, Y}[11]. For sequence S, the position-specific scoring matrix (PSSM) was generated by implementing the PSI-BLAST program[12]. PSSM is a L*20 matrix[13] and can be expressed as follows:
| (2) |
where each row represents the corresponding position of S (e.g., the 1st row refers to s1, the 2nd row refers to s2, and so forth). Each column represents the corresponding residue type of the amino acid alphabet (e.g., the 1st column refers to “A,” the 2nd row refers to “C,” and so forth). pi,j(1 ≤ i ≤ L, j = 1,2,…, 20) is a score that represents the odds of si being mutated to residue type j during evolutionary processes; for example, p1,1 represents the odds of s1 being mutated to residue type “A”. A high score for pi,j usually indicates that the mutation occurs frequently and that the corresponding residue in that position may be functional.
ACC feature representation algorithm
The framework consists of two feature models denoted as AC and CC. By using the PSSM of Eq (2), the enzyme sequence is formulated into a 20-D feature vector. The 20-D feature vector is calculated as
| (3) |
where represents the average score of the amino acids in the enzyme sequence, which indicates the general odds of the sequence being muted to residue j during the evolutionary process.
In the model of AC, the enzyme sequence is computed as
| (4) |
As shown in Eq (4), FAC measures the average correlation between two amino acids separated by a distance of λ in the enzyme sequence. The dimension of the feature vector FAC is λ * 20.
In the model of CC, the enzyme sequence is computed as
| (5) |
As shown in Eq (5), FCC measures the average correlation between two amino acids separated by a distance of λ in the enzyme sequence among 20 types of standard amino acids. The dimension of the feature vector FCC is λ * 380.
Combining FAC and FCC generates a (400 * λ)−D feature vector to represent the enzyme sequence, as represented by
| (6) |
The ACC feature representation algorithm fully employs the influence of the position correlation among sequence amino acids on protein homology detection. Secondary structure features[14, 15] were considered in other protein classification works. However, it is too time consuming for constructing web server.
Classifier selection and tools
KNN algorithm
The K-nearest neighbors (KNN) algorithm is a mature method and is one of the simplest machine learning algorithms in theory. It is widely used for classification and regression. The key idea in this algorithm is that an object can be assigned to a class if the majority of its k nearest neighbors belong to this class. If k equals 1, then the object is simply assigned to the class of that single nearest neighbor.
For instance, in Fig 1, the objective is to classify the test sample (star) either to the first class of triangles or to the second class of squares. If k equals three, we assign it to the second class according to dashed line circle because two squares and only one triangle exist inside the circle. If k equals five, we assign it to the first class according to the solid line circle because three triangles and only two squares exist inside the circle.
Fig 1. KNN algorithm diagram.
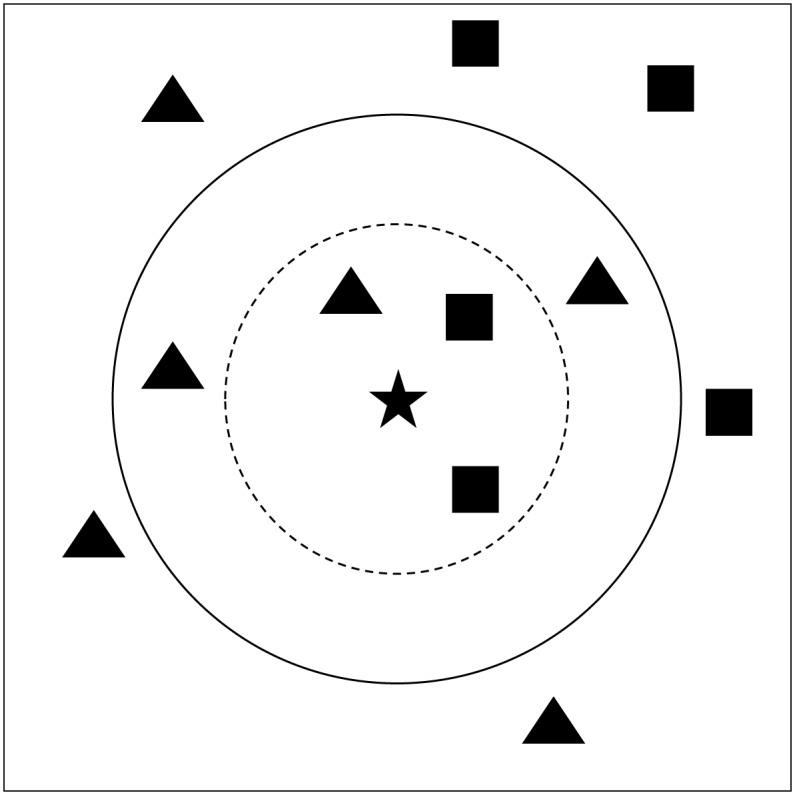
The choice of parameter k in this algorithm is important and depends on the data mostly. Generally, a large value of k dilutes the effect of noise in the classification but renders the boundaries between the categories less distinct. In our experiments, a large k value does not perform well.
KNN has been extensively utilized for the classification task in bioinformatics. Many recent studies have proven its high efficiency. In our experiments, we implemented a host of underlying classification algorithms and found that KNN is 20% more accurate than others.
WEKA and MULAN
Two of the main tools we utilized are Waikato environment for knowledge analysis (WEKA) and multi-label learning (MULAN). WEKA is an ensemble Java package with numerous machine learning algorithms and a graphical user interface. Several standard data mining tasks, including data preprocessing, feature selection, clustering, classification, regression, and visualization, are supported. MULAN is a Java library for learning from multi-label data. WEKA and MULAN contain an evaluation framework that calculates a rich variety of performance measures. They provide a convenient means to compare performance on different data using different classifiers.
Measurement
Single-label measurement
Given multi-label test datasets S = {(xi,yi)|1≤ i ≤ n}, for class yi where 1≤ j ≤ m, the binary classification performance of a predictor is presented by the four variables below.
TPj indicates the number of true positive instances, FPj indicates the number of false positive instances, TNj indicates the number of true negative instances, and FNj indicates the number of false negative instances. h(xi) indicates the classification results of sample xi predicted by classifier h.
We obtained four evaluation performance indicators according to these four variables as shown below[1, 16–22].
| (7) |
| (8) |
| (9) |
| (10) |
Multi-label measurement
We employed two evaluation indicators[23], namely, example-based and label-based metrics. For example-based metrics, we calculated the classification results for each sample first and then obtained the average value for the entire dataset.
We considered multi-label classifier h and multi-label dataset S = {(xi,Yi)|1≤ i ≤ n}, where Yi is the label collection of sample xi. Yi = {0,1,1,0,1,0} denotes that sample xi belongs to classes 1, 2, and 4 simultaneously.
| (11) |
This index indicates the performance of the relevance tag emerging before a certain tag in the sorted class label sequences. The higher average precision is, the better the performance is; the best value is 1.
For label-based metrics, we calculated the binary classification results for each class first and then obtained the average value for all classes.
Based on single-label measurement, we supposed that B(TPi, FPi, TNi, FNi) represents the binary classification indicator. The following are defined.
| (12) |
| (13) |
Bmacro measures the classification capability in each class and obtains the average of all classes as the final result. Its main idea is that each class shares the same weight. However, Bmicro endows each sample the same weight. It calculates the sum of values in all classes and then utilizes the value to obtain classification capability as the final result. Such is the difference between these two indicators.
Multi-label classification ensemble algorithm
Suppose that m classifiers solve an n-class classification problem. We define score matrix scoreVectors, and scoreVectors(i,j) indicates the possibility of the sample being classified into class j by classifier i, where 0≤scoreVectors(i,j)≤1, 1≤i≤n, 1≤j≤m.
Similarly, we define binary matrix bipartitionVectors, and bipartitionVectors(i,j) represents whether the sample is classified into class j by classifier i, where bipartitionVectors(i,j)∈{0,1}, 1≤i≤n, 1≤j≤m.
Below are three ensemble methods.
| (14) |
| (15) |
| (16) |
where Sort(scoreVectors(i,j)) represents the scores being sorting in descending order.
Result and Discussion
Monofunctional enzyme classification
First, we evaluated the importance of distance parameter λ in the ACC feature representation algorithm; 94.1% accuracy is attained for the dataset with similarity below 65% when λ is set to 1. With the increase in parameter λ, the improvement is not evident (only 0.1% increase), but time consumption is multiplied. This condition implies that the homology among adjacent amino acids is high. Second, we compared the performance of ACC method in different classifiers. IB1, which was built by KNN where neighbor k was set to 1, yielded the best results. The comparison results are shown in Fig 2.
Fig 2. Results of ACC method on different classifiers.

We also compared ACC with other popular protein prediction methods, such as 188D[24] (which considers the constitution, physicochemical properties[25], and distribution of amino acids), liu_feature (820D)[26] (which combines evolution information extracted from frequency profiles with sequence-based kernels for protein remote homology detection), n-gram (20D)[27] proposed by Browm et al. (which denotes the feature vectors by probability calculation), Pse-AAC (420D) originally proposed by Chou[28, 29] (which has been comprehensively applied for diverse biological sequence analyses as an effective protein descriptor[30–38], and DNA descriptor[39–42]. As shown in Fig 3, the advantage of the ACC algorithm is obvious.
Fig 3. Results of fivefeaturerepresentationmethods on IB1 classifier.

Aside from these five feature representation methods, we also tested two other enzyme-oriented online platforms. The first one is EzyPred. We randomly extracted 10 enzyme sequences from each class within one multifunctional enzyme as the test dataset and obtained 80% accuracy, which is lower than the 93.7% accuracy mentioned in the paper. The public test website http://www.csbio.sjtu.edu.cn/bioinf/EzyPred/EzyPred is free to the public. The second platform is EFICAz2.5[11, 43]. We obtained 86.4% accuracy with the code obtained from the link http://cssb.biology.gatech.edu/skolnick/webservice/EFICAz2/index.html. This accuracy value is lower than the 92% accuracy mentioned in the paper.
Multifunctional enzyme classification
We applied the ACC method to multifunctional enzyme classification according to the results of monofunctional enzyme prediction. Given that KNN works well in monofunctional enzyme classification, we focused on classifiers (IBLR_ML[44]/MLkNN[45]/BRkNN[46]) whose kernel is the KNN algorithm with the aid of MULAN. Two other classifiers (RakEL[47]/HOMER) were also tested. From Table 3, we can see that the classifier IBLR_ML obtained the best average precision of 95.54%. Classifiers MLkNN and BRkNN also produced good results.
Table 3. Cross-validation results of Multi-Label classifiers.
| IBLR_ML | MLkNN | BRkNN | RAkEL | HOMER | |
|---|---|---|---|---|---|
| Micro-averaged Precision | 0.9239 | 0.9202 | 0.9251 | 0.9117 | 0.9070 |
| Micro-averaged Recall | 0.9128 | 0.919 | 0.9159 | 0.9117 | 0.8869 |
| Micro-averaged F-Measure | 0.9183 | 0.9196 | 0.9205 | 0.8628 | 0.8968 |
| Macro-averaged Precision | 0.9176 | 0.9134 | 0.9189 | 0.9181 | 0.9006 |
| Macro-averaged Recall | 0.9021 | 0.9103 | 0.907 | 0.8039 | 0.8759 |
| Macro-averaged F-Measure | 0.9097 | 0.9118 | 0.9128 | 0.8559 | 0.8879 |
| Average Precision | 0.9554 | 0.9542 | 0.9442 | 0.9267 | 0.9305 |
To test the classification performance of the multifunctional enzyme further, we performed cross validation on the multifunctional enzyme only. To ensure data reliability and experimental accuracy, the threshold of data redundancy was set to 0.9. Then, we obtained the dataset in Table 4. Table 5 shows that 89.4% average precision was obtained.
Table 4. Distribution of multifunctional enzyme after de-redundance (0.9).
| EC 1 | EC 2 | EC 3 | EC 4 | EC 5 | EC 6 | Total |
|---|---|---|---|---|---|---|
| 861 | 994 | 1426 | 927 | 290 | 91 | 4589 |
Table 5. Cross-validation results of Multi-Label classification on multifunctional enzymes only.
| IBLR_ML | MLkNN | BRkNN | RAkEL | HOMER | |
|---|---|---|---|---|---|
| Micro-averaged Precision | 0.8406 | 0.8374 | 0.8279 | 0.8090 | 0.7519 |
| Micro-averaged Recall | 0.8178 | 0.8209 | 0.8285 | 0.8126 | 0.8233 |
| Micro-averaged F-Measure | 0.8290 | 0.8290 | 0.8282 | 0.8108 | 0.7859 |
| Macro-averaged Precision | 0.6792 | 0.6746 | 0.7341 | 0.7364 | 0.6056 |
| Macro-averaged Recall | 0.6705 | 0.6761 | 0.7379 | 0.6917 | 0.6619 |
| Macro-averaged F-Measure | 0.6737 | 0.6747 | 0.7347 | 0.7004 | 0.6305 |
| Average Precision | 0.8940 | 0.8930 | 0.8583 | 0.8910 | 0.8407 |
To obtain good results, the five classifiers shown in Table 5 are combined into one. Precision increased to 91.25% with the TOP3 combination rule.
In statistical prediction, the independent dataset test, subsampling or K-fold crossover test and jackknife test are the three cross-validation methods often used to check a predictor for its accuracy[48]. However, among the three test methods, the jackknife test is deemed the least arbitrary that can always yield a unique result for a given benchmark dataset[49]. Accordingly, the jackknife test has been increasingly used and widely recognized by investigators to examine the quality of various predictors[31, 32, 34, 39, 40, 50–54]. However, for saving computational time, the 5-fold cross-validation was used in this study.
Conclusion
We have explored a new method of multifunctional enzyme prediction. Considering the position relation and homology among amino acids[55], we extracted sequence features by using ACC method and performed prediction by using the KNN algorithm. The cross-validation test results indicate that our method outperforms other existing algorithms in datasets with similarity below 65%. Accuracy values of 94.1% in monofunctional enzyme classification and 95.54% in multifunctional enzyme classification were achieved. Compared with other existing prediction methods in the field of multifunctional enzyme class prediction, our method demonstrates better versatility and effectiveness. A public prediction—recognition platform is provided at http://server.malab.cn/MEC/. Our work is expected to be helpful for enzyme prediction in the future.
Our work just focused on the features and multi-label classifier. Some other machine learning techniques, such as feature selection[56], training sample selection[57, 58], ensemble learning[59–61], network features[62–64], imbalance classification[65, 66], ought to be considered in the next step. It is worth noting that there are many other potential tools for enzyme prediction, such as, evolutionary computation[67, 68] and spiking neural models[69–76]. Furthermore, parallel techniques, such as Map Reduce[77, 78], should also be considered for big testing data in the future.
Data Availability
Data are available for download from Figshare (https://figshare.com/s/7c8fe7f9bb4176015bfa).
Funding Statement
Natural Science Foundation of China (No. 61302139), http://www.nsfc.gov.cn/.
References
- 1.Cheng X-Y, Huang W-J, Hu S-C, Zhang H-L, Wang H, Zhang J-X, et al. , A global characterization and identification of multifunctional enzymes. PLoS One, 2012. 7(6): p. e38979 10.1371/journal.pone.0038979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liu B, Liu F, Wang X, Chen J, Fang L and Chou K-C, Pse-in-One: a web server for generating various modes of pseudo components of DNA, RNA, and protein sequences. Nucleic Acids Research, 2015. 43(W1): p. W65–W71. 10.1093/nar/gkv458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Huang WL, Chen HM, Hwang SF and Ho SY, Accurate prediction of enzyme subfamily class using an adaptive fuzzy k-nearest neighbor method. Biosystems, 2007. 90(2): p. 405–13. [DOI] [PubMed] [Google Scholar]
- 4.Shen HB and Chou KC, EzyPred: a top-down approach for predicting enzyme functional classes and subclasses. Biochem Biophys Res Commun, 2007. 364(1): p. 53–9. [DOI] [PubMed] [Google Scholar]
- 5.Tian W, Arakaki AK and Skolnick J, EFICAz: a comprehensive approach for accurate genome-scale enzyme function inference. Nucleic Acids Res, 2004. 32(21): p. 6226–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goryanin I, DF L, A S, vH J, EnzML: multi-label prediction of enzyme classes using InterPro signatures. 2012. 13(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zou Q, Chen W, Huang Y, Liu X and Jiang Y, Identifying Multi-Functional Enzyme by Hierarchical Multi-Label Classifier. Journal of Computational and Theoretical Nanoscience, 2013. 10(4): p. 1038–1043. [Google Scholar]
- 8.Fu L, Niu B, Zhu Z, Wu S and Li W, CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics, 2012. 28(23): p. 3150–2. 10.1093/bioinformatics/bts565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dong Q, Zhou S and Guan J, A new taxonomy-based protein fold recognition approach based on autocross-covariance transformation. Bioinformatics, 2009. 25(20): p. 2655–62. 10.1093/bioinformatics/btp500 [DOI] [PubMed] [Google Scholar]
- 10.Liu B, Liu F, Fang L, Wang X and Chou K-C, repDNA: a Python package to generate various modes of feature vectors for DNA sequences by incorporating user-defined physicochemical properties and sequence-order effects. Bioinformatics, 2015. 31(8): p. 1307–1309. 10.1093/bioinformatics/btu820 [DOI] [PubMed] [Google Scholar]
- 11.Arakaki AK, Huang Y and Skolnick J, EFICAz2: enzyme function inference by a combined approach enhanced by machine learning. BMC Bioinformatics, 2009. 10: p. 107 10.1186/1471-2105-10-107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Altschul Stephen F., Madden Thomas L., Schäffer Alejandro A., Zhang Jinghui, Zhang Zheng, Miller Webb, et al. , Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Research, 1997. 25(17): p. 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu B, Wang S and Wang X, DNA binding protein identification by combining pseudo amino acid composition and profile-based protein representation. Scientific Reports, 2015. 5: p. 15479 10.1038/srep15479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wei L, Liao M, Gao X and Zou Q, Enhanced Protein Fold Prediction Method through a Novel Feature Extraction Technique. IEEE Transactions on Nanobioscience, 2015. 14(6): p. 649–659. 10.1109/TNB.2015.2450233 [DOI] [PubMed] [Google Scholar]
- 15.Wei L, Liao M, Gao X and Zou Q, An Improved Protein Structural Prediction Method by Incorporating Both Sequence and Structure Information. IEEE Transactions on Nanobioscience, 2015. 14(4): p. 339–349. [DOI] [PubMed] [Google Scholar]
- 16.Liu B, Fang L, Long R, Lan X and Chou K-C, iEnhancer-2L: a two-layer predictor for identifying enhancers and their strength by pseudo k-tuple nucleotide composition. Bioinformaitcs, 2016. 32(3): p. 362–369. [DOI] [PubMed] [Google Scholar]
- 17.Chen J, Wang X and Liu B, iMiRNA-SSF: Improving the Identification of MicroRNA Precursors by Combining Negative Sets with Different Distributions. Scientific Reports, 2016. 6: p. 19062 10.1038/srep19062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu B, Fang L, Liu F, Wang X and Chou K-C, iMiRNA-PseDPC: microRNA precursor identification with a pseudo distance-pair composition approach. Journal of Biomolecular Structure and Dynamics, 2016. 34(1): p. 220–232. [DOI] [PubMed] [Google Scholar]
- 19.Cai S, Yang S, Zheng F, Lu M, Wu Y and Krishnan S, Knee joint vibration signal analysis with matching pursuit decomposition and dynamic weighted classifier fusion. Computational and Mathematical Methods in Medicine, 2013. 2013: p. 904267 10.1155/2013/904267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen W, Feng PM, Deng EZ, Lin H and Chou KC, iTIS-PseTNC: a sequence-based predictor for identifying translation initiation site in human genes using pseudo trinucleotide composition. Anal Biochem, 2014. 462: p. 76–83. 10.1016/j.ab.2014.06.022 [DOI] [PubMed] [Google Scholar]
- 21.Chen W, Feng PM, Lin H and Chou KC, iRSpot-PseDNC: identify recombination spots with pseudo dinucleotide composition. Nucleic Acids Res, 2013. 41(6): p. e68 10.1093/nar/gks1450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen W, Feng PM, Lin H and Chou KC, iSS-PseDNC: identifying splicing sites using pseudo dinucleotide composition. Biomed Res Int, 2014. 2014: p. 623149 10.1155/2014/623149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang Min-Ling and Zhou Z-H, A Review on Multi-Label Learning Algorithms. IEEE Transactions on Knowledge & Data Engineering, 2014. 26(8): p. 1. [Google Scholar]
- 24.Lin C, Zou Y, Qin J, Liu X, Jiang Y, Ke C, et al. , Hierarchical classification of protein folds using a novel ensemble classifier. PLoS One, 2013. 8(2): p. e56499 10.1371/journal.pone.0056499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu B, Wang X, Chen Q, Dong Q and Lan X, Using Amino Acid Physicochemical Distance Transformation for Fast Protein Remote Homology Detection. PLoS ONE, 2012. 7(9): p. e46633 10.1371/journal.pone.0046633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu B, Zhang D, Xu R, Xu J, Wang X, Chen Q, et al. , Combining evolutionary information extracted from frequency profiles with sequence-based kernels for protein remote homology detection. Bioinformatics, 2013. 30(4): p. 472–479. 10.1093/bioinformatics/btt709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brown Peter F., Della Pietra Vincen t J., deSouza Peter V., Lai Jenifer C. and Mercer ReL, Class-based n-gram models of natural language. Computational linguistics, 1992. 18(4): p. 467–479. [Google Scholar]
- 28.Chou KC, Using amphiphilic pseudo amino acid composition to predict enzyme subfamily classes. Bioinformatics, 2005. 21(1): p. 10–9. [DOI] [PubMed] [Google Scholar]
- 29.Liu B, Xu J, Fan S, Xu R, Zhou J and Wang X, PseDNA-Pro: DNA-Binding Protein Identification by Combining Chou's PseAAC and Physicochemical Distance Transformation. Molecular Informatics, 2015. 34(1): p. 8–17. [DOI] [PubMed] [Google Scholar]
- 30.Liu B, Xu J, Lan X, Xu R, Zhou J, Wang X, et al. , iDNA-Prot|dis: Identifying DNA-Binding Proteins by Incorporating Amino Acid Distance-Pairs and Reduced Alphabet Profile into the General Pseudo Amino Acid Composition. PLoS ONE, 2014. 9(9): p. e106691 10.1371/journal.pone.0106691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ding H, Liu L, Guo FB, Huang JA and Lin H, Identify Golgi Protein Types with Modified Mahalanobis Discriminant Algorithm and Pseudo Amino Acid Composition. Protein And Peptide Letters, 2011. 18(1): p. 58–63. [DOI] [PubMed] [Google Scholar]
- 32.Ding H, Luo LF and Lin H, Prediction of Cell Wall Lytic Enzymes Using Chou's Amphiphilic Pseudo Amino Acid Composition. Protein And Peptide Letters, 2009. 16(4): p. 351–355. [DOI] [PubMed] [Google Scholar]
- 33.Lin H, Ding H, Guo FB, Zhang AY and Huang J, Predicting subcellular localization of mycobacterial proteins by using Chou's pseudo amino acid composition. Protein And Peptide Letters, 2008. 15(7): p. 739–744. [DOI] [PubMed] [Google Scholar]
- 34.Zhu PP, Li WC, Zhong ZJ, Deng EZ, Ding H, Chen W, et al. , Predicting the subcellular localization of mycobacterial proteins by incorporating the optimal tripeptides into the general form of pseudo amino acid composition. Molecular Biosystems, 2015. 11(2): p. 558–563. 10.1039/c4mb00645c [DOI] [PubMed] [Google Scholar]
- 35.Ding H, Deng EZ, Yuan LF, Liu L, Lin H, Chen W, et al. , iCTX-Type: A Sequence-Based Predictor for Identifying the Types of Conotoxins in Targeting Ion Channels. Biomed Research International, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Feng P, Jiang N and Liu N, Prediction of DNase I hypersensitive sites by using pseudo nucleotide compositions. ScientificWorldJournal, 2014. 2014: p. 740506 10.1155/2014/740506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Feng P, Lin H, Chen W and Zuo Y, Predicting the types of J-proteins using clustered amino acids. Biomed Res Int, 2014. 2014: p. 935719 10.1155/2014/935719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Feng PM, Chen W, Lin H and Chou KC, iHSP-PseRAAAC: Identifying the heat shock protein families using pseudo reduced amino acid alphabet composition. Anal Biochem, 2013. 442(1): p. 118–25. 10.1016/j.ab.2013.05.024 [DOI] [PubMed] [Google Scholar]
- 39.Guo SH, Deng EZ, Xu LQ, Ding H, Lin H, Chen W, et al. , iNuc-PseKNC: a sequence-based predictor for predicting nucleosome positioning in genomes with pseudo k-tuple nucleotide composition. Bioinformatics, 2014. 30(11): p. 1522–1529. 10.1093/bioinformatics/btu083 [DOI] [PubMed] [Google Scholar]
- 40.Lin H, Deng EZ, Ding H, Chen W and Chou KC, iPro54-PseKNC: a sequence-based predictor for identifying sigma-54 promoters in prokaryote with pseudo k-tuple nucleotide composition. Nucleic Acids Research, 2014. 42(21): p. 12961–12972. 10.1093/nar/gku1019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen W, Feng P, Ding H, Lin H and Chou KC, iRNA-Methyl: Identifying N(6)-methyladenosine sites using pseudo nucleotide composition. Anal Biochem, 2015. 490: p. 26–33. 10.1016/j.ab.2015.08.021 [DOI] [PubMed] [Google Scholar]
- 42.Feng P, Chen W and Lin H, Prediction of CpG island methylation status by integrating DNA physicochemical properties. Genomics, 2014. 104(4): p. 229–33. 10.1016/j.ygeno.2014.08.011 [DOI] [PubMed] [Google Scholar]
- 43.Kumar N and Skolnick J, EFICAz2.5: application of a high-precision enzyme function predictor to 396 proteomes. Bioinformatics, 2012. 28(20): p. 2687–8. 10.1093/bioinformatics/bts510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cheng Wei-Wei and Hullermeier E, Combining instance-based learning and logistic regression for multilabel classification. Machine Learning, 2009. 76(2–3): p. 211–225. [Google Scholar]
- 45.Zhang Min-ling and Zhou Z, ML-KNN: A lazy learning approach to multi-label learning. Pattern Recognition, 2007. 40: p. 2038–2048. [Google Scholar]
- 46.E. Spyromitros, G. Tsoumakas and Vlahavas. I, An empirical study of lazy multilabel classification algorithms. Proc. 5th Hellenic Conference on Artificial Intelligence, 2008.
- 47.Tsoumakas Grigorios and Vlahavas I, Random k-Labelsets An Ensemble Method for Multilabel Classification. Pattern Recognition, 2007. 4701: p. 406–417. [Google Scholar]
- 48.Chou KC and Zhang CT, Prediction of Protein Structural Classes. Critical Reviews in Biochemistry and Molecular Biology, 1995. 30(4): p. 275–349. [DOI] [PubMed] [Google Scholar]
- 49.Chou KC, Some remarks on protein attribute prediction and pseudo amino acid composition. Journal of Theoretical Biology, 2011. 273(1): p. 236–247. 10.1016/j.jtbi.2010.12.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yuan LF, Ding C, Guo SH, Ding H, Chen W and Lin H, Prediction of the types of ion channel-targeted conotoxins based on radial basis function network. Toxicology in Vitro, 2013. 27(2): p. 852–856. 10.1016/j.tiv.2012.12.024 [DOI] [PubMed] [Google Scholar]
- 51.Lin H, The modified Mahalanobis Discriminant for predicting outer membrane proteins by using Chou's pseudo amino acid composition. Journal of Theoretical Biology, 2008. 252(2): p. 350–356. 10.1016/j.jtbi.2008.02.004 [DOI] [PubMed] [Google Scholar]
- 52.Lin H, Ding C, Song Q, Yang P, Ding H, Deng KJ, et al. , The prediction of protein structural class using averaged chemical shifts. Journal of Biomolecular Structure & Dynamics, 2012. 29(6): p. 643–649. [DOI] [PubMed] [Google Scholar]
- 53.Feng P, Chen W and Lin H, Identifying Antioxidant Proteins by Using Optimal Dipeptide Compositions. Interdiscip Sci, 2015. [DOI] [PubMed] [Google Scholar]
- 54.Tang H, Chen W and Lin H, Identification of immunoglobulins using Chou's pseudo amino acid composition with feature selection technique. Molecular BioSystems, 2016. [DOI] [PubMed] [Google Scholar]
- 55.Liu B, Chen J and Wang X, Application of Learning to Rank to protein remote homology detection. Bioinformatics, 2015. 31(21): p. 3492–3498. 10.1093/bioinformatics/btv413 [DOI] [PubMed] [Google Scholar]
- 56.Zou Q, Zeng J, Cao L and Ji R, A Novel Features Ranking Metric with Application to Scalable Visual and Bioinformatics Data Classification. Neurocomputing, 2016. 173: p. 346–354. [Google Scholar]
- 57.Wei L, Liao M, Gao Y, Ji R, He Z and Zou Q, Improved and Promising Identification of Human MicroRNAs by Incorporating a High-quality Negative Set. IEEE/ACM Transactions on Computational Biology and Bioinformatics, 2014. 11(1): p. 192–201 10.1109/TCBB.2013.146 [DOI] [PubMed] [Google Scholar]
- 58.Zeng X, Yuan S, Huang X and Zou Q, Identification of cytokine via an improved genetic algorithm. Frontiers of Computer Science, 2015. 9(4): p. 643–651. [Google Scholar]
- 59.Wang C, Hu L, Guo M, Liu X and Zou Q, imDC: an ensemble learning method for imbalanced classification with miRNA data. Genetics and Molecular Research, 2015. 14(1): p. 123–133. 10.4238/2015.January.15.15 [DOI] [PubMed] [Google Scholar]
- 60.Zou Q, Wang Z, Guan X, Liu B, Wu Y and Lin Z, An approach for identifying cytokines based on a novel ensemble classifier. BioMed research international, 2013. 2013(2013): p. 686090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lin C, Chen W, Qiu C, Wu Y, Krishnan S and Zou Q, LibD3C: Ensemble Classifiers with a Clustering and Dynamic Selection Strategy. Neurocomputing, 2014. 123: p. 424–435. [Google Scholar]
- 62.Zou Q, Li J, Song L, Zeng X and Wang G, Similarity computation strategies in the microRNA-disease network: A Survey. Briefings in Functional Genomics, 2016. 15(1): p. 55–64. 10.1093/bfgp/elv024 [DOI] [PubMed] [Google Scholar]
- 63.Zeng X, Zhang X and Zou Q, Integrative approaches for predicting microRNA function and prioritizing disease-related microRNA using biological interaction networks. Briefings in Bioinformatics, 2016. 17(2): p. 193–203. 10.1093/bib/bbv033 [DOI] [PubMed] [Google Scholar]
- 64.Zeng X, Liao Y, Liu Y and Zou Q, Prediction and validation of disease genes using HeteSim Scores. IEEE/ACM Transactions on Computational Biology and Bioinformatics, 2016. [DOI] [PubMed] [Google Scholar]
- 65.Zou Q, Xie S, Lin Z, Wu M and Ju Y, Finding the best classification threshold in imbalanced classification. Big Data Research, 2016. [Google Scholar]
- 66.Song L, Li D, Zeng X, Wu Y, Guo L and Zou Q, nDNA-prot: Identification of DNA-binding Proteins Based on Unbalanced Classification. BMC Bioinformatics, 2014. 15: p. 298 10.1186/1471-2105-15-298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhang Xingyi, T Y, Cheng Ran, Jin Yaochu, An efficient approach to non-dominated sorting for evolutionary multi-objective optimization. IEEE Transactions on Evolutionary Computation, 2015. 19(2): p. 201–213. [Google Scholar]
- 68.Zhang Xingyi, T Y, Jin Yaochu, A knee point driven evolutionary algorithm for many-objective optimization. IEEE Transactions on Evolutionary Computation, 2015. 19(6): p. 761–776. [Google Scholar]
- 69.Zeng Xiangxiang, P L, Pérez-Jiménez Mario J., Small Universal Simple Spiking Neural P System with Weights. SCIENCE CHINA: Information Science, 2014. 57(9): p. 92–102. [Google Scholar]
- 70.Zeng Xiangxiang, Z X, Song Tao, Pan Linqiang, Spiking Neural P Systems with Thresholds. Neural Computation, 2014. 26(7): p. 1340–1361. 10.1162/NECO_a_00605 [DOI] [PubMed] [Google Scholar]
- 71.Zhang Xingyi, P L, Pǎun Andrei , On universality of axon P systems. IEEE Transactions on Neural Networks and Learning Systems, 2015. 26(11): p. 2816–2829. [DOI] [PubMed] [Google Scholar]
- 72.Zhang Xingyi, Z X, Luo Bin, Pan Linqiang, On some classes of sequential spiking neural P systems. Neural Computation, 2014. 26(5): p. 974–997. 10.1162/NECO_a_00580 [DOI] [PubMed] [Google Scholar]
- 73.Zhang Xingyi, L Y, Luo Bin, Pan Linqiang, Computational power of tissue P systems for generating control languages. Information Sciences, 2014. 278(10): p. 285–297. [Google Scholar]
- 74.Chen Xu, P-J MJ, Valencia-Cabrera Luis, Wang Beizhan, Zeng Xiangxiang, Computing with viruses. Theoretical Computer Science, 2015. [Google Scholar]
- 75.Song Tao, P L, On the Universality and Non-universality of Spiking Neural P Systems with Rules on Synapses. IEEE Trans on Nanobioscience, 2015. [DOI] [PubMed] [Google Scholar]
- 76.Song Tao, X J, Pan Linaqing, Spiking Neural P Systems with Request Rules. Neurocomputing, 2016. [Google Scholar]
- 77.Zou Q, Li X, Jiang W, Lin Z, Li G and Chen K, Survey of MapReduce Frame Operation in Bioinformatics. Briefings in Bioinformatics, 2014. 15(4): p. 637–647. 10.1093/bib/bbs088 [DOI] [PubMed] [Google Scholar]
- 78.Zou Q, Hu Q, Guo M and Wang G, HAlign: Fast Multiple Similar DNA/RNA Sequence Alignment Based on the Centre Star Strategy. Bioinformatics, 2015. 31(15): p. 2475–2481. 10.1093/bioinformatics/btv177 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data are available for download from Figshare (https://figshare.com/s/7c8fe7f9bb4176015bfa).