

Summary
MicroRNAs (miRNAs) regulate diverse biological processes by repressing mRNAs, but their modest effects on direct targets, together with their participation in larger regulatory networks, make it challenging to delineate miRNA-mediated effects. Here, we describe an approach to characterizing miRNA-regulatory networks by systematically profiling transcriptional, post-transcriptional and epigenetic activity in a pair of isogenic murine fibroblast cell lines with and without Dicer expression. By RNA sequencing (RNA-seq) and CLIP (crosslinking followed by immunoprecipitation) sequencing (CLIP-seq), we found that most of the changes induced by global miRNA loss occur at the level of transcription. We then introduced a network modeling approach that integrated these data with epigenetic data to identify specific miRNA-regulated transcription factors that explain the impact of miRNA perturbation on gene expression. In total, we demonstrate that combining multiple genome-wide datasets spanning diverse regulatory modes enables accurate delineation of the downstream miRNA-regulated transcriptional network and establishes a model for studying similar networks in other systems.
Graphical Abstract
Introduction
MicroRNAs (miRNAs) are ~22 nucleotide regulatory RNAs that guide the RNA-induced silencing complex (RISC) to the 3′ untranslated region (3′UTR) of messenger RNAs (mRNAs) to inhibit translation and promote degradation (Baek et al., 2008; Guo et al., 2010; Selbach et al., 2008). miRNA activity is pleiotropic, with each miRNA repressing numerous targets that can be identified computationally using sequence features of mRNAs (Garcia et al., 2011; Grimson et al., 2007; Pasquinelli, 2012) or experimentally by individual nucleotide cross-linking followed by immunoprecipitation (iCLIP) of Argonaute, a member of the RISC (Chi et al., 2009; König et al., 2012; Sugimoto et al., 2012). Misregulation of miRNAs can lead to strong phenotypes in development (Chen et al., 2004) and disease (Lu et al., 2005; Mendell and Olson, 2012) despite the finding that most direct targets are only modestly (~2-fold) repressed (Baek et al., 2008).
Recent studies have found that miRNAs can have more profound effects when acting within larger regulatory networks, either alongside other miRNAs or together with transcription factors (Gurtan and Sharp, 2013; Herranz and Cohen, 2010; Schmiedel et al., 2015). When miRNAs regulate transcription factors, they can affect cellular phenotype, as demonstrated by miR-134 regulation of differentiation through interactions with mRNAs encoding Nanog and LRH1 transcription factors (Tay et al., 2008), let-7 regulation of HMGA2 (Mayr et al., 2007) or miR-145 regulation of SOX9 (Rani et al., 2013). Some studies have suggested that miRNAs preferentially target transcription factors (Lewis et al., 2003) and cause widespread changes in transcriptional activation (Gurtan et al., 2013). Additionally, miRNAs are often found within network motifs containing transcription factors, suggesting that they act alongside transcription factors to buffer gene expression (Gerstein et al., 2012; Shalgi et al., 2007; Tsang et al., 2007).
Despite the known biological importance of studying miRNA-transcription factor interactions, to date it is still challenging to distinguish direct miRNA-mediated effects from transcriptional effects by measuring mRNA alone, via arrays or with RNA sequencing (RNA-seq). While there are both experimental (Chi et al., 2009; Wen et al., 2011) and computational (Agarwal et al., 2015; Chiu et al., 2015; Garcia et al., 2011) methods to identify miRNA targets, identifying miRNA-regulated transcriptional changes is more challenging. Numerous computational approaches have used computational target prediction algorithms with transcription factor binding prediction tools to model the downstream effects of miRNAs through transcription factors (Afshar et al., 2014; Bisognin et al., 2012; Friard et al., 2010; Naeem et al., 2011; Tu et al., 2009). Recent advances in RNA sequencing efforts have enabled the use of total RNA measurements to capture both intronic and exonic changes. While this has been used as an additional way to identify genes that show evidence of post-transcriptional rather than transcriptional regulation (Du et al., 2014; Gaidatzis et al., 2015), it can still conflate transcriptional and post-transcriptional regulation.
Recently, the use of epigenetic data such as DNase I hypersensitivity assays (Song and Crawford, 2010) and histone post-translational modification marks (Ernst and Kellis, 2010) has improved characterization of transcriptional regulatory changes. These assays can measure specific changes to chromatin configuration near transcription start sites, providing accurate identification of genes with altered transcriptional regulation in a condition of interest. Incorporating these data into transcription factor binding predictions can improve the identification of genes that are transcriptionally regulated (Heintzman et al., 2009), as well as the transcription factors that are regulating the genes (Cuellar-Partida et al., 2012; Pique-Regi et al., 2011). To date, however, measurement of epigenetic perturbations alongside miRNA perturbation has been studied only in context of general changes (Gurtan et al., 2013) and not used to characterize miRNA regulatory networks.
In this work, we describe a comprehensive approach to study the relationship between miRNAs and transcription factors through integrative analysis of epigenetic, transcriptional and post-transcriptional changes. We apply this approach to immortalized Dicerf/f (wild-type; WT) and Dicer−/− (knockout; KO) murine fibroblast cell lines (Gurtan et al., 2013) to study the impact of global miRNA loss in a stable system. We collect and analyze miRNA expression, RNA expression and epigenetic data in both cell lines to fully quantify the contribution of transcriptional regulatory changes compared to post-transcriptional regulation. Then, introduce a network-based computational approach that takes advantage of these diverse high-throughput measurements to enable the identification of transcription factors likely to contribute to miRNA-mediated changes. Given the widespread availability of epigenetic and transcriptional data across various diseases, tissues and cell line models, this approach is highly applicable to study the effect of miRNAs in many different contexts.
Results
Decoupling Post-transcriptional and Transcriptional Regulation Reveals Global Changes in Transcription upon miRNA Loss
We first collected RNA from an isogenic clonal pair of immortalized Dicerf/f WT and Dicer−/− KO murine fibroblast cell lines (Gurtan et al., 2013) to quantify the changes in gene expression observed upon global miRNA loss. We sequenced two distinct RNA libraries: (1) ribo-depleted total-RNA libraries from WT and KO cells (see Experimental Procedures) to compare changes in exonic reads upon Dicer KO (Table S1A) with intronic read changes (Table S1B), which are unaffected by direct miRNA-mRNA interactions and (2) poly(A)-selected libraries (see Experimental Procedures, Table S1C). The exonic read changes from the total RNA library were highly correlated with reads from the polyA library (Fig. S1A; Spearman’s ρ =0.88) as well as individually-selected low-throughput targets via qPCR (see Experimental Procedures, Fig. S1B; Pearson’s r=0.96).
We then used intronic reads to estimate how much of the observed changes in mature mRNA expression were caused by changes in transcription. Since introns are spliced out of transcripts before export to the cytoplasm, comparisons of intronic and exonic read changes have been used as a way to isolate post-transcriptional changes of interest, as changes in intronic reads, which represent changes in pre-mRNA expression, can be ‘subtracted’ from mature mRNA changes represented by exonic reads (Du et al., 2014; Gaidatzis et al., 2015). We first confirmed that the intronic measurements of gene expression changes were accurate in a low-throughput manner using qPCR (Fig. S1C,D). We then used the intronic measurements to ask what fraction of mature mRNA changes observed upon Dicer loss could be attributed to changes in transcription. We found that changes in exonic reads were highly correlated with intronic reads within the same library (Fig. 1A; Spearman’s ρ =0.94). We repeated the same analysis with gene expression changes measured in polyA-selected library and found a statistically significant strong correlation (Fig. 1B; Spearman’s ρ =0.83). Together, these correlations strongly suggest that most significant mRNA expression changes observed after miRNA perturbation can be explained by changes in gene transcription and not miRNA-mediated degradation
Fig 1.

Transcription drives gene expression changes following miRNA loss. (A) Log2 fold change of genes that exhibit significant (q<0.05) change in ribo-depleted exon-aligned reads from WT vs. KO cells (x-axis) compared to fold change of intronic reads aligned to same genes (y-axis); regression line drawn in blue. (B) Log2 fold change of polyA-collected reads from WT vs. KO cells (x-axis) from significantly (q<0.05) changing genes compared to changes in reads aligned to introns from the ribo-depleted libraries (y-axis).
miRNA Target Identification
To identify those genes that exhibited evidence of post-transcriptional regulation, we used previously published iCLIP data (Gurtan et al., 2013) to identify bound targets of stably expressed Flag-hemagglutinin (HA)-tagged Ago2 in Dicer WT fibroblasts (Bosson et al., 2014; Zhang and Darnell, 2011) and small RNA-Seq measurements (Gurtan et al., 2013) (see Experimental Procedures) from the same cells. Using these two datasets, we identified high-confidence miRNA targets as those that showed evidence of a significant (q<0.05) iCLIP binding event in the 3′ UTR as well as a 7-mer or 8-mer seed match of an expressed miRNA, for a total of 2,754 miRNA-targeted genes in the poly(A) data and 2,729 miRNA-targeted genes in the ribo-depleted data (see Experimental Procedures for more details, Table S2). As expected, these biochemically-identified targets were enriched in genes that were up-regulated upon Dicer loss (p=1.16e-11 in the polyA data, p=1.66e-40 in the ribo-depleted data) and had a statistically significant impact on global mRNA expression change in both libraries (Fig. 2A,B). Furthermore, iCLIP activity was positively correlated with an increase in expression of those targets upon Dicer KO in both RNA libraries (Pearson’s r=0.24, p=2.36e-38 in the polyA data; Pearson’s r=0.27, p=4.09e-48 in the ribo-depleted data, Fig. S2B,C).
Fig. 2.

CDFs of miRNA targets upon global miRNA loss. (A) Mature mRNA expression changes according to exonic reads of total RNA of direct miRNA targets (blue) compared to non-targets (grey) upon Dicer KO. (B) Mature mRNA expression of direct miRNA targets by polyA-tagged mRNA (C) Δexon-Δintron values of direct miRNA targets representing differences in exonic measurements versus intronic measurements of miRNA targets (blue) compared to non-targets (grey).
We evaluated the post-transcriptional gene expression changes of biochemically identified miRNA targets by comparing exonic read changes (Δexon, defined as log2 ExonWT/ExonKO) and intronic read changes (Δintron, defined as log2 IntronWT/IntronKO) between Dicer WT and KO cells. This approach was recently introduced (Gaidatzis et al., 2015) and uses a generalized linear model using DESeq2 (Love et al., 2014) to assess the changes between exonic and intronic reads in the same sample (see Experimental Procedures). Genes with a greater difference between exonic and intronic changes are likely to be altered post-transcriptionally; therefore this metric can be used to assess miRNA-mediated repression of transcripts. mRNAs that are post-transcriptionally repressed by miRNAs will exhibit greater repression at the exonic level compared to the intronic level causing the Δexon-Δintron values of these genes to be negative. As expected, Δexon-Δintron values of genes that are miRNA targets are significantly (p=7.60×10e-107) more negative than non-targets, as depicted in Fig. 2C. This shift confirms the post-transcriptional effect of global Dicer loss on gene expression of direct miRNA targets.
Epigenetic Data Integration Identifies Transcriptional Regulatory Changes
To identify genes that were transcriptionally modulated upon Dicer deletion, we measured histone modifications, which are altered during transcription factor activity (Ernst and Kellis, 2010). We analyzed reads from previously-collected histone 3 lysine 4 tri-methylation (H3K4me3) marks (Gurtan et al., 2013), present on active promoters, and histone 3 lysine 36 tri-methylation (H3K36me3) marks (Gurtan et al., 2013), present on active gene bodies. We additionally collected data from histone 3 lysine 27 acetylation (H3K27ac), a mark associated with transcriptional promoters and enhancers (Creyghton et al., 2010). We used all three marks to identify regions that showed significant (q<0.05 for each mark) enrichment in WT (WT-specific) or KO (KO-specific) cells (see Experimental Procedures, Table S3A–F). By pairing changes in histone marks to nearby genes (see Experimental Procedures), we identified genes with gain or loss of transcriptional activity in the KO (1,187 and 2,259 genes respectively, Fig. S3A,B). After eliminating the 66 genes that showed evidence of gain of activation with one mark and loss of activation with another mark, we found a total of 3,314 genes with evidence of altered transcriptional regulation, representing ~25% of the total number of expressed genes. We confirmed that each of the three histone marks represent changes in transcriptional activity by measuring correlations between changes in histone modifications and mature mRNA expression of proximal genes (Pearson’s r=0.61, 0.68, 0.79 for H3K4me3, H3K27ac and H3K36me3 respectively), shown in Figs. S3C–E, as well as intronic expression of proximal genes, shown in Figs. S3F–H (Pearson’s r=0.56, 0.67, 0.66 for H3K4me3, H3K27ac and H3K36me3 respectively).
Then, we compared impact of transcriptional regulation with post-transcriptional regulation by dividing the gene population according to its mode of regulation (transcriptional, post-transcriptional, or both; Figures 3A and 3C, insets) and then computing the cumulative distribution functions (CDFs) of the mRNA log fold change of each set. The CDFs of all five sets of genes are depicted in Figure 3A, together with the genes that show no evidence of either transcriptional or post-transcriptional regulation (Figure 3A, gray curve). While genes regulated only post-transcriptionally exhibit a statistically significant shift in distribution (Figure 3A, blue curve; p = 1.64e-63), much greater shifts were observed for the CDFs of genes that are activated only transcriptionally (Figure 3A, yellow curve) or are both regulated post-transcriptionally and activated transcriptionally (Figure 3A, green curve). Approximately 60% of mRNAs exhibiting a >4-fold increase in expression in the KO cells show evidence of transcriptional activity (Figure 3B, yellow and green bars), which dwarfs the impact of miRNAs, whose targets constitute fewer than 5% of genes showing a >4-fold increase in expression (Figure 3B). Thus, transcriptional changes are far greater in both magnitude and number than post-transcriptional changes.
Fig. 3.
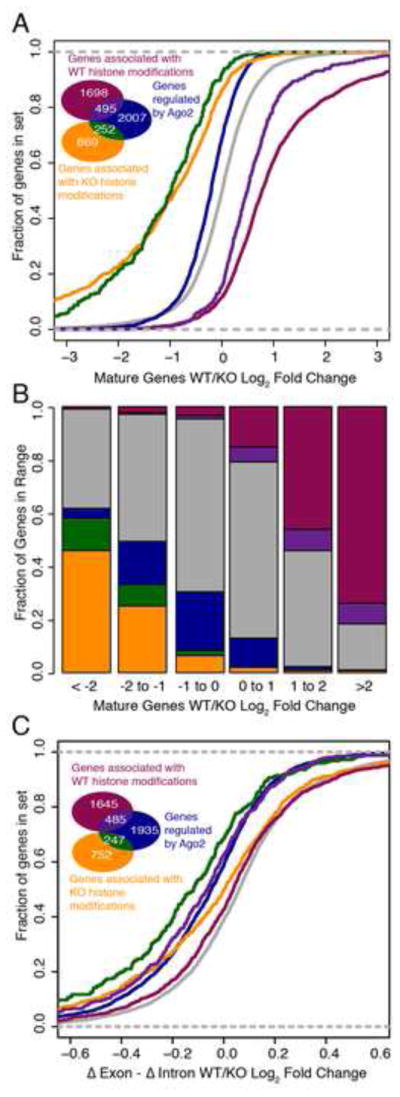
Histone marks illustrate collaboration between transcriptional and post-transcriptional regulatory modes with distinct impacts of gene expression. (A) Cumulative distribution plot of groups of genes defined by their mode of regulation, with colors and counts indicated by inset Venn diagram; grey represents genes without evidence of transcriptional or miRNA regulation. (B) Fraction of genes within each fold change bracket belonging to each class. (C) Distribution of Δexon - Δintron log2 fold change values of genes according to mode of regulation.
Lastly, we also compared Δexon - Δintron measurements changed among genes that were transcriptionally regulated and post-transcriptionally regulated. As we described earlier, genes that are post-transcriptionally repressed in the WT will have lower Δexon values than Δintron values, which would cause the distribution of Δexon - Δintron values to be more negative. We plotted these values in cumulative distribution in Fig. 3C for each of the same groups of genes described in Fig. 3A. Genes with evidence of iCLIP activity without transcriptional activity (blue curve, Fig. 3C) indeed exhibit a negative shift in cumulative distribution of Δexon - Δintron values compared to genes without any evidence of transcriptional or post-transcriptional regulation (grey curve). Additionally, we see changes in Δexon - Δintron between genes that are both transcriptionally and post-transcriptionally regulated (green, purple curves in Fig. 3C) compared to those that are only transcriptionally regulated (yellow, magenta curves in Fig. 3C). The distinct distribution of genes that are co-regulated by miRNAs and transcription factors (green, purple) both in Fig. 3C and Fig. 3A, underscore the importance of characterizing the transcriptional regulatory changes that occur downstream of miRNA perturbation, as miRNA and mRNA measurements alone fail to fully characterize the impact that miRNAs can have on regulatory networks. Measuring epigenetic changes that occur upon global Dicer loss greatly increases the ability to characterize the broader impacts of post-transcriptional regulation.
Hierarchical Network Algorithm Integrates All Data to Characterize Transcriptional Programs Activated upon miRNA Loss
We then used the epigenetic information provided by the histone marks to enumerate the transcriptional regulatory network activated upon miRNA perturbation. Specifically, we built an algorithm that explicitly modeled transcriptional activity in a network framework together with miRNA abundance and binding activity to identify which miRNA-regulated transcription factors best explain the observed global expression changes. The algorithm consists of two primary steps: assembling the diverse high throughput data into a graph (Fig. S4), and reducing the graph to the smallest set of nodes and edges that best explains the observed data (see Experimental Procedures for details).
The graph structure, summarized in Fig. 4, consists of nodes and edges that represent the individual data sets measuring changes between WT and KO fibroblasts. The nodes of the graph represent miRNAs (squares), transcription factors (triangles), predicted transcription factor binding regions (hexagons), mRNA (circles) and two dummy nodes dubbed the ‘Source’ (S) and the ‘Sink’ (T). Each edge is weighted by the likelihood of an interaction between two of the nodes in the network, and every possible path between the source and the sink represents a putative way in which miRNAs can affect mRNA changes (as measured at intronic level, see Experimental Procedures, Fig. S4). Thus, if a miRNA affects transcription of an mRNA, a green edge is shown between that miRNA and a transcription factor, a gold edge is shown between that transcription factor and a binding site upstream of the gene that encodes the mRNA, and a red or blue edge is shown between the binding site and the mRNA.
Fig. 4.
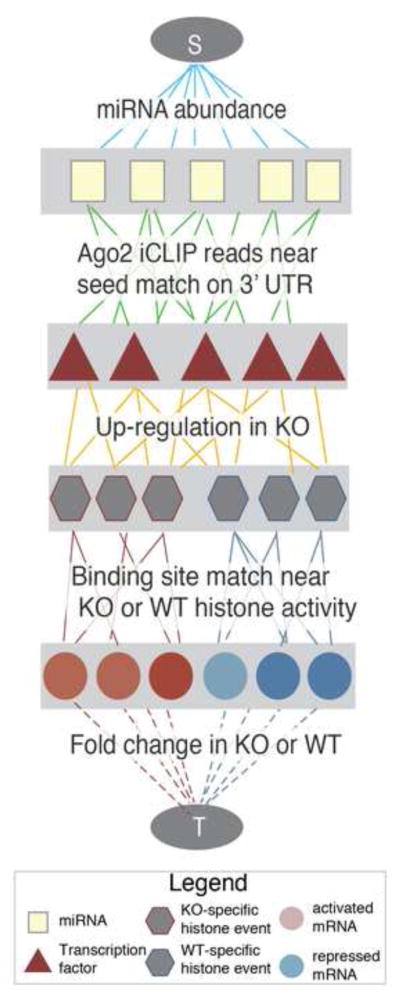
Implementation of hierarchical network algorithm. The network structure weights edges according to five types of data representing changes incurred upon miRNA loss. Legend inset.
To reduce the space of thousands of putative interactions between miRNAs, mRNAs that encode transcription factors, DNA-binding proteins and DNA binding sites, we applied a graph reduction step that uses the SAMNet constrained optimization algorithm (Gosline et al., 2012) to select the minimum number of edges in the graph that connect the source to the sink while ensuring to select the combination of edges with the highest total weight. SAMNet uses a ‘network flow’ approach that attempts to find the best path from the Source node to the Sink node using the fewest total edges while maximizing the sum of the weight on all the edges. Once a suitable solution is found from the source to the sink, no additional nodes are selected.
The resulting network, depicted in Fig. 5, maps a subset of the observed intronic mRNA changes (85 activated genes and 26 repressed) via 6 miRNAs and 14 distinct transcription factors. Given the algorithmic goal of minimizing the selection of nodes and edges while maximizing total weight, only the mRNAs that exhibit the largest absolute fold change are selected. As such, we focused our analysis on selected transcription factors (triangles in Fig. 5), their predicted number of targets (indicated by size), and up-regulation in the KO (indicated by the degree of red coloring), described in Table S4. As a control to address the possibility that the Argonaute protein complex can affect mRNA expression without a precise miRNA seed match (Chi et al., 2012), we allowed the algorithm to consider the possibility that a transcription factor can be repressed without the presence of an exact seed match (‘No Seed’ in Fig. 5). In this analysis, we identified two putatively active transcription factors, Foxd1 and Klf5, but each had few of their own targets predicted (Table S4), suggesting that these factors are less biologically relevant than those with seed evidence of miRNA binding. The complete list of transcription factors agrees with previous miRNA-transcription factor studies: let-7 represses Hmga2 (Lee and Dutta, 2007; Mayr et al., 2007) and Nr6a1(Gurtan et al., 2013) and miR-145 has been shown to regulate Sox9 (Rani et al., 2013).
Fig. 5.

The predicted network implicates 14 transcription factors (triangles) in activating mRNA (red circles) and repressing mRNA (blue circles) downstream of expressed miRNAs (squares). Legend inset.
To validate the robustness of the algorithm to the method by which miRNA targets were selected, we explored the possibility of applying the network approach with miRNA target predictions rather than iCLIP data. Given the steady improvements in the accuracy of miRNA target prediction tools such as TargetScan (Agarwal et al., 2015) and the difficulty of executing the iCLIP protocol, it was important evaluate the performance of the network algorithm using computational predictions. To do so, we applied the network approach using the TargetScan 6.2 mouse miRNA context+ scores as weights on the edges between miRNA and transcription factor nodes (see Experimental Procedures). The resulting network is depicted in Fig. S5. The network identified 28 transcription factors regulated by 7 miRNAs. Of these 28 transcription factors, 10 were found in the original iCLIP-derived network (that predicted only 14 transcription factors, Fig. 5). The ten common transcription factors include those that were experimentally validated below. The increased number of transcription factors in the network using TargetScan is likely due to a combination of false negatives in the iCLIP data and false positives in the TargetScan predictions.
Model Assessment and Validation
To assess the predictions made by the model, we applied both a computational and experimental approach. The computational approach ensured that the predictions made by the algorithm were due to the experimental data and not based on other biases in the prediction algorithm. The experimental validation showed that the transcription factors selected can partially recover the transcriptional changes observed upon Dicer deletion.
To computationally assess the predictions, we re-ran the algorithm on 1,000 different graphs, each graph comprised of the same nodes as the original network but with shuffled weights on each of the edges (see Experimental Procedures). If a node in the graph that represents a transcription factor is frequently identified in a random network, it reduces our overall confidence that the transcription factor is truly represented by the data. Therefore we favor transcription factors that show up with lower frequency in the random networks. The frequency of each transcription factor selected by the model is shown in black in Fig. 6A. These results suggest that the transcription factors selected by the network algorithm are specific to the experimental data, as they all appear in less than 5% of the graphs in which data were perturbed.
Fig. 6.

Computational and experimental validation of selected transcriptional factors. (A) Results of computational network perturbation indicating how frequently the algorithm-selected transcription factors showed up when either individual (colored) data sources were perturbed or all (black) data sources were perturbed. (B–D) Genes with significant (q<0.05) changes in intronic RNA levels that are activated (red) and repressed (blue) upon over-expression of (B) Flag-HA-Tead4, (C) Flag-HA-Sox9, and (D) Flag-HA-Pbx3 are significantly enriched in genes with intronic log2 RNA changes > 0.5 (pink) or less than −0.5 (cyan) in the Dicer KO. P-values computed via Fisher’s exact test.
We also used the network randomization to assess precisely which type of data contributed to each transcription factor prediction. To do this we perturbed only one type of data by randomizing the edge weights and then measured how frequently the resulting transcription factors appeared in the random networks. For example, when only miRNA-targeting data is randomized, the network algorithm cannot predict most transcription factors with a high degree of accuracy (orange points, Fig. 6A), suggesting that miRNA target information is critical to these predictions. When other types of data are perturbed, however, nodes are still selected by the network, suggesting that the predictions in the final network do not rely on each type of data equally. Specific examples of this include predictions of Ahr, Nr6a1 and Glis2, which rely heavily on miRNA targeting data (orange) but not on mRNA expression data (yellow). Therefore, we can use this data-specific perturbation to assess the quality of the predictions made in the final network.
To confirm the efficacy of the network algorithm in identifying transcription factors that regulate miRNA-mediated response, we validated specific transcription factors supported by the computational model and subsequent randomizations. Specifically, we selected Pbx3, Tead4, and Sox9 for over-expression in Dicer WT cells to observe changes in gene expression. We transduced Dicer WT cells with N-terminally Flag-HA-tagged retroviral expression constructs for either Tead4, Sox9, Pbx3 or an empty vector negative control. We isolated ribo-depleted total RNA and carried out RNA-seq in duplicate for each construct (see Experimental Procedures, Fig. S6A) and then measured changes in intronic regions to assay transcriptional changes (Table S6) without the confounding post-transcriptional effect of endogenous miRNAs (Khan et al., 2009) and then compared the genes that were activated and repressed in each of the over-expression experiments to those genes activated and repressed in the Dicer KO cells. We found the overlap to be statistically significant according Fisher’s exact test, as shown in Fig. 6B–D, confirming that each transcription factor significantly contributed to the transcriptional changes measured in the Dicer KO cells.
Discussion
In this work, we present a comprehensive approach to deconvoluting the impact of miRNAs on gene expression by the identification of miRNA-regulated transcription factors. We use ribo-depleted RNA-Seq, poly(A) mRNA-Seq, small RNA-Seq, iCLIP, and histone chromatin immunoprecipitation (ChIP-seq) to delineate the impact of global miRNA loss on both transcriptional and post-transcriptional regulation. Changes in mRNA expression levels are highly correlated with changes in total RNA-Seq reads mapping to introns, indicating that most genes that change in expression after Dicer loss are altered transcriptionally. We showed experimentally that the magnitude of changes caused by transcription, as indicated by epigenetic measurements of histone marks, is greater than changes caused by miRNA-mediated repression. We then introduced a robust computational method to identify the transcription factors that explain these transcriptional changes downstream of miRNA loss and experimentally validate three of these transcription factors as amplifying the effects of miRNAs.
Given the pronounced role of transcription factors in mediating miRNA-mediated effects, the graphical modeling approach introduced here enables reverse engineering of the regulatory network from gene expression and epigenetic data. This approach advances the field of miRNA analysis by leveraging valuable epigenetic data to deconvolute the pleiotropic effects of miRNAs and filter for miRNAs consequential to gene expression. By incorporating miRNA activity upstream and intronic RNA changes downstream of transcription, our approach also builds upon transcription factor prediction tools that employ epigenetic and expression data (Foat et al., 2006; Sherwood et al., 2014). The algorithm presented here is flexible and can be applied widely to any matched miRNA/mRNA/epigenetic such as those in large repositories NIH Roadmap Epigenomics Mapping project (Bernstein et al., 2010) or ENCODE (Rosenbloom et al., 2012) together with miRNA target prediction algorithms.
miRNAs have now been implicated in a stunningly wide range of biological processes and diseases (Zadran et al, 2013; Chen et al, 2004) and lead to large global changes in mRNA expression (Garcia et al, 2011) while causing only moderate repression of most direct targets. This study demonstrates that decoupling transcriptional changes from post-transcriptional changes and integrating them with epigenetic alterations in a computational framework can elucidate the transcriptional network that tunes and amplifies the effect of miRNA loss. The computational framework introduced here may benefit studies of miRNAs by shifting emphasis to the rewired transcriptional networks that cause the majority of the transcript-level changes.
Experimental Procedures
miRNA Target Identification
iCLIP reads were collected from GSE45828 and aligned to mm9 using Bowtie (Langmead and Salzberg, 2012). iCLIP events were assigned significance using GEM v1.1 (Guo et al., 2012) together with a custom read distribution derived from reads surrounding let-7 binding sites. Significant (q<10e-5) events were then filtered for the presence of a 7mer or 8mer seed match of a miRNA family that represented at least 1% of the reads from the small RNA population of the Dicer WT cells from GSE44156 as described in the Supplemental Experimental Procedures. To compare the effect of iCLIP-defined targets, we used Context+ scores from TargetScan 6.2 (Garcia et al., 2011) from http://www.targetscan.org/mmu_61/.
RNA expression measurements
This work included a total of six RNA-Seq datasets, each collected in duplicate. Mature mRNA was collected from untreated KO and WT fibroblasts via traditional polyA-collected library preparation, total RNA was collected from the same cells using Illumina Ribo-Zero Kit. Total RNA was also collected from Dicer WT fibroblasts transduced retrovirally with vector control, Flag-HA-PBX3, Flag-HA-TEAD4, or Flag-HA-SOX9 (see Supplemental Experimental Procedures). DESeq v1.10.1(Anders and Huber, 2010) was used for all data normalization and differential expression calls – a minimum of 2 DESeq-normalized reads (in both conditions) was required to call a gene expressed. The polyA data contained 13,413 genes expressed in both KO and WT cells, while the total RNA data contained 12,638 genes in both genotypes at the exonic level and 14,487 genes in both genotypes at the intronic level. Quantitative PCR measurements of seven genes were used to confirm the exon and intron reads from the total RNA measurements, described in the Supplemental Experimental Procedures. Δexon-Δintron values were computed using DESeq 2 (Love et al., 2014) according to the exon-intron split analysis (EISA) method previously described (Gaidatzis et al., 2015). Data from each of the 12 sequencing runs is available under the GEO accession GSE61035.
Histone data collection and event-calling
Chromatin immunoprecipitation (ChIP) assays for the H3K27ac mark were performed as previously described (Macisaac and Fraenkel, 2010) and other marks were collected from previously published data (Gurtan et al., 2013), available under GEO accession GSE44159. For H3K27ac and H3K4me3 marks, custom read distributions were used to call significant (q=0.05) events between WT and KO marks using GEM v1. (Guo et al., 2012), as described in the Supplemental Experimental Procedures while the default read distribution was used for the H3K36me3 marks. H3K4me3 and H3K27ac marks were associated with a gene if they fell with 10kb of a transcription start site, while H3K36me3 marks were associated with a gene if they fell within the gene body. See Supplemental Experimental Procedures for more details.
Network integration
Small RNA expression levels, iCLIP binding levels, mature mRNA expression levels, ChIP-Seq binding data and intronic RNA expression changes were encoded in a graphical model depicted in Figs. 3,S4 that was reduced using a version of the SAMNet algorithm (Gosline et al., 2012) as described in great detail in the Supplemental Experimental Procedures. The Garnet module of the OmicsIntegrator package (http://fraenkel.mit.edu/OmicsIntegrator) was used to predict transcription factor binding sites using the histone data. Network inputs and additional details are described in the Supplemental Experimental Procedures, and the code used to implement the algorithm is freely available at http://github.com/sgosline/topaz.
Experimental validation and target identification
N-terminally Flag-HA-tagged Pbx3, Sox9, and Tead4 were PCR-amplified from mouse cDNA generated from Dicer KO fibroblasts. Transduced cells were sequenced in duplicate together with a vector control and DESeq v1.1 was used to compare intronic reads between conditions. Genes that were significantly (p<0.05) up-regulated upon transfection and also up-regulated in the Dicer KO cells were considered activated the transcription factor while genes that were significantly (p<0.05) down-regulated upon transfection that were down-regulated in the Dicer WT were considered repressed. Details are described in Supplemental Experimental Procedures.
Supplementary Material
Acknowledgments
The authors thank members of the Fraenkel and Sharp labs for useful discussions about this work. The work was supported by the National Institutes of Health via grants U54-CA112967 (EF), R01-GM089903 (EF), U01-CA184898 (EF), R01-CA133404 (PAS), PO1-CA042063 (PAS) and RO1-GM34277 (PAS) and partially by the Koch Institute Support (core) grant P30-CA14051 from the National Cancer Institute. Computing resources were funded by the National Science Foundation under Award No. DB1-0821391, and AMG was supported by the Leukemia and Lymphoma Society grant 5198-09. All data can be found under GEO accession GSE61035.
Footnotes
Author Contributions:
SJCG, AMG, EF and PAS designed the study. SD performed ChIP-Seq. CKJ performed over-expression experiments. AB performed the iCLIP. PM, BM and YSY prepared various samples for sequencing. Sequencing was performed at the BioMicroCenter at MIT. SJCG performed all the analysis. SJCG, AMG, EF and PAS wrote the manuscript.
Accession Numbers:
The data presented here are avialable under GEO SuperSeries Accession: GSE61035. Total RNA-Seq is available under GSE61033 and the H3K27ac data is available under GSE61034. This work also reference previously collected ChIP-Seq data under GSE44159 and previously collected iCLIP data under GSE45828.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Afshar AS, Xu J, Goutsias J. Integrative Identification of Deregulated MiRNA/TF-Mediated Gene Regulatory Loops and Networks in Prostate Cancer. PLoS One. 2014;9:e100806. doi: 10.1371/journal.pone.0100806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal V, Bell GW, Nam JW, Bartel DP. Predicting effective microRNA target sites in mammalian mRNAs. Elife. 2015;4:e05005. doi: 10.7554/eLife.05005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11:R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baek D, Villén J, Shin C, Camargo FD, Gygi SP, Bartel DP. The impact of microRNAs on protein output. Nature. 2008;455:64–71. doi: 10.1038/nature07242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein BE, Stamatoyannopoulos JA, Costello JF, Ren B, Milosavljevic A, Meissner A, Kellis M, Marra MA, Beaudet AL, Ecker JR, Farnham PJ, Hirst M, Lander ES, Mikkelsen TS, Thomson JA. The NIH Roadmap Epigenomics Mapping Consortium. Nat Biotechnol. 2010;28:1045–8. doi: 10.1038/nbt1010-1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisognin A, Sales G, Coppe A, Bortoluzzi S, Romualdi C. MAGIA2: from miRNA and genes expression data integrative analysis to microRNA-transcription factor mixed regulatory circuits (2012 update) Nucleic Acids Res. 2012;40:W13–21. doi: 10.1093/nar/gks460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosson AD, Zamudio JR, Sharp PA. Endogenous miRNA and Target Concentrations Determine Susceptibility to Potential ceRNA Competition. Mol Cell. 2014;56:347–359. doi: 10.1016/j.molcel.2014.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CZ, Li L, Lodish HF, Bartel DP. MicroRNAs modulate hematopoietic lineage differentiation. Science. 2004;303:83–6. doi: 10.1126/science.1091903. [DOI] [PubMed] [Google Scholar]
- Chi SW, Hannon GJ, Darnell RB. An alternative mode of microRNA target recognition. Nat Struct Mol Biol. 2012;19:321–327. doi: 10.1038/nsmb.2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi SW, Zang JB, Mele A, Darnell RB. Argonaute HITS-CLIP decodes microRNA-mRNA interaction maps. Nature. 2009;460:479–86. doi: 10.1038/nature08170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu H-S, Llobet-Navas D, Yang X, Chung W-J, Ambesi-Impiombato A, Iyer A, Kim HR, Seviour EG, Luo Z, Sehgal V, Moss T, Lu Y, Ram P, Silva J, Mills GB, Califano A, Sumazin P. Cupid: simultaneous reconstruction of microRNA-target and ceRNA networks. Genome Res. 2015;25:257–67. doi: 10.1101/gr.178194.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, Hanna J, Lodato MA, Frampton GM, Sharp PA, Boyer LA, Young RA, Jaenisch R. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci U S A. 2010;107:21931–6. doi: 10.1073/pnas.1016071107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuellar-Partida G, Buske FA, McLeay RC, Whitington T, Noble WS, Bailey TL. Epigenetic priors for identifying active transcription factor binding sites. Bioinformatics. 2012;28:56–62. doi: 10.1093/bioinformatics/btr614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du NH, Arpat AB, De Matos M, Gatfield D. MicroRNAs shape circadian hepatic gene expression on a transcriptome-wide scale. Elife. 2014;3:e02510. doi: 10.7554/eLife.02510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J, Kellis M. Discovery and characterization of chromatin states for systematic annotation of the human genome. Nat Biotechnol. 2010;28:817–25. doi: 10.1038/nbt.1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foat BC, Morozov AV, Bussemaker HJ. Statistical mechanical modeling of genome-wide transcription factor occupancy data by MatrixREDUCE. Bioinformatics. 2006;22:e141–9. doi: 10.1093/bioinformatics/btl223. [DOI] [PubMed] [Google Scholar]
- Friard O, Re A, Taverna D, De Bortoli M, Corá D. CircuitsDB: a database of mixed microRNA/transcription factor feed-forward regulatory circuits in human and mouse. BMC Bioinformatics. 2010;11:435. doi: 10.1186/1471-2105-11-435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaidatzis D, Burger L, Florescu M, Stadler MB. Analysis of intronic and exonic reads in RNA-seq data characterizes transcriptional and post-transcriptional regulation. Nat Biotechnol. 2015 doi: 10.1038/nbt.3269. [DOI] [PubMed] [Google Scholar]
- Garcia DM, Baek D, Shin C, Bell GW, Grimson A, Bartel DP. Weak seed-pairing stability and high target-site abundance decrease the proficiency of lsy-6 and other microRNAs. Nat Struct Mol Biol. 2011;18:1139–46. doi: 10.1038/nsmb.2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstein MB, Kundaje A, Hariharan M, Landt SG, Yan KK, Cheng C, Mu XJ, Khurana E, Rozowsky J, Alexander R, Min R, Alves P, Abyzov A, Addleman N, Bhardwaj N, Boyle AP, Cayting P, Charos A, Chen DZ, Cheng Y, Clarke D, Eastman C, Euskirchen G, Frietze S, Fu Y, Gertz J, Grubert F, Harmanci A, Jain P, Kasowski M, Lacroute P, Leng J, Lian J, Monahan H, O’Geen H, Ouyang Z, Partridge EC, Patacsil D, Pauli F, Raha D, Ramirez L, Reddy TE, Reed B, Shi M, Slifer T, Wang J, Wu L, Yang X, Yip KY, Zilberman-Schapira G, Batzoglou S, Sidow A, Farnham PJ, Myers RM, Weissman SM, Snyder M. Architecture of the human regulatory network derived from ENCODE data. Nature. 2012;489:91–100. doi: 10.1038/nature11245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosline SJ, Spencer SJ, Ursu O, Fraenkel E. SAMNet: a network-based approach to integrate multi-dimensional high throughput datasets. Integr Biol. 2012 doi: 10.1039/c2ib20072d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimson A, Farh KKH, Johnston WK, Garrett-Engele P, Lim LP, Bartel DP. MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol Cell. 2007;27:91–105. doi: 10.1016/j.molcel.2007.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo H, Ingolia NT, Weissman JS, Bartel DP. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature. 2010;466:835–840. doi: 10.1038/nature09267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y, Mahony S, Gifford DK. High resolution genome wide binding event finding and motif discovery reveals transcription factor spatial binding constraints. PLoS Comput Biol. 2012;8:e1002638. doi: 10.1371/journal.pcbi.1002638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurtan AM, Ravi A, Rahl PB, Bosson AD, Jnbaptiste CK, Bhutkar A, Whittaker CA, Young RA, Sharp PA. Let-7 represses Nr6a1 and a mid-gestation developmental program in adult fibroblasts. Genes Dev. 2013;27:941–54. doi: 10.1101/gad.215376.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurtan AM, Sharp PA. The role of miRNAs in regulating gene expression networks. J Mol Biol. 2013;425:3582–600. doi: 10.1016/j.jmb.2013.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heintzman ND, Hon GC, Hawkins RD, Kheradpour P, Stark A, Harp LF, Ye Z, Lee LK, Stuart RK, Ching CW, Ching KA, Antosiewicz-Bourget JE, Liu H, Zhang X, Green RD, Lobanenkov VV, Stewart R, Thomson JA, Crawford GE, Kellis M, Ren B. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature. 2009;459:108–12. doi: 10.1038/nature07829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herranz H, Cohen SM. MicroRNAs and gene regulatory networks: managing the impact of noise in biological systems. Genes Dev. 2010;24:1339–1344. doi: 10.1101/gad.1937010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan AA, Betel D, Miller ML, Sander C, Leslie CS, Marks DS. Transfection of small RNAs globally perturbs gene regulation by endogenous microRNAs. Nat Biotechnol. 2009;27:549–55. doi: 10.1038/nbt.1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- König J, Zarnack K, Luscombe NM, Ule J. Protein–RNA interactions: new genomic technologies and perspectives. Nat Rev Genet. 2012;13:77–83. doi: 10.1038/nrg3141. [DOI] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–9. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YS, Dutta A. The tumor suppressor microRNA let-7 represses the HMGA2 oncogene. Genes Dev. 2007;21:1025–30. doi: 10.1101/gad.1540407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis BP, Shih I, Jones-Rhoades MW, Bartel DP, Burge CB. Prediction of mammalian microRNA targets. Cell. 2003;115:787–98. doi: 10.1016/s0092-8674(03)01018-3. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/PREACCEPT-8897612761307401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Getz G, Miska EA, Alvarez-Saavedra E, Lamb J, Peck D, Sweet-Cordero A, Ebert BL, Mak RH, Ferrando AA, Downing JR, Jacks T, Horvitz HR, Golub TR. MicroRNA expression profiles classify human cancers. Nature. 2005;435:834–8. doi: 10.1038/nature03702. [DOI] [PubMed] [Google Scholar]
- Macisaac KD, Fraenkel E. Sequence analysis of chromatin immunoprecipitation data for transcription factors. Methods Mol Biol. 2010;674:179–93. doi: 10.1007/978-1-60761-854-6_11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayr C, Hemann MT, Bartel DP. Disrupting the pairing between let-7 and Hmga2 enhances oncogenic transformation. Science. 2007;315:1576–9. doi: 10.1126/science.1137999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendell JT, Olson EN. MicroRNAs in Stress Signaling and Human Disease. Cell. 2012;148:1172–1187. doi: 10.1016/j.cell.2012.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naeem H, Küffner R, Zimmer R. MIRTFnet: analysis of miRNA regulated transcription factors. PLoS One. 2011;6:e22519. doi: 10.1371/journal.pone.0022519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasquinelli AE. MicroRNAs and their targets: recognition, regulation and an emerging reciprocal relationship. Nat Rev Genet. 2012;13:271–82. doi: 10.1038/nrg3162. [DOI] [PubMed] [Google Scholar]
- Pique-Regi R, Degner JF, Pai AA, Gaffney DJ, Gilad Y, Pritchard JK. Accurate inference of transcription factor binding from DNA sequence and chromatin accessibility data. Genome Res. 2011;21:447–55. doi: 10.1101/gr.112623.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rani SB, Rathod SS, Karthik S, Kaur N, Muzumdar D, Shiras AS. MiR-145 functions as a tumor-suppressive RNA by targeting Sox9 and adducin 3 in human glioma cells. Neuro Oncol. 2013;15:1302–16. doi: 10.1093/neuonc/not090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbloom KR, Sloan CA, Malladi VS, Dreszer TR, Learned K, Kirkup VM, Wong MC, Maddren M, Fang R, Heitner SG, Lee BT, Barber GP, Harte RA, Diekhans M, Long JC, Wilder SP, Zweig AS, Karolchik D, Kuhn RM, Haussler D, Kent WJ. ENCODE Data in the UCSC Genome Browser: year 5 update. Nucleic Acids Res. 2012;41:D56–63. doi: 10.1093/nar/gks1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmiedel JM, Klemm SL, Zheng Y, Sahay A, Bluthgen N, Marks DS, van Oudenaarden A. MicroRNA control of protein expression noise. Science (80-) 2015;348:128–132. doi: 10.1126/science.aaa1738. [DOI] [PubMed] [Google Scholar]
- Selbach M, Schwanhäusser B, Thierfelder N, Fang Z, Khanin R, Rajewsky N. Widespread changes in protein synthesis induced by microRNAs. Nature. 2008;455:58–63. doi: 10.1038/nature07228. [DOI] [PubMed] [Google Scholar]
- Shalgi R, Lieber D, Oren M, Pilpel Y. Global and local architecture of the mammalian microRNA-transcription factor regulatory network. PLoS Comput Biol. 2007;3:e131. doi: 10.1371/journal.pcbi.0030131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherwood RI, Hashimoto T, O’Donnell CW, Lewis S, Barkal AA, van Hoff JP, Karun V, Jaakkola T, Gifford DK. Discovery of directional and nondirectional pioneer transcription factors by modeling DNase profile magnitude and shape. Nat Biotechnol. 2014;32:171–8. doi: 10.1038/nbt.2798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song L, Crawford GE. DNase-seq: a high-resolution technique for mapping active gene regulatory elements across the genome from mammalian cells. Cold Spring Harb Protoc. 2010;2010 doi: 10.1101/pdb.prot5384. pdb.prot5384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimoto Y, König J, Hussain S, Zupan B, Curk T, Frye M, Ule J. Analysis of CLIP and iCLIP methods for nucleotide-resolution studies of protein-RNA interactions. Genome Biol. 2012;13:R67. doi: 10.1186/gb-2012-13-8-r67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tay Y, Zhang J, Thomson AM, Lim B, Rigoutsos I. MicroRNAs to Nanog, Oct4 and Sox2 coding regions modulate embryonic stem cell differentiation. Nature. 2008;455:1124–8. doi: 10.1038/nature07299. [DOI] [PubMed] [Google Scholar]
- Tsang J, Zhu J, van Oudenaarden A. MicroRNA-mediated feedback and feedforward loops are recurrent network motifs in mammals. Mol Cell. 2007;26:753–67. doi: 10.1016/j.molcel.2007.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu K, Yu H, Hua Y-J, Li Y-Y, Liu L, Xie L, Li Y-X. Combinatorial network of primary and secondary microRNA-driven regulatory mechanisms. Nucleic Acids Res. 2009;37:5969–80. doi: 10.1093/nar/gkp638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen J, Parker BJ, Jacobsen A, Krogh A. MicroRNA transfection and AGO-bound CLIP-seq data sets reveal distinct determinants of miRNA action. RNA. 2011;17:820–34. doi: 10.1261/rna.2387911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zadran S, Remacle F, Levine RD. miRNA and mRNA cancer signatures determined by analysis of expression levels in large cohorts of patients. Proc Natl Acad Sci. 2013;110:19160–19165. doi: 10.1073/pnas.1316991110. http://dx.doi.org/10.1073/pnas.1316991110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Darnell RB. Mapping in vivo protein-RNA interactions at single-nucleotide resolution from HITS-CLIP data. Nat Biotechnol. 2011;29:607–14. doi: 10.1038/nbt.1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.