

Yeast Hsp70 nucleotide exchange factor Fes1 is expressed by rare alternative splicing as two isoforms. Fes1L is targeted to the nucleus, and Fes1S localizes to the cytosol and is required for the efficient proteasomal degradation of cytosolic misfolded proteins, as well as of species that are imported into the nucleus for degradation.
Abstract
Cells maintain proteostasis by selectively recognizing and targeting misfolded proteins for degradation. In Saccharomyces cerevisiae, the Hsp70 nucleotide exchange factor Fes1 is essential for the degradation of chaperone-associated misfolded proteins by the ubiquitin-proteasome system. Here we show that the FES1 transcript undergoes unique 3′ alternative splicing that results in two equally active isoforms with alternative C-termini, Fes1L and Fes1S. Fes1L is actively targeted to the nucleus and represents the first identified nuclear Hsp70 nucleotide exchange factor. In contrast, Fes1S localizes to the cytosol and is essential to maintain proteostasis. In the absence of Fes1S, the heat-shock response is constitutively induced at normally nonstressful conditions. Moreover, cells display severe growth defects when elevated temperatures, amino acid analogues, or the ectopic expression of misfolded proteins, induce protein misfolding. Importantly, misfolded proteins are not targeted for degradation by the ubiquitin-proteasome system. These observations support the notion that cytosolic Fes1S maintains proteostasis by supporting the removal of toxic misfolded proteins by proteasomal degradation. This study provides key findings for the understanding of the organization of protein quality control mechanisms in the cytosol and nucleus.
INTRODUCTION
The folding of polypeptides into their native three-dimensional structures is a prerequisite for function. The cellular folding process is not perfect and is compromised by genetic disposition, transcriptional and translational imprecision, and stress conditions. To counteract the buildup of potentially cytotoxic aberrant proteins that threaten proteostasis, cells use protein quality control mechanisms that selectively recognize and remove misfolded proteins (Chen et al., 2011).
Molecular chaperones of the Hsp70 class promote folding and prevent aggregation of proteins by binding and shielding exposed hydrophobic segments of non–natively folded substrates (Hartl et al., 1994; Mayer and Bukau, 2005). Binding of the hydrophobic segments is regulated by the intrinsic ATPase activity of Hsp70. When ATP is bound, substrate binds and releases with fast kinetics. ATP hydrolysis is stimulated by cochaperones of the Hsp40 class, resulting in ADP-bound Hsp70, which attains a closed conformation with high affinity for substrate. The interactions with nucleotide exchange factors (NEFs) accelerate ADP–ATP exchange, resulting in release of bound substrate and exposure of its hydrophobic segments.
The function of Hsp70 in protein folding is coordinated with protein quality control mechanisms so that non–natively folded proteins are triaged between folding and degradation pathways. In budding yeast Saccharomyces cerevisiae, a functional Hsp70 system is required for the ubiquitylation and proteasomal degradation of misfolded proteins (McClellan et al., 2005; Park et al., 2007; Metzger et al., 2008). Specifically, Hsp70 is required to keep misfolded proteins soluble, facilitate their nuclear import, and promote their association with the ubiquitin E3 ligases (Park et al., 2007; Heck et al., 2010; Prasad et al., 2010; Guerriero et al., 2013; Shiber et al., 2013). San1 and Ubr1 are ubiquitin ligases required for cytosolic and nuclear degradation of misfolded proteins (Eisele and Wolf, 2008; Heck et al., 2010; Nillegoda et al., 2010). San1 resides in the nucleus and recognizes exposed hydrophobic segments of misfolded proteins by direct interactions via its intrinsically unfolded N-terminal tail (Rosenbaum et al., 2011). Ubr1 recognizes proteins by direct binding of N-terminal methionine, followed by a hydrophobic residue (Kim et al., 2014). Thus the exposure of hydrophobic segments on the surface of misfolded proteins is a key character that determines their interaction with both Hsp70 and the ubiquitin ligases.
Fes1 is an HspBP1-class Hsp70 NEF that is essential in protein quality control. Its interactions with Hsp70s are well understood (Shomura et al., 2005; Andréasson et al., 2008a). Recently we found that Fes1 is essential for ubiquitin-dependent degradation of misfolded proteins that are associated with Hsp70 (Gowda et al., 2013). Fes1 triggers the release of the misfolded proteins from Hsp70 and thereby facilitates their interaction with ubiquitin ligase Ubr1. Studies indicate that import of misfolded cytosolic proteins into the nucleus is a prominent degradation pathway (Heck et al., 2010; Prasad et al., 2010; Park et al., 2013). It is not clear, however, whether Fes1 is required for the degradation of misfolded proteins that are imported into the nucleus. One such misfolded model protein, ΔssCPY‡–green fluorescent protein (GFP), does not depend on Fes1 for its degradation (Abrams et al., 2014). However, this single example might not be representative for the entire class of misfolded proteins that are imported into the nucleus for degradation.
The distribution of Fes1 in the cytosol and nucleus is not well described. Overexpressed Fes1 tagged with N-terminal GFP appeared to be cytosolic, but the signal was also detected in the nucleus (Kabani et al., 2002). Moreover, the subcellular distribution of Fes1 might be regulated by alternative splicing; ab initio computer annotation of the yeast transcriptome identified FES1 transcripts that extend far beyond the 3′ end of the previously annotated single open reading frame (ORF) and undergo alternative splicing (Yassour et al., 2009). The nucleocytoplasmic distribution of Fes1 affects our understanding of both Fes1 function and the general function of the Hsp70 system in protein quality control.
Here we show that heat shock–regulated alternative splicing of FES1 transcripts results in the expression of two functional isoforms, Fes1L and Fes1S. Fes1L is actively targeted to the nucleus, whereas Fes1S localizes to the cytosol and is required for the efficient proteasomal degradation of cytosolic misfolded proteins, as well as of species that are imported into the nucleus for degradation.
RESULTS
The FES1 transcript is alternatively spliced and expresses splice isoforms Fes1S and Fes1L
We inspected the FES1 locus downstream of the previously annotated ORF and found sequence elements associated with an intron and a second exon (Figure 1A). Specifically, the putative intron is flanked by canonical 5′ (5′-GTATGT-3′) and 3′ (5′-TAG-3′) splice sites and a branch point sequence element was identifiable (Supplemental Figure S1A). Alignment of FES1 locus sequences from sensu stricto Saccharomyces species revealed that the functional elements of putative intron and the coding sequence of exon II are conserved (Supplemental Figure S1B).
FIGURE 1:

Heat shock–regulated alternative splicing of FES1 transcripts results in the expression of Fes1L and Fes1S. (A) The heat shock element–promoted (HSE) FES1 locus contains sequence elements that encode two exons (I and II) separated by an intron (dashed line). Use of the promoter-proximal site of polyadenylation in the intron (▼) supports the expression of Fes1S from a transcript that encompasses exon I. Alternative splicing removes the promoter- proximal polyadenylation site in the intron and supports expression of Fes1L from exons I and II. (B) RT-PCR analysis of transcripts that encompass exon I and II, using primers F and R as indicated in A. The longer transcript (Pre) contains the intron, and the shorter transcript is splicing (Spliced) and supports the expression of Fes1L. (C) Western analysis of protein extracts from WT and fes1Δ with rabbit serum raised against recombinant Fes1S. Both Fes1S (32.6 kDa) and Fes1L (34.2 kDa) are expressed from the endogenous FES1 locus. Parallel blotting against Pgk1 functions as a loading control. (D) Western analysis as in C of fes1Δ cells transformed with vector control (VC) or derivatives that carry either the WT FES1 locus or a version with a 9.4- kDa 6×HA (HA) fused in-frame to the 3′ end of exon II. (E) Western analysis as in C of WT and splicing-impaired mutants prp18Δ and mud2Δ. (F) Western analysis as in C of fes1Δ cells transformed with VC or derivatives that carry either WT FES1 locus or a derivative with a T-to-C substitution mutation at the polyadenylation site indicated in A. (G) Quantification of FES1 transcript variants from Illumina 1G Analyzer reads under growth of WT yeast at 22°C in YPD medium and after transient heat shock (HS); 37°C, 15 min. Data are presented as reads per lane per kilobase for mRNA. Error bars indicate SD of data from two biological replicates. (H) Quantification of Fes1L and Fes1S expression levels using Western analysis under the same conditions as in G. Error bars indicate SD (N = 3). *p < 0.05, **p < 0.01, ns > 0.05.
The bulk of the FES1 transcripts have been experimentally determined to become polyadenylated 11 base pairs downstream of the ORF, corresponding to 24 base pairs downstream of the putative 5′ splice site (Supplemental Figure S1A; Nagalakshmi et al., 2008). We examined whether full-length FES1 transcripts could extend beyond this known promoter-proximal polyadenylation site and into exon II. Reads from next-generation sequencing of FES1 mRNA showed a signature typical for alternative splicing (Supplemental Figure S1C). Direct amplification of exon I, intron, and parts of exon II mRNA by reverse transcriptase PCR (RT-PCR; primer positions in Figure 1A) resulted in amplification of two products with sizes corresponding to the unspliced precursor mRNA (Pre) and the 128 base pair–smaller spliced mRNA (Spliced; Figure 1B). Sequencing of each product confirmed their identities, verifying splicing (Supplemental Figure S1D).
Splicing between exon I and exon II results in the replacement of the five last codons of the ORF with 17 codons from exon II and thus extends the protein with 12 C-terminal amino acids (Supplemental Figure S1B). We raised rabbit antibodies against the heterologously expressed Fes1 protein encoded by the FES1 ORF (no intron or exon II; see Materials and Methods). Western analysis of total protein lysates prepared from wild-type (WT) and isogenic fes1Δ strains demonstrated that Fes1 is expressed as two isoforms that migrate as distinct bands on gels (Figure 1C). The predominant form migrates faster and was named Fes1 Short isoform (Fes1S). The slower-migrating form was named Fes1 Long isoform (Fes1L). Judged from SDS–PAGE size standards, both putative isoforms migrate close to the predicated sizes of 32.6 kDa (Fes1S) and 34.2 kDa (Fes1L).
We directly tested whether Fes1L represents the alternative spliced isoform by extending exon II in the context of the endogenous locus on single-copy plasmids with sequences encoding a 9.4-kDa tag (six-times-reiterated hemagglutinin epitope [6xHA]). Fes1S migration was unaltered by the extension of exon II, but the position of Fes1L on the gel shifted to a position ∼10 kDa above its normal migration (Figure 1D). Plasmids carrying the FES1 locus encoding 6xHA-tagged Fes1L expressed functional Fes1 activity, as judged by complementation of fes1Δ thermosensitivity for growth (Supplemental Figure S2). In all, our analysis indicates that Fes1S is expressed from the unspliced mRNA with a predicted size of 290 amino acids (32.6 kDa), and Fes1L is expressed from spliced exon I and exon II with a predicted size of 302 amino acids (34.1 kDa).
FES1 alternative splicing is regulated by competition between promoter-proximal polyadenylation and intron excision
We investigated the mechanism that regulates relative Fes1S and Fes1L expression levels. First, we analyzed Fes1S and Fes1L expression in strains that lack nonessential components of the spliceosome (Abovich et al., 1994); Mud2 functions in early spliceosome assembly and mediates branch point recognition (Wang et al., 2008), and Prp18 is involved in second step of pre-mRNA splicing and interacts with U5 snRNA to facilitate splicing (Vijayraghavan and Abelson, 1990; Crotti et al., 2007). Fes1L expression was reduced in mud2Δ and prp18Δ strains, whereas Fes1S expression remained unaltered (Figure 1E). This result indicates that Fes1L but not Fes1S expression is dependent on the activity of core components of the spliceosome.
Next we asked whether polyadenylation of the FES1 transcript at the promoter-proximal polyadenylation site in the intron competes with production of longer and spliced mRNAs that express Fes1L. Consistent with this notion, we found that introduction of a single T-to-C point mutation at the promoter-proximal polyadenylation site (10 base pairs downstream of the Fes1S STOP codon) results in increased expression of Fes1L (Figure 1F). Thus, in light of the genetic structure of the FES1 locus, our data indicate that promoter-proximal polyadenylation and splicing of the intron are competing processes that determine the levels of Fes1L and Fes1S expression.
Heat stress modulates FES1 alternative splicing and splice-isoform expression
FES1 is transcriptionally activated by stress-responsive Hsf1, which directly binds to canonical HSE in the promoter (Hahn et al., 2004; Yamamoto et al., 2005). We investigated whether Fes1S and Fes1L expression is altered in response to protein folding stress in the form of transient heat shock, the defining inducing condition for Hsf1. We analyzed next-generation sequencing reads of transcripts (Yassour et al., 2009) and determined the levels of transcripts specific for Fes1S and Fes1L under nonstressful and transient heat-shock conditions. Under nonstressful conditions (22°C), Fes1S-specific mRNA dominated and was expressed at 4.4-fold higher level than Fes1L-specific mRNA (Figure 1G). After heat shock at 37°C, Fes1S-specific mRNA was induced 12-fold and Fes1L-specific mRNA 6-fold. The initially higher levels of Fes1S-specific mRNA and the preferential induction during heat shock resulted in 15-fold-higher levels of Fes1S-specific mRNA relative to Fes1L-specific mRNA during transient heat shock. The transcript analysis suggests that Fes1S is preferentially induced during acute heat stress, either by direct regulation or as the result of secondary affects, for example, limiting spliceosome activity.
To assess the effect of the isoform-specific transcriptional changes on protein level, we performed quantitative Western analysis. At nonstressful conditions, Fes1S was expressed at 2.5-fold higher levels than Fes1L (Figure 1H). After heat shock at 37°C, the ratio increased to a fourfold difference. Both splice isoforms are very stable proteins and were not significantly turned over in a 2 h time-course experiment (Supplemental Figure S3). These results suggest that during acute heat stress, primarily Fes1S expression is induced, which suggests a role for this splice isoform in maintaining proteostasis under stressful conditions.
Fes1 isoforms have similar NEF activity
Fes1S binds to Hsp70, thereby triggering accelerated nucleotide release from Hsp70 (Kabani et al., 2002; Dragovic et al., 2006). We investigated whether purified Fes1S and Fes1L promote nucleotide release from Hsp70 with similar kinetics. Preloaded complexes consisting of Hsp70 (Ssa1) and the fluorescent nucleotide derivative MABA-ADP were rapidly mixed with Fes1S and Fes1L, and dissociation of MABA-ADP was followed by decrease in its fluorescence signal in stopped-flow measurements. The basal nucleotide dissociation rate of Ssa1 was determined to 0.39 s−1, and both Fes1S and Fes1L accelerated the release rate 10-fold, to 3.7 and 3.9 s−1, respectively (Figure 2). The basal and induced release rates are in agreement with previously published experiments for Fes1S (Dragovic et al., 2006; Raviol et al., 2006b). The data indicate that Fes1S and Fes1L are both functional Hsp70 NEFs and have similar activity.
FIGURE 2:
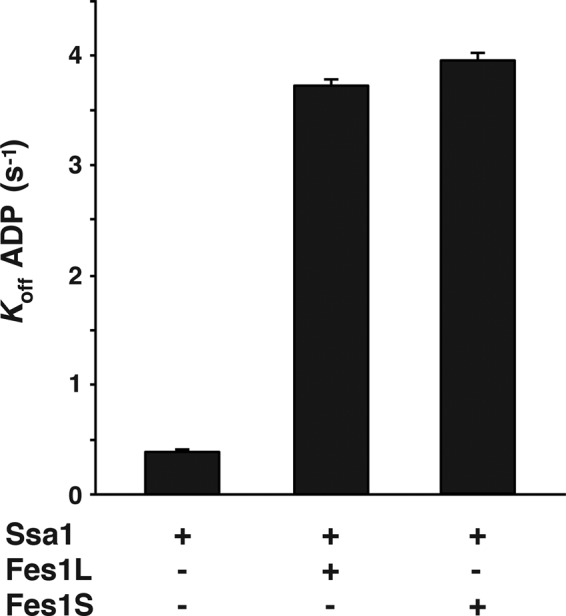
Fes1L and Fes1S are isoforms with similar NEF activity. Basal and induced rates of ADP dissociation from Ssa1. Ssa1-MABA-ADP (0.25 μM) was rapidly mixed with ATP in a stopped-flow instrument in the presence or absence of Fes1L (1 μM) or Fes1S (1 μM).
Fes1S localizes to cytosol and Fes1L is targeted to the nucleus
We analyzed the 16 C-terminal amino acids that are specific for Fes1L and found a motif similar to known monopartite nuclear localization signals (NLSs) with the consensus sequence K[K/R]X[K/R] (Hodel et al., 2001; Figure 3A). The NLS-like motif was conserved in Fes1L orthologues in other Saccharomycetaceae species (Supplemental Figure S1E). No other putative NLS sequences were found in the Fes1S or Fes1L sequences.
FIGURE 3:

Fes1S is a cytosolic protein, and Fes1L is targeted to the nucleus by a C-terminal NLS. (A) An NLS-like sequence in the C-terminus of Fes1L and the changes in amino acid sequence of the Fes1L mutant K/R → E. (B) Western analysis of C-terminally GFP-tagged Fes1S, Fes1L, and K/R → E expressed from plasmids with the endogenous FES1 promoter in fes1Δ cells. (C) Fluorescence microscopy localization of the GFP fusions in B. The nucleus is labeled with mCherry-tagged Histone 2B (Htb2). Scale bar, 10 μM.
To clarify the isoform-specific subcellular localization, we analyzed individually expressed, C-terminally GFP-tagged Fes1S and Fes1L (Figure 3B). Both proteins were functional when expressed from the endogenous FES1 promoter on single-copy plasmids (Supplemental Figure S4A). Fluorescence microscopy showed that Fes1S localized evenly throughout the cytosol, with no enrichment in the nucleus (Figure 3C and Supplemental Figure S4B). In contrast, Fes1L was enriched in the nucleus, with some signal left in the cytosol (Figure 3C and Supplemental Figure S4C). Substitution of the five basic amino acids of the putative NLS in Fes1L to glutamates (K/R → E) resulted in cytosolic localization of the protein. Lysis of spheroplasts at low detergent concentrations to avoid disruption of the nuclear envelope resulted in release of 55% of Fes1S and 45% of Fes1L (Supplemental Figure S4D). The data support the notion that Fes1S resides in the cytosol, whereas Fes1L carries an NLS that targets the protein to the nucleus.
Fes1S is required for growth at conditions that induce proteotoxic stress
We asked whether the cellular functions of Fes1 depend on the expression of both isoforms by directly modifying the FES1 locus to either express Fes1S (fes1ΔL, exon II removed) or Fes1L (fes1ΔS,L↑, intron removed and exon I and II fused; Figure 4A). To counteract the overexpression of Fes1L from the fes1ΔS,L↑ allele, which lacks the expression-limiting intron, the endogenous promoter was exchanged to the weaker ADH1 promoter (fes1ΔS; Figure 4B).
FIGURE 4:

Temperature tolerance requires Fes1S but not Fes1L activity. (A) Schematic representation of the chromosomal modifications made in the WT FES1 locus to obtain the following strains: no expression of Fes1L or Fes1S (fes1Δ), only expression of Fes1S (fes1ΔL), only expression of Fes1L (fes1ΔS), and only expression of Fes1L but at higher than WT levels (fes1ΔS,L↑). (B) Western analysis of the expression of Fes1L and Fes1S in the strains in A. Quantification of Fes1 isoform expression levels (bottom). Error bar indicates SD (N = 3). (C) Analysis of the fes1 temperature-sensitive growth phenotype of the strains in A. Cell suspensions were 10-fold serially diluted, spotted onto solid SC or SC supplemented with 1 mg/l l-canavanine (Can) medium, and incubated at the indicated temperature.
The strains were subjected to phenotypic growth tests based on the fes1Δ sensitivity to induced protein misfolding by increased temperatures or amino acid analogues (Kabani et al., 2002; Gowda et al., 2013). The fes1ΔL strain grew indistinguishably from WT cells both at elevated temperature (39°C) and in the presence of l-canavanine (Figure 4C). In contrast, the fes1ΔS strain exhibited clear growth defects at both conditions, similar to how the fes1Δ strain behaved. Overexpression of Fes1L in the absence of Fes1S (fes1ΔS,L↑) resulted in complementation of the phenotype, likely by titrating up the cytosolic pool of Fes1L (Figure 4C and Supplemental Figure S4, C and D). Indeed, introduction of the K/R → E NLS mutation on the chromosome of the Fes1L-expressing strain (fes1ΔS) resulted in suppression of the thermosensitivity phenotype (Supplemental Figure S4E). The phenotypes indicate that Fes1S performs the major functions required for growth under conditions that induce protein misfolding.
Fes1S keeps a repressed heat shock response
The heat shock response is induced under conditions that perturb the proteostasis system (Verghese et al., 2012). A striking phenotype associated with fes1Δ is the activation of an unusually strong and constitutive heat shock response at the normally repressive temperature of 25°C (Gowda et al., 2013). We assessed transcriptional changes in our Fes1-isoform specific strains grown at 25°C by RNA sequencing (RNA-Seq; Supplemental Table S1). We found 157 genes to be differentially expressed (p < 0.01, q < 0.1, absolute log2-fold change >0.5) in the fes1 mutants compared with the WT strain (Figure 5A and Supplemental Table S2). The transcriptional profiles of WT and fes1ΔL strains matched closely, and only 4 genes were differentially expressed. The transcriptional profiles of the fes1Δ and fes1ΔS strains clustered and were different from the profiles of WT and fes1ΔL strains. In total, 157 genes were differentially expressed (96 induced and 61 repressed) in the fes1Δ (133 unique genes) and fes1ΔS (102 unique genes) strains compared with the WT strain. These included 35 genes of 78 known to be induced by the heat shock response (Hahn et al., 2004), providing strong evidence for induction of the heat shock response in the fes1Δ (t test; p = 4.9e−10) and fes1ΔS (p = 7.9e−13) strains but not in the fes1ΔL strain (p = 0.09; Figure 5B). The Fes1S-dependent repression of Hsf1 activity was also evident using a heat shock–regulated promoter driving the LacZ reporter gene (Figure 5C). Cells expressing only Fes1L at low or high levels did not keep the reporter repressed (Figure 5C and Supplemental Figure S4F). The data indicate that Fes1S keeps the heat-shock response repressed.
FIGURE 5:
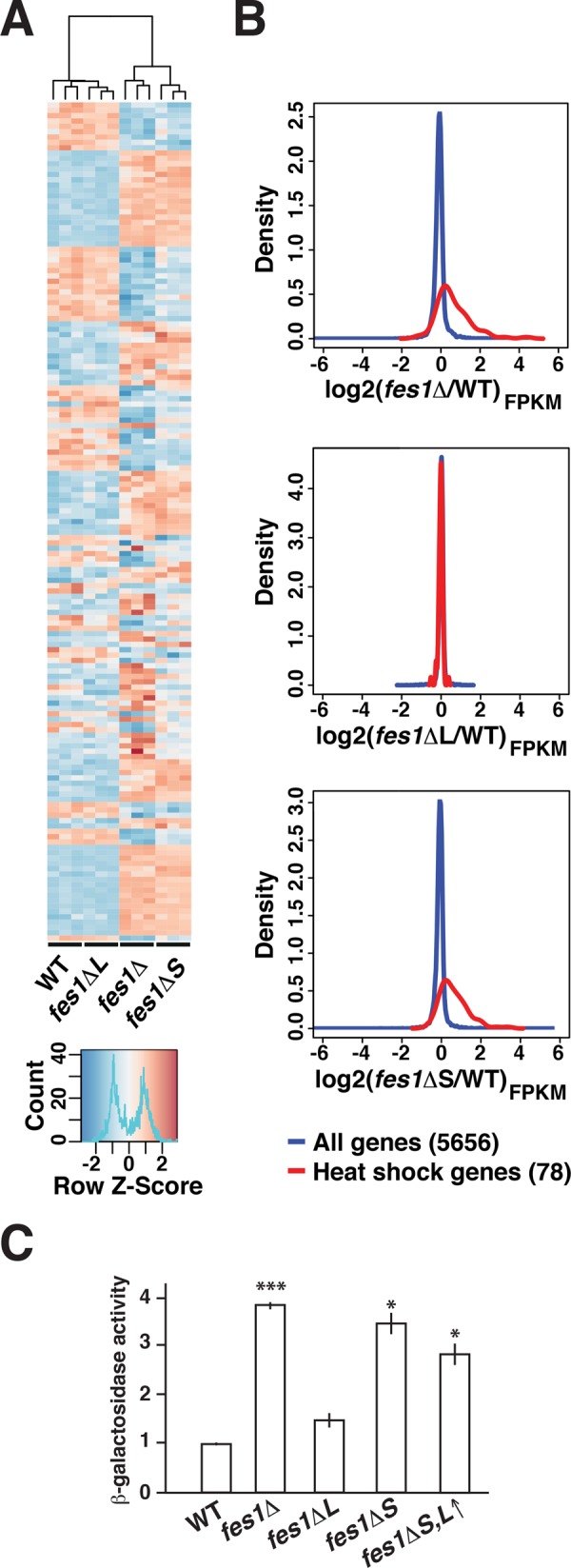
Fes1S keeps the heat shock response. (A) Heat map representing the relative expression of 157 differentially expressed genes over triplicate RNA-Seq samples from WT, fes1Δ, fes1ΔL, and fes1ΔS strains. The dendrogram displays how the samples cluster based on the differential expression. (B) Density plots that display the expression of all 5656 genes (blue) and 78 heat shock–regulated genes (red) in fes1Δ, fes1ΔL, and fes1ΔS relative to WT cells. (C) Relative β-galactosidase activity expressed from the heat shock–responsive reporter PHSP104-LacZ. Error bars indicate the SD of three biological replicates. t test relative to WT: *p ≤ 0.05, ***p ≤ 0.001.
Fes1 is not involved in the reactivation of thermally denatured firefly luciferase
Contradictory results have been obtained regarding the involvement of Fes1 in reactivation of heat-denatured firefly luciferase in the cytosol (Raviol et al., 2006a; Abrams et al., 2014). We fused firefly luciferase to GFP together with either a nuclear export signal (NES) or an NLS (Supplemental Figure S5A), subjected our fes1 mutant strains expressing these reporters to transient heat stress at 43°C, and followed recovery of the activity. Recovery of cytosolic FFL-GFP-NES, as well as nuclear FFL-GFP-NLS, activity was not dependent on Fes1 (Supplemental Figure S5B). We conclude that Fes1 isoforms are not involved in the reactivation of thermally denatured cytosolic firefly luciferase.
Fes1S is required for the degradation of misfolded proteins
Fes1 has an essential function as a NEF in the degradation of misfolded proteins by the ubiquitin-proteasome system (Gowda et al., 2013). We investigated the isoform-specific involvement of Fes1S and Fes1L in the degradation of well-characterized misfolded model proteins that require Hsp70 for ubiquitin-dependent degradation. First, we tested Rpo41*, an internal misfolded fragment of mitochondrial RNA polymerase that mistargets to the cytoplasm, where its degradation requires Fes1 and Hsp70 (Gowda et al., 2013). Turnover of Rpo41* after arrest of translation with cycloheximide was dependent on Fes1S (p ≤ 0.05 compared with WT) but not on Fes1L (Figure 6A). Next we monitored the degradation of Ste6* C, a soluble and misfolded C-terminal fragment of the transmembrane protein Ste6 that requires Hsp70 and, when fused to a C-terminal HA tag, mainly Ubr1 and to some extent San1 for its degradation (Prasad et al., 2012). Ste6* C was stabilized in fes1Δ cells (p ≤ 0.001) and required Fes1S (p ≤ 0.001) but not Fes1L for its degradation (Figure 6B). Finally, we used vacuolar protease A without its signal sequence (ΔssPrA), which mislocalizes to the cytosol and is targeted to the nucleus for San1-dependent degradation (Prasad et al., 2010). Again, Fes1S (p ≤ 0.001), but not Fes1L, was required for turnover of ΔssPrA (Figure 6C). Overexpression of Fes1L as the sole source of Fes1 restored the degradation of Ste6* C and partially of ΔssPrA, likely due to increased cytosolic pools of the protein (Figure 6D). Inactivating the NLS of Fes1L expressed at low endogenous levels (K/R → E mutation) did not result in sufficiently high levels of Fes1 activity in the cytosol to suppress the degradation defect (Supplemental Figure S4G). The degradation of Rpo41* requires Fes1 interaction with Hsp70 for nucleotide exchange (Gowda et al., 2013). In addition, the degradation of Ste6* C and ΔssPrA depended on residues required for the interaction of Fes1 with Hsp70 (Supplemental Figure S6; Shomura et al., 2005). Consistent with a recent study, we found that not all misfolded proteins depend on Fes1 for their degradation (Abrams et al., 2014). Sed1*-GFP and Rad16*-GFP are well-characterized nuclear San1 substrates (Fredrickson et al., 2011) and do not depend on Fes1 for turnover (Supplemental Figure S7). We conclude that specifically the NEF activity of isoform Fes1S is required for degradation of toxic misfolded proteins.
FIGURE 6:

Fes1S is essential for ubiquitin-proteasome system–dependent degradation of misfolded proteins. (A) Western analysis of the turnover of the misfolded model protein Rpo41* after the addition of cycloheximide (CHX). Strains as in Figure 4A. Error bars represent SD (N = 3). (B) Western analysis as in A but of misfolded model protein Ste6* C. (C) Western analysis as in A but of the turnover of the nuclear-targeted misfolded model protein ΔssPrA. (D) Cell suspensions of the strains expressing empty vector control (VC) or Ste6* C or ΔssPrA were serially diluted 10-fold and spotted onto selective medium. Photographs were taken after 3 d of incubation at 25°C. Bottom, Western analysis of the steady-state expression of ΔssPrA.
Fes1S alleviates toxicity of misfolded proteins
High-level expression of misfolded proteins is toxic to yeast cells (Heck et al., 2010; Oling et al., 2014). Because the degradation of misfolded proteins depends on Fes1S, we reasoned that these proteins should be hypertoxic to cells that lack Fes1S. Indeed, the expression of Ste6* C and ΔssPrA inhibited growth of specifically the fes1Δ and fes1ΔS strains but not the WT and fes1ΔL strains (Figure 6D). When Fes1L was overexpressed, the protein could alleviate toxicity of both Ste6* C and ΔssPrA. By determining the steady-state levels of ΔssPrA, we found that the level of growth inhibition that we observed correlated with the ΔssPrA expression level. In conclusion, Fes1S is essential to alleviate the toxicity of misfolded proteins.
DISCUSSION
To summarize our findings regarding the cellular function of the two isoforms of Fes1 and put them in context with the literature, we propose the model outlined in Figure 7. Briefly, Hsp70 associates transiently with exposed hydrophobic patches of misfolded proteins and keeps these aggregation-prone proteins soluble. Hsp70 facilitates the folding of misfolded proteins in ATPase-driven cycles of transient interactions with the misfolded proteins. Proteins resilient to folding will remain associated with Hsp70 in futile chaperone cycles. In the cytosol, Fes1S triggers nucleotide exchange of the Hsp70, which results in the release of the misfolded protein from Hsp70 and facilitates interactions with downstream ubiquitin E3 ligases such as Ubr1 and nuclear San1. Degradation of misfolded proteins that are delivered to the nucleus does not depend on the nuclear isoform Fes1L. Genetic removal of Fes1S results in failure to degrade the misfolded proteins and their accumulation. The buildup of misfolded proteins is toxic and makes the cells hypersensitive to protein-misfolding conditions. Moreover, proteostasis breaks down and induces the heat shock response. The model highlights key roles for Fes1S in the compartment-specific delivery of misfolded protein from the Hsp70 system to the downstream ubiquitin proteasome system.
FIGURE 7:

Model for the function of Fes1L and Fes1S. See Discussion for details.
We documented a requirement for Fes1S for the ubiquitin-dependent degradation of five misfolded model substrates (Rpo41*, DHFRMutC-HA-Ura3, DHFRMutD-HA-Ura3, Ste6* C, and ΔssPrA; this study; Gowda et al., 2013). These misfolded proteins require Hsp70 (SSA class) and ubiquitin E3 ligases San1 and Ubr1 for their degradation. Of interest, another misfolded ubiquitin proteasome system substrate, CPY‡-GFP, has recently been reported not to depend on Fes1 for its degradation (Abrams et al., 2014). In this study, we document that nuclear and San1-dependent substrates Sed1*-GFP and Rad16*-GFP also do not depend on Fes1. These observations suggest that yet-undetermined characteristics of particular misfolded proteins target them for degradation via different pathways. In light of the fact that Fes1S functions in misfolded protein degradation as an Hsp70 nucleotide exchange factor, the transient and dynamic interactions of a given misfolded protein and Hsp70 could influence its dependence on Fes1. We are investigating what characteristics of a misfolded protein control its Fes1S dependence for degradation.
Three lines of evidence support the idea that Fes1L has evolved to provide novel functions for the FES1 gene. First, the intron and the amino acid sequences encoded by exon II are conserved within Saccharomycetaceae. Second, Fes1L is an active Hsp70 NEF in vitro and can, when overexpressed in the cytosol, replace Fes1S function. Third, Fes1L is differentially targeted to the nucleus by an NLS. Of importance, even though Fes1L is targeted to the nucleus by its NLS, it is not excluded from the cytosol. Thus it is hindered in executing the functions of Fes1S in the cytosol in part by its nuclear targeting but also in part by its lower expression levels. The smaller volume of the nucleoplasm compared with that of the cytosol might explain how Fes1L reaches sufficiently high levels for activity. However, the exact cell biological role that Fes1L plays as an Hsp70 NEF in the nucleus remains to be determined. To this end, our data show that its removal does not severely impair proteostasis, the heat shock response, and the clearance of misfolded proteins. Based on the well-understood biochemical role of Hsp70 NEFs, however, it is likely that Fes1L functions to release protein substrates bound by Hsp70 in the nucleus. According to this scenario, the hydrophobic patches of the substrate become available for interaction with downstream factors (e.g., chaperones and ubiquitin E3 ligases), and, at the same time, Hsp70 becomes available to engage new substrates.
The FES1 gene is activated via a canonical HSE by Hsf1 and responds to heat shock by increased transcription. The increased transcription results in preferential induction of transcripts that ends at the promoter-proximal polyadenylation site. Consistent with the preferential induction of Fes1S over Fes1L, this splice isoform is the most abundant and also displays the most severe phenotypes when removed. Thus Fes1S can be viewed as the stress-inducible form of Fes1.
For the FES1 transcript, polyadenylation at the intron-localized promoter-proximal polyadenylation site competes with excision of the intron. The stress-regulated alternative splicing can principally occur at the level of polyadenylation or at the level of splicing. Both processes are affected by heat shock. Specifically, splicing is inactivated by heat shock, and Hsp70 is involved in its recovery (Vogel et al., 1995). Recently a mechanism for the regulation of polyadenylation during heat shock has been described involving poly(ADP-ribosyl)ation of poly(A) polymerase (Di Giammartino et al., 2013).
Alternative splicing is very rare in yeast, and the previously described examples that two functional mRNAs being produced are limited to PTC7 and SKI7/HBS1 (Juneau et al., 2009; Marshall et al., 2013). To the best of our knowledge, the FES1 transcript appears to be unique in having a 3′ intron that carries a polyadenylation site that competes with intron excision to produce two alternative splice isoforms. In addition, alternative splicing of PTC7 leads to subfunctionalization of the expressed proteins (Juneau et al., 2009). When the PTC7 intron is spliced, the expressed protein is targeted to mitochondria, and when instead the intron is retained, the protein is targeted to the nuclear envelope. Thus alternative splicings of both FES1 and PTC7 result in isoforms that localize to different cellular compartments. The evolutionary origins of intron-driven subfunctionalization of FES1 and PTC7 also exhibit similarities (Marshall et al., 2013). The introns of both FES1 and PTC7 appear to have been gained by an ancestor of the Saccharomycetaceae, since homologues outside of the family do not carry introns. Thus the evolution of Saccharomycetaceae includes at least two examples of subfunctionalization by gain of introns.
The finding that cytosolic Fes1S rather than nuclear Fes1L is required for the degradation of misfolded proteins that are targeted for nuclear San1-dependent degradation indicates that the function of Hsp70 in misfolded protein degradation is mainly cytosolic. Specifically, for the set of misfolded model proteins investigated here, Hsp70-Fes1S is spatially separated from the ubiquitin ligase San1. The downstream factor responsible for the delivery of the misfolded proteins to the nucleus is likely the Hsp40 Sis1 (Park et al., 2013; Summers et al., 2013). Curiously, Fes1L is not required for misfolded protein degradation in the nucleus. A simple explanation for this phenomenon is that Hsp70 does not interact with misfolded proteins en route to degradation in the nucleus. Instead, they are directly targeted to the ubiquitin ligase San1 or transit a solid-phase intranuclear quality control compartment (Miller et al., 2015). Thus phase transitions in the form of reversible aggregation of misfolded proteins may play a key role in the intranuclear management of misfolded proteins, whereas the same misfolded proteins are managed by the Hsp70 system in the cytosol.
MATERIALS AND METHODS
Yeast strains, media, and plasmids
Yeast strains used in this study are listed in Supplemental Table S3. The strains were grown in yeast extract/peptone medium (YPD) or synthetic complete medium (SC) to select for plasmids. The plasmids used in this study are listed in Supplemental Table S4. Details regarding their construction are available upon request.
RT-PCR analysis
Total RNA was extracted from yeast cells exponentially growing in YPD at 30°C by mixing the cells with equal volumes of 10 mM Tris HCl, pH 7.5, 10 mM EDTA, 0.5% SDS, and hot acid phenol. After incubation at 65°C for 30 min, the water phase was extracted against acid phenol and chloroform and precipitated using ethanol and sodium acetate. The RNA was reverse transcribed using oligo(dT) primer and Superscript III Reverse Transcriptase (Thermo Fisher Scientific, Waltham, MA) and used as a template for PCR amplification (primers, 5′-ATGGAAAAGCTATTACAGTGG-3′ and 5′-TAATGAATTCCATTTTACAG-3′).
Western analysis
Total protein extraction was performed using NaOH treatment, followed by trichloroacetic acid precipitation (Silve et al., 1991). Proteins were loaded on to NuPAGE gels (Invitrogen) and transferred to nitrocellulose membranes. Membranes were blocked for 1 h at room temperature with 5% nonfat dry milk in TBST (Tris-buffered saline/0.1% Tween) before incubation with primary antibody in TBST for 1 h at room temperature: anti-Fes1 (rabbit serum), 1:5000; anti-GFP (anti-GFP, clones 7.1 and 13.1; Roche Life Science, Roche Diagnostics Corporation, Indianapolis, IN), 1:1000; anti-HA (Anti-HA High Affinity, clone 9F10; Roche Life Science), 1:10,000; anti–Histone H3 (Anti-Histone H3, nuclear loading control; ab1791; Abcam, Cambridge, UK), 1:1000; and α-Pgk1 (Thermo Fisher Scientific), 1:10,000. Serum reactive toward Fes1 was obtained from a rabbit immunized with purified Fes1S (see Protein expression and purification). Membranes were washed three times with TBST for 45 min at room temperature and then incubated with secondary antibody in TBST. Membranes were washed as before, and signals were visualized with chemiluminescence using horseradish peroxidase substrate (SuperSignal West Dura Extended-Duration Substrate; Thermo Fisher Scientific) or near-infrared fluorescence detection (Odyssey Fc; LI-COR Biosciences, Lincoln, NE). Quantification was performed using Image Studio Lite software (LI-COR Biosciences) by normalizing background-corrected signals to the Pgk1 loading control.
Quantification of FES1 mRNA levels
Data for FES1 transcript levels were exported from the University of California, Santa Cruz, database (http://genome.ucsc.edu/; Yassour et al., 2009) as cumulative reads per lane per thousand base pairs. Levels of Fes1S-encoding mRNA were calculated by subtracting the reads from exon II from the reads from exon I. Spliced mRNA levels encoding Fes1L were calculated by subtracting intronic reads from exon II reads.
Preparation of cytosolic fractions
Cytosolic fractions were prepared according to Xu et al. (2010). Briefly, spheroplasts were lysed at low levels of Triton X-100, and cytosolic fractions were separated from cells and nuclei by centrifugation over a sucrose cushion. Signals from Western analysis of Fes1, Histone 3 (nuclear marker protein), and Pgk1 (cytosolic marker protein) were quantified, and the Fes1 signals were normalized to the Pgk1 signal. Signal from the nuclear marker in the cytosolic fractions was used to compensate for leakage from nuclei during the preparation. Average Fes1 signal in the cytosol was calculated from three independent experiments.
RNA-Seq
Cells were grown in YPD at 25°C until log phase and harvested by centrifugation, and RNA was extracted using the RiboPure RNA Purification Kit for Yeast (Thermo Fisher Scientific). Libraries were prepared using Illumina TruSeq Stranded mRNA (polyA selection) and quality checked using Quant-iT (DNA BR) and CaliperGX. Minimum required concentration was 5 ng/μl and a size of >300 base pairs. RNA-Seq was performed on Illumina HiSeq2500 (2 × 101–base pair paired end read). Sequencing reads were mapped to the sacCer2 genome with Ensembl annotation Saccharomyces cerevisiae EF2.62 using Tophat (version 2.0.4). Gene counts and FPKM values were estimated using HTSeq (version 0.6.1) and Cufflinks (version 2.1.1), respectively. The heat map plot was generated by hierarchical clustering based on the regularized log-transformed count matrix of filtered and differentially expressed genes using R (version 3.1.1). Poorly expressed genes were removed (total counts across 12 samples, ≤70, and genes that only were expressed in one replicate sample for each strain). Of the 5778 genes remaining, 157 were found to be differentially expressed in fes1Δ versus WT, fes1ΔL versus WT, or fes1ΔS versus WT using the DESeq2 R package (version 1.6.3; Love et al., 2014). The density plots and associated statistical analysis were performed using R (version 3.1.1) based on the FPKM tables. Poorly expressed genes were removed (total FPKM value across 12 samples, ≤20, and genes that only were expressed in one replicate sample for each strain). For each of the 5656 genes that passed the quality filtering, log2 (µfes1∆ / µWT), log2 (µfes1∆L / µWT), and log2 (µfes1∆S / µWT) were calculated, where μ is the mean FPKM of each strain, averaging over the three replicates. The data have been submitted to the Gene Expression Omnibus (GEO) database under accession number GSE78136.
Protein expression and purification
Proteins (Ssa1, Fes1S, and Fes1L) were expressed with N-terminal–cleavable 6xHis-SUMO tags in Escherichia coli. Details regarding the expression and purification are given elsewhere (Andréasson et al., 2008b; Holmberg et al., 2014). Briefly, expression in strain BL21-SI/pCodonPlus (Thermo Fisher Scientific) was induced at room temperature by the addition 0.3 M NaCl and 0.5 mM isopropyl-β-d-thiogalactoside. After overnight expression, cells were harvested and mechanically disrupted, and the cell-free protein lysates were subjected to Ni–iminodiacetic acid chromatography. The 6xHis-SUMO tags were removed by digestion with Ulp1 protease.
Nucleotide release analysis
Stopped-flow analysis was carried out using an Applied Photophysics SX20 instrument (Applied Photophysics, Leatherhead, United Kingdom) as previously described (Theyssen et al., 1996; Andréasson et al., 2010). For basal dissociation rate, 0.5 μM Ssa1 was incubated with MABA-ATP in 25 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid–KOH, pH 7.5, 150 mM KCl, and 5 mM MgCl2 for 30 min at 30°C. Basal and induced rates were determined by mixing equal volumes of Ssa1-MABA-ADP solution with either 250 μM ATP solution or the same buffer supplemented with 2 μM of either Fes1L or Fes1S.
Fluorescence microscopy
Live cells in 2% glucose solution were visualized, and images were captured using a Zeiss Axiovert 200 M inverted fluorescence microscope (Carl Zeiss, Jena, Germany), 63× oil immersion, DG4 light source (Sutter Instruments, Novato, CA) equipped with an AxioCam MRm camera (Carl Zeiss) and SlideBook 5.0 software (Intelligent Imaging Innovations, GmbH, Göttingen, Germany). Alternatively, for confocal microscopy, an inverted Zeiss LSM 780 microscope with a plan-apochromatic 63×/1.4 numerical aperture oil immersion lens was used. For confocal excitation of GFP, a 488-nm argon laser (25 mW) was set at 2.0%, and emission was detected between wavelengths 495 and 540 nm, and for mCherry, a 561-nm laser line (DPSS 561-10) was used at 2.0%, and emission was detected between 580 and 695 nm. Images were acquired and processed using SlideBook Reader or ZEN 2011 software. Image quantification was done using ImageJ software (National Institutes of Health, Bethesda, MD).
In vivo luciferase refolding assay
Strains expressing FFL-GFP-NES and FFL-GFP-NLS were grown at 25°C to logarithmic phase. The cultures were treated with cycloheximide (100 mg/l) before a 30-min heat shock at 43°C and allowed to recover at 25°C. Collected samples were mixed with d-luciferin (Sigma-Aldrich Sweden AB, Stockholm, Sweden) at a final concentration of 455 mg/l, and luminescence was determined using an Orion II Microplate Luminometer (Berthold Detection Systems GmbH, Pforzheim, Germany).
Heat shock response reporter assays
Cells were grown to logarithmic phase in synthetic complete medium lacking uracil to select for the PHSP104-LacZ reporter plasmid and harvested by centrifugation. β-Galactosidase activity was determined in 0.2% N-lauroyl sarcosine–permeabilized cells in Z buffer (60 mM Na2HPO4, 40 mM NaH2PO4, 10 mM KCl, 1 mM MgSO4, 50 mM β-mercaptoethanol), using ortho-nitrophenyl-β-galactoside as a substrate (Kippert, 1995). For NanoLuc luciferase assays (Masser et al., 2016), Nano-Glo substrate (Promega Corporation, Madison, WI) was diluted 1:100 with the supplied lysis buffer and mixed 1:10 with the cell culture in a white, 96-well plate. Bioluminescence was determined immediately using an Orion II Microplate Luminometer (Berthold Detection Systems).
Supplementary Material
Acknowledgments
We thank D. Ng and R. G. Gardner for plasmids and M. Ott and P. O. Ljungdahl for fruitful discussions throughout the work. We also thank P. Brzezinski for help with the stopped-flow measurements. Support from the Imaging Facility at Stockholm University is acknowledged. This work was supported by the Swedish Research Council and Carl Tryggers Stiftelse för Vetenskaplig Worskning. We also acknowledge support from the Science for Life Laboratory, the National Genomics Infrastructure, Sweden, and the Knut and Alice Wallenberg Foundation, as well as the Uppsala Multidisciplinary Center for Advanced Computational Science for providing assistance in massively parallel DNA sequencing and computational infrastructure. W.K. and M.R.F. acknowledge funding from the Strategic Research Area Program of the Swedish Research Council through Stockholm University.
Abbreviations used:
- ADP
adenosine diphosphate
- ATP
adenosine triphosphate
- CHX
cycloheximide
- C-termini
carboxyl termini
- FPKM
fragments per kilobase of exon per million fragments mapped
- GFP
green fluorescent protein
- HSE
heat shock element
- MABA-ADP
8-[(4-amino)butyl]-amino-adenosine-5’-triphosphate, labeled with MANT
- NEF
nucleotide exchange factor
- NES
nuclear export signal
- NLS
nuclear localization signal
- N-terminal
amino terminal
- ORF
open reading frame
- PremRNA
precursor mRNA
- RNA-Seq
RNA sequencing
- SC
synthetic complete medium
- TBST
Tris-buffered saline and Tween 20
- VC
vector control
- WT
wild type
- YPD
yeast extract–peptone medium
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E15-10-0697) on February 24, 2016.
REFERENCES
- Abovich N, Liao XC, Rosbash M. The yeast MUD2 protein: an interaction with PRP11 defines a bridge between commitment complexes and U2 snRNP addition. Genes Dev. 1994;8:843–854. doi: 10.1101/gad.8.7.843. [DOI] [PubMed] [Google Scholar]
- Abrams JL, Verghese J, Gibney PA, Morano KA. Hierarchical functional specificity of cytosolic heat shock protein 70 (Hsp70) nucleotide exchange factors in yeast. J Biol Chem. 2014;289:13155–13167. doi: 10.1074/jbc.M113.530014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andréasson C, Fiaux J, Rampelt H, Druffel-Augustin S, Bukau B. Insights into the structural dynamics of the Hsp110-Hsp70 interaction reveal the mechanism for nucleotide exchange activity. Proc Natl Acad Sci USA. 2008a;105:16519–16524. doi: 10.1073/pnas.0804187105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andréasson C, Fiaux J, Rampelt H, Mayer MP, Bukau B. Hsp110 is a nucleotide-activated exchange factor for Hsp70. J Biol Chem. 2008b;283:8877–8884. doi: 10.1074/jbc.M710063200. [DOI] [PubMed] [Google Scholar]
- Andréasson C, Rampelt H, Fiaux J, Druffel-Augustin S, Bukau B. The endoplasmic reticulum Grp170 acts as a nucleotide exchange factor of Hsp70 via a mechanism similar to that of the cytosolic Hsp110. J Biol Chem. 2010;285:12445–12453. doi: 10.1074/jbc.M109.096735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B, Retzlaff M, Roos T, Frydman J. Cellular strategies of protein quality control. Cold Spring Harb Perspect Biol. 2011;3:a004374. doi: 10.1101/cshperspect.a004374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crotti LB, Bacikova D, Horowitz DS. The Prp18 protein stabilizes the interaction of both exons with the U5 snRNA during the second step of pre-mRNA splicing. Genes Dev. 2007;21:1204–1216. doi: 10.1101/gad.1538207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Giammartino DC, Shi Y, Manley JL. PARP1 represses PAP and inhibits polyadenylation during heat shock. Mol Cell. 2013;49:7–17. doi: 10.1016/j.molcel.2012.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dragovic Z, Shomura Y, Tzvetkov N, Hartl FU, Bracher A. Fes1p acts as a nucleotide exchange factor for the ribosome-associated molecular chaperone Ssb1p. Biol Chem. 2006;387:1593–1600. doi: 10.1515/BC.2006.198. [DOI] [PubMed] [Google Scholar]
- Eisele F, Wolf DH. Degradation of misfolded protein in the cytoplasm is mediated by the ubiquitin ligase Ubr1. FEBS Lett. 2008;582:4143–4146. doi: 10.1016/j.febslet.2008.11.015. [DOI] [PubMed] [Google Scholar]
- Fredrickson EK, Rosenbaum JC, Locke MN, Milac TI, Gardner RG. Exposed hydrophobicity is a key determinant of nuclear quality control degradation. Mol Biol Cell. 2011;22:2384–2395. doi: 10.1091/mbc.E11-03-0256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gowda NK, Kandasamy G, Froehlich MS, Dohmen RJ, Andréasson C. Hsp70 nucleotide exchange factor Fes1 is essential for ubiquitin-dependent degradation of misfolded cytosolic proteins. Proc Natl Acad Sci USA. 2013;110:5975–5980. doi: 10.1073/pnas.1216778110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerriero CJ, Weiberth KF, Brodsky JL. Hsp70 targets a cytoplasmic quality control substrate to the San1p ubiquitin ligase. J Biol Chem. 2013;288:18506–18520. doi: 10.1074/jbc.M113.475905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn JS, Hu Z, Thiele DJ, Iyer VR. Genome-wide analysis of the biology of stress responses through heat shock transcription factor. Mol Cell Biol. 2004;24:5249–5256. doi: 10.1128/MCB.24.12.5249-5256.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartl FU, Hlodan R, Langer T. Molecular chaperones in protein folding: the art of avoiding sticky situations. Trends Biochem Sci. 1994;19:20–25. doi: 10.1016/0968-0004(94)90169-4. [DOI] [PubMed] [Google Scholar]
- Heck JW, Cheung SK, Hampton RY. Cytoplasmic protein quality control degradation mediated by parallel actions of the E3 ubiquitin ligases Ubr1 and San1. Proc Natl Acad Sci USA. 2010;107:1106–1111. doi: 10.1073/pnas.0910591107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodel MR, Corbett AH, Hodel AE. Dissection of a nuclear localization signal. J Biol Chem. 2001;276:1317–1325. doi: 10.1074/jbc.M008522200. [DOI] [PubMed] [Google Scholar]
- Holmberg MA, Gowda NK, Andréasson C. A versatile bacterial expression vector designed for single-step cloning of multiple DNA fragments using homologous recombination. Protein Expr Purif. 2014;98:38–45. doi: 10.1016/j.pep.2014.03.002. [DOI] [PubMed] [Google Scholar]
- Juneau K, Nislow C, Davis RW. Alternative splicing of PTC7 in Saccharomyces cerevisiae determines protein localization. Genetics. 2009;183:185–194. doi: 10.1534/genetics.109.105155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabani M, Beckerich JM, Brodsky JL. Nucleotide exchange factor for the yeast Hsp70 molecular chaperone Ssa1p. Mol Cell Biol. 2002;22:4677–4689. doi: 10.1128/MCB.22.13.4677-4689.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HK, Kim RR, Oh JH, Cho H, Varshavsky A, Hwang CS. The N-terminal methionine of cellular proteins as a degradation signal. Cell. 2014;156:158–169. doi: 10.1016/j.cell.2013.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kippert F. A rapid permeabilization procedure for accurate quantitative determination of beta-galactosidase activity in yeast cells. FEMS Microbiol Lett. 1995;128:201–206. doi: 10.1111/j.1574-6968.1995.tb07523.x. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall AN, Montealegre MC, Jimenez-Lopez C, Lorenz MC, van Hoof A. Alternative splicing and subfunctionalization generates functional diversity in fungal proteomes. PLoS Genet. 2013;9:e1003376. doi: 10.1371/journal.pgen.1003376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masser AE, Kandasamy G. Luciferase NanoLuc as a reporter for gene expression and protein levels in Saccharomyces cerevisiae. Yeast (in press) 2016 doi: 10.1002/yea.3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer MP, Bukau B. Hsp70 chaperones: cellular functions and molecular mechanism. Cell Mol Life Sci. 2005;62:670–684. doi: 10.1007/s00018-004-4464-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClellan AJ, Scott MD, Frydman J. Folding and quality control of the VHL tumor suppressor proceed through distinct chaperone pathways. Cell. 2005;121:739–748. doi: 10.1016/j.cell.2005.03.024. [DOI] [PubMed] [Google Scholar]
- Metzger MB, Maurer MJ, Dancy BM, Michaelis S. Degradation of a cytosolic protein requires endoplasmic reticulum-associated degradation machinery. J Biol Chem. 2008;283:32302–32316. doi: 10.1074/jbc.M806424200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller SB, Ho CT, Winkler J, Khokhrina M, Neuner A, Mohamed MY, Guilbride DL, Richter K, Lisby M, Schiebel E, et al. Compartment-specific aggregases direct distinct nuclear and cytoplasmic aggregate deposition. EMBO J. 2015;34:778–797. doi: 10.15252/embj.201489524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagalakshmi U, Wang Z, Waern K, Shou C, Raha D, Gerstein M, Snyder M. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science. 2008;320:1344–1349. doi: 10.1126/science.1158441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nillegoda NB, Theodoraki MA, Mandal AK, Mayo KJ, Ren HY, Sultana R, Wu K, Johnson J, Cyr DM, Caplan AJ. Ubr1 and Ubr2 function in a quality control pathway for degradation of unfolded cytosolic proteins. Mol Biol Cell. 2010;21:2102–2116. doi: 10.1091/mbc.E10-02-0098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oling D, Eisele F, Kvint K, Nystrom T. Opposing roles of Ubp3-dependent deubiquitination regulate replicative life span and heat resistance. EMBO J. 2014;33:747–761. doi: 10.1002/embj.201386822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SH, Bolender N, Eisele F, Kostova Z, Takeuchi J, Coffino P, Wolf DH. The cytoplasmic Hsp70 chaperone machinery subjects misfolded and endoplasmic reticulum import-incompetent proteins to degradation via the ubiquitin-proteasome system. Mol Biol Cell. 2007;18:153–165. doi: 10.1091/mbc.E06-04-0338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SH, Kukushkin Y, Gupta R, Chen T, Konagai A, Hipp MS, Hayer-Hartl M, Hartl FU. PolyQ proteins interfere with nuclear degradation of cytosolic proteins by sequestering the Sis1p chaperone. Cell. 2013;154:134–145. doi: 10.1016/j.cell.2013.06.003. [DOI] [PubMed] [Google Scholar]
- Prasad R, Kawaguchi S, Ng DT. A nucleus-based quality control mechanism for cytosolic proteins. Mol Biol Cell. 2010;21:2117–2127. doi: 10.1091/mbc.E10-02-0111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad R, Kawaguchi S, Ng DT. Biosynthetic mode can determine the mechanism of protein quality control. Biochem Biophys Res Commun. 2012;425:689–695. doi: 10.1016/j.bbrc.2012.07.080. [DOI] [PubMed] [Google Scholar]
- Raviol H, Bukau B, Mayer MP. Human and yeast Hsp110 chaperones exhibit functional differences. FEBS Lett. 2006a;580:168–174. doi: 10.1016/j.febslet.2005.11.069. [DOI] [PubMed] [Google Scholar]
- Raviol H, Sadlish H, Rodriguez F, Mayer MP, Bukau B. Chaperone network in the yeast cytosol: Hsp110 is revealed as an Hsp70 nucleotide exchange factor. EMBO J. 2006b;25:2510–2518. doi: 10.1038/sj.emboj.7601139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbaum JC, Fredrickson EK, Oeser ML, Garrett-Engele CM, Locke MN, Richardson LA, Nelson ZW, Hetrick ED, Milac TI, Gottschling DE, et al. Disorder targets misorder in nuclear quality control degradation: a disordered ubiquitin ligase directly recognizes its misfolded substrates. Mol Cell. 2011;41:93–106. doi: 10.1016/j.molcel.2010.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiber A, Breuer W, Brandeis M, Ravid T. Ubiquitin conjugation triggers misfolded protein sequestration into quality control foci when Hsp70 chaperone levels are limiting. Mol Biol Cell. 2013;24:2076–2087. doi: 10.1091/mbc.E13-01-0010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shomura Y, Dragovic Z, Chang HC, Tzvetkov N, Young JC, Brodsky JL, Guerriero V, Hartl FU, Bracher A. Regulation of Hsp70 function by HspBP1: structural analysis reveals an alternate mechanism for Hsp70 nucleotide exchange. Mol Cell. 2005;17:367–379. doi: 10.1016/j.molcel.2004.12.023. [DOI] [PubMed] [Google Scholar]
- Silve S, Volland C, Garnier C, Jund R, Chevallier MR, Haguenauer-Tsapis R. Membrane insertion of uracil permease, a polytopic yeast plasma membrane protein. Mol Cell Biol. 1991;11:1114–1124. doi: 10.1128/mcb.11.2.1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Summers DW, Wolfe KJ, Ren HY, Cyr DM. The Type II Hsp40 Sis1 cooperates with Hsp70 and the E3 ligase Ubr1 to promote degradation of terminally misfolded cytosolic protein. PloS One. 2013;8:e52099. doi: 10.1371/journal.pone.0052099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theyssen H, Schuster HP, Packschies L, Bukau B, Reinstein J. The second step of ATP binding to DnaK induces peptide release. J Mol Biol. 1996;263:657–670. doi: 10.1006/jmbi.1996.0606. [DOI] [PubMed] [Google Scholar]
- Verghese J, Abrams J, Wang Y, Morano KA. Biology of the heat shock response and protein chaperones: budding yeast (Saccharomyces cerevisiae) as a model system. Microbiol Mol Biol Rev. 2012;76:115–158. doi: 10.1128/MMBR.05018-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijayraghavan U, Abelson J. PRP18, a protein required for the second reaction in pre-mRNA splicing. Mol Cell Biol. 1990;10:324–332. doi: 10.1128/mcb.10.1.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel JL, Parsell DA, Lindquist S. Heat-shock proteins Hsp104 and Hsp70 reactivate mRNA splicing after heat inactivation. Curr Biol. 1995;5:306–317. doi: 10.1016/s0960-9822(95)00061-3. [DOI] [PubMed] [Google Scholar]
- Wang Q, Zhang L, Lynn B, Rymond BC. A BBP-Mud2p heterodimer mediates branchpoint recognition and influences splicing substrate abundance in budding yeast. Nucleic Acids Res. 2008;36:2787–2798. doi: 10.1093/nar/gkn144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu T, Johnson CA, Gestwicki JE, Kumar A. Conditionally controlling nuclear trafficking in yeast by chemical-induced protein dimerization. Nat Protoc. 2010;5:1831–1843. doi: 10.1038/nprot.2010.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto A, Mizukami Y, Sakurai H. Identification of a novel class of target genes and a novel type of binding sequence of heat shock transcription factor in Saccharomyces cerevisiae. J Biol Chem. 2005;280:11911–11919. doi: 10.1074/jbc.M411256200. [DOI] [PubMed] [Google Scholar]
- Yassour M, Kaplan T, Fraser HB, Levin JZ, Pfiffner J, Adiconis X, Schroth G, Luo S, Khrebtukova I, Gnirke A, et al. Ab initio construction of a eukaryotic transcriptome by massively parallel mRNA sequencing. Proc Natl Acad Sci USA. 2009;106:3264–3269. doi: 10.1073/pnas.0812841106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.