

Steady-state and transiently perturbed nitrogen-limited chemostats show that nitrogen abundance is a primary signal controlling nitrogen-responsive gene expression. When cells experience an increase in nitrogen, some transcripts are rapidly degraded, suggesting that accelerated mRNA degradation contributes to remodeling of gene expression.
Abstract
Cell growth rate is regulated in response to the abundance and molecular form of essential nutrients. In Saccharomyces cerevisiae (budding yeast), the molecular form of environmental nitrogen is a major determinant of cell growth rate, supporting growth rates that vary at least threefold. Transcriptional control of nitrogen use is mediated in large part by nitrogen catabolite repression (NCR), which results in the repression of specific transcripts in the presence of a preferred nitrogen source that supports a fast growth rate, such as glutamine, that are otherwise expressed in the presence of a nonpreferred nitrogen source, such as proline, which supports a slower growth rate. Differential expression of the NCR regulon and additional nitrogen-responsive genes results in >500 transcripts that are differentially expressed in cells growing in the presence of different nitrogen sources in batch cultures. Here we find that in growth rate–controlled cultures using nitrogen-limited chemostats, gene expression programs are strikingly similar regardless of nitrogen source. NCR expression is derepressed in all nitrogen-limiting chemostat conditions regardless of nitrogen source, and in these conditions, only 34 transcripts exhibit nitrogen source–specific differential gene expression. Addition of either the preferred nitrogen source, glutamine, or the nonpreferred nitrogen source, proline, to cells growing in nitrogen-limited chemostats results in rapid, dose-dependent repression of the NCR regulon. Using a novel means of computational normalization to compare global gene expression programs in steady-state and dynamic conditions, we find evidence that the addition of nitrogen to nitrogen-limited cells results in the transient overproduction of transcripts required for protein translation. Simultaneously, we find that that accelerated mRNA degradation underlies the rapid clearing of a subset of transcripts, which is most pronounced for the highly expressed NCR-regulated permease genes GAP1, MEP2, DAL5, PUT4, and DIP5. Our results reveal novel aspects of nitrogen-regulated gene expression and highlight the need for a quantitative approach to study how the cell coordinates protein translation and nitrogen assimilation to optimize cell growth in different environments.
INTRODUCTION
Regulated cell growth (increase in biomass and cell size) requires the coordination of diverse cellular processes, ranging from macromolecular synthesis to metabolism (Ingraham et al., 1983; Hall et al., 2004). The rate at which cells grow is determined both by factors that are intrinsic to the cell and factors in the external environment. In all eukaryotic cells, essential nutrients including carbon, nitrogen, and phosphorus are required for growth. The extracellular abundance and molecular form of these nutrients are critical determinants of the growth rate of the cell. As nutrient concentration decreases, bacteria (Monod, 1949) and budding yeast (Ziv et al., 2013) decrease their growth rates with Michaelis–Menten-type kinetics. Different molecular forms of essential nutrients such as carbon (e.g., glucose and galactose) or nitrogen (e.g., glutamine and proline) support different growth rates and are metabolized with difference preferences for reasons that are poorly understood. How cells integrate information about the environmental status of essential nutrients, regulate preferential metabolism of particular compounds containing essential nutrients, and coordinate diverse cellular functions with cell growth and division remains one of the central problems in systems biology (Klumpp et al., 2009; Scott et al., 2010, 2014).
Differences in cell growth rate profoundly affect the physiology of cells. Classic studies in bacteria showed that the mass of cells and the absolute abundance of cellular components, including RNA, proteins, and ribosomes, increase as cells grow faster (Kjeldgaard et al., 1958; Schaechter et al., 1958). More recently, studies of global gene expression using chemostats, in which growth rate is controlled by changing the dilution rate (Monod, 1950; Novick and Szilard, 1950; Kubitschek, 1970), have shown that the rate at which populations of bacterial (Ishii et al., 2007) or budding yeast (Regenberg et al., 2006; Castrillo et al., 2007; Brauer et al., 2008) cells grow is a primary determinant of the relative abundance of a large fraction of mRNAs. In budding yeast, the relative expression of one-fourth of all mRNAs can be explained by a simple linear relationship with population growth rate across a wide range of conditions (Brauer et al., 2008). Growth rate–correlated transcripts encode products with functions ranging from energy metabolism to ribosome biogenesis, suggesting that multiple cellular functions and processes are systematically modulated as cell growth rate varies.
The stereotypic relationship between the relative abundance of many mRNAs and growth rate enables estimation of cell growth rates on the basis of gene expression using a compendium of diagnostic transcripts (Brauer et al., 2008; Airoldi et al., 2009). This “instantaneous growth rate” estimation provides a means of detecting changes in a population’s growth rate on time scales that are not typically amenable to direct measurement. Our understanding of the signaling networks that coordinate cell growth rate with gene expression remains incomplete. However, ectopic activation of the Ras/protein kinase A (PKA) pathway using an activated Ras allele recapitulates some of the gene expression changes associated with increased growth rate despite the absence of an actual increase in biomass (Airoldi et al., 2009). This suggests a model in which the cell establishes a gene expression program that reflects its perception of the growth potential of the current environment, transmitted via nutrient-responsive signaling pathways, rather than the gene expression program being determined by growth rate.
The rate at which budding yeast cells grow is sensitive to the molecular form of nitrogen in the environment. Yeast cells are able to use and discriminate between different nitrogen sources (Cooper, 1982; Magasanik and Kaiser, 2002). When a variety of nitrogen sources are available, a yeast cell will preferentially transport and metabolize particular nitrogen-containing compounds by decreasing levels of transcripts and proteins required for use of nonpreferred nitrogen sources (Cooper, 1982; Magasanik and Kaiser, 2002). A study of yeast cells growing in the presence of different individual nitrogen sources provided a genome-wide view of nitrogen-regulated gene expression and suggested that >500 genes are differentially expressed as a function of environmental nitrogen source (Godard et al., 2007). On the basis of differential gene expression, promoter sequence elements, and published literature, Godard et al. (2007) assigned membership of many of these transcripts to five regulons that are responsive to environmental nitrogen: the nitrogen catabolite repression A (NCR-A) regulon, which includes bona fide NCR targets; the potential NCR target (NCR-P) regulon; the general amino acid control (GAAC) regulon; the unfolded protein response (UPR) regulon; and the SSY1-PTR3-SSY5 (SPS) regulon.
Transcriptional control of the NCR regulon (i.e., both NCR-A and NCR-P regulons) is mediated by the transcription factors GLN3, GAT1, DAL80, and GZF3, which bind to the 5′-GATAA-3′ consensus sequence in target gene promoter regions (Cooper, 2002; Magasanik and Kaiser, 2002). Whereas DAL80 and GZF3 act as repressors of NCR transcription, GLN3 and GAT1 activate the transcription of NCR genes in a nitrogen source–dependent manner. The evolutionarily conserved TOR complex 1 (TORC1) is believed to be an upstream regulator of NCR expression, as it promotes the nuclear exclusion of GLN3 by physical association with URE2 in a phosphorylation-dependent manner (Beck and Hall, 1999). TORC1 effects nitrogen-responsive gene expression more broadly, as it promotes expression of the SPS regulon by stabilizing the transcription factor STP1 (Shin et al., 2009) and affects GAAC expression by regulating GCN2, which regulates translation of the transcription factor GCN4 (Loewith and Hall, 2011).
In addition to its role in nitrogen-use regulation, TORC1 coordinates myriad growth-related processes, ranging from ribosome biogenesis to autophagy (Loewith and Hall, 2011). TORC1 directly phosphorylates SFP1, leading to the activation of transcripts required for ribosome biogenesis (the RiBi regulon), and promotes the transcription of mRNAs encoding ribosomal proteins (the RP regulon) by phosphorylation of IFH1 (Loewith and Hall, 2011). TORC1 also has important roles in the posttranslational regulation of protein production through phosphorylation of the S6-kinase homologue, SCH9 (Urban et al., 2007). The centrality of TORC1 in the biogenesis and regulation of ribosomes and protein translation required for cell growth and the regulation of nitrogen utilization suggests that these two processes are intimately entwined (Cardenas et al., 1999).
To study the effect on mRNA expression of environmental nitrogen source variation in nitrogen-limited, growth rate–controlled conditions, we studied cells growing in chemostats using six different nitrogen sources at four different dilution rates. We show that differential expression of the NCR and SPS regulons is primarily a function of growth in a nitrogen-limited environment, with the molecular form of nitrogen having minimal effect on differential gene expression when cells are limited for nitrogen. By contrast, the GAAC and UPR regulons do not respond specifically to nitrogen limitation compared with other nutrient-limited conditions. To study the dynamics of nitrogen-responsive gene expression, we performed transient perturbation experiments in which different quantities and sources of nitrogen were added to cells growing in nitrogen-limited chemostats. The addition of either the preferred nitrogen source, glutamine, or the nonpreferred nitrogen source, proline, to cells growing in nitrogen-limited conditions results in rapid repression of the NCR regulon in a dose-dependent manner. Surprisingly, a sudden increase in environmental nitrogen does not correspond to a detectable increase in biomass production or cell number, consistent with a time delay between activation of the transcriptional growth program and its manifestation in an increased rate of cell growth. To compare global gene expression in dynamic conditions with mRNA expression in steady-state conditions, we used computational estimation of instantaneous growth rate from gene expression profiles (Brauer et al., 2008; Airoldi et al., 2009) and defined gene expression responses to growth rate in both steady-state and dynamic conditions using linear regression. We find that the response of transcripts required for protein translation (RP and RiBi) in cells provided with an increase in nitrogen exceeds the response to growth rate in cells growing in steady-state conditions consistent with a transient overproduction of RP and RiBi transcripts. Finally, we show that accelerated degradation of some NCR transcripts underlies gene expression remodeling in response to sudden relief from nitrogen limitation, indicating the activity of a posttranscriptional mechanism controlling nitrogen-responsive gene expression.
RESULTS
Yeast cells convert different nitrogen compounds to biomass with similar yields
The abundance and molecular form of environmental nitrogen are important determinants of growth rate and gene expression in yeast cells (Cooper, 1982; Magasanik and Kaiser, 2002). We investigated the physiological and transcriptional response of yeast cells growing in different nitrogen sources, using batch and chemostat cultures. Although the maximal growth rate attained by cells depends on the molecular form of environmental nitrogen (Cooper, 1982), we found that when nitrogen is the growth-limiting agent (i.e., the first nutrient exhausted in the growth medium) and provided at equimolar concentrations in batch cultures, the biomass yield is equivalent from different nitrogen sources (Figure 1A and Supplemental Figure S1). Thus different nitrogen sources support a range of maximal growth rates, but, when present in limiting concentrations, yeast cells convert nitrogen to biomass with similar final yields regardless of its molecular form.
FIGURE 1:

Physiology of yeast cultures grown in nitrogen-limited batch and chemostat cultures. (A) When normalized for total nitrogen concentration, the biomass yield is the same regardless of nitrogen source. Values are determined by regression analysis of cell yield, measured using a Klett colorimeter, in batch cultures containing different concentrations of nitrogen. Error bars represent 95% confidence interval of the regression coefficient. (B) Approach to steady-state kinetics for glutamine- and proline-limited chemostats. Each chemostat culture was inoculated with 106 cell/ml and monitored over an 80-h period. Experimental measurements for proline (black triangles) and glutamine (red circles) are in good agreement with a mathematical model of the chemostat (lines).
Studying the effect of different nitrogen sources on cell physiology and molecular processes, such as gene expression, may be confounded by variation in growth rate. In contrast to growth in batch cultures, the chemostat enables experimental control of population growth rates. In a steady-state chemostat, the dilution rate is equal to the exponential growth rate constant (Monod, 1950; Novick and Szilard, 1950; Kubitschek, 1970). We established independent chemostat cultures limited for one of six different nitrogen sources, including both preferred and nonpreferred nitrogen sources, using media containing equimolar concentrations of nitrogen. We found that when the growth medium contains a concentration of 800 μM nitrogen, the steady-state cell density in a chemostat maintained at a very low dilution rate of 0.06 culture volumes/h (V/h) is only marginally less than the final density in batch cultures that have exhausted all nitrogen from the medium (Supplemental Figure S2). This is consistent with a chemostat culture being most similar to a batch culture just before an essential nutrient is exhausted (Saldanha et al., 2004). We systematically increased the rate at which each nitrogen-limited chemostat was diluted, establishing rates of D = 0.06, 0.12, 0.16, and 0.21 V/h, which correspond to steady-state cultures with doubling times of 11.6, 5.8, 4.3, and 3.3 h, respectively. Consistent with chemostat theory (Kubitschek, 1970), in all nitrogen-limiting conditions, steady-state culture density declined as the dilution rate increased (Supplemental Figure S2), which results in a concomitant increase in the steady-state nitrogen concentration. Surprisingly, and in contrast to theoretical expectations (Kubitschek, 1970), by sequentially increasing the dilution rate starting at a very low dilution rate, we were able to maintain proline-limited chemostats at dilution rates that exceed the reported maximal growth rate of yeast cells in batch cultures that contain proline as the sole nitrogen source.
In batch cultures containing a single nitrogen source, proline supports one of the slowest maximal growth rates, whereas glutamine supports one of the fastest (Cooper, 1982). Therefore we repeated our analyses of batch and chemostat cultures growing in proline- and glutamine-limited media and measured cell counts. We found that cell yields in batch cultures are slightly higher in proline-limited media (36,069 ± 3521 cells/ml per μmol of nitrogen) compared with glutamine-limited conditions (32,445 ± 3422 cells/ml per μmol of nitrogen), although the difference is not significant. From steady-state glutamine- and proline-limited chemostats growing at different dilution rates, we estimated s, the residual nitrogen concentration (Supplemental Methods) and used these values to estimate the half-maximal growth rate constant (Ks) for both glutamine and proline media (Supplemental Methods). Using these estimated parameters, and published maximal growth rates (Cooper, 1982), we modeled the approach to steady state in chemostats and found that the model is in good agreement with experimental data (Figure 1B). Thus, in steady-state chemostats containing equimolar concentrations of different nitrogen sources at growth-limiting concentrations, growth rates and culture densities are equivalent (Supplemental Figure S2), and all other environmental factors are essentially identical, enabling the dissection of growth rate–specific and nitrogen-specific effects on gene expression.
Nitrogen limitation is a primary determinant of nitrogen-regulated gene expression
To identify the set of genes that are differentially expressed when nitrogen in the form of ammonium sulfate is the growth rate–limiting nutrient, we first analyzed the data of Brauer et al. (2008), in whose study populations of cells were limited in chemostats for one of six different essential nutrients (glucose, ammonium, phosphorus, sulfur, leucine, and uracil) at six different growth rates (ranging from 0.05 to 0.3 h−1), using an analysis of covariance (ANCOVA) in which we modeled the expression of each transcript as a function of the limiting nutrient and dilution rate (Materials and Methods). We compared this more complex model to the original model used in Brauer et al. (2008), in which a growth rate effect on gene expression was modeled without consideration of the limiting nutrient (Supplemental Table S1). The majority of mRNAs (4726 of 5537 at a 10% false discovery rate [FDR]) are modeled significantly better by the more complex two-factor model, suggesting that nutrient-specific effects contribute to expression variation of many yeast genes.
We identified 105 genes for which ammonium limitation contributes significantly to variation in expression (10% FDR), including 66 transcripts that are increased in expression and 39 transcripts that are decreased in expression compared with growth limitation by the five other nutrients used by Brauer et al. (2008; Supplemental Table S1). The majority of transcripts belonging to the NCR-A (28 of 39 measured transcripts, p < 0.05) regulon and many belonging to the NCR-P (11 of 42 measured transcripts, p < 0.05) regulon are significantly increased in expression in nitrogen-limiting conditions compared with other nutrient-limited conditions (Figure 2A). By contrast, transcripts in the GAAC and UPR regulons do not show evidence of a specific response to ammonium limitation (Figure 2A). The SPS regulon is specifically down-regulated in ammonium-limited chemostats (six of seven measured transcripts; p < 0.05) compared with other nutrient limitations (Figure 2A), as are specific permeases that transport nitrogen sources other than ammonia (Supplemental Table S1). These results show that the NCR-A and NCR-P regulons are derepressed in nitrogen-limiting conditions when ammonium is the nitrogen source, whereas the SPS regulon is repressed. This confirms and extends previous studies in which growth in ammonium-limited chemostats resulted in derepression of NCR genes (Boer et al., 2003; Usaite et al., 2006).
FIGURE 2:

Differential gene expression as a function of nitrogen source in nutrient-limited chemostats. (A) The nitrogen-responsive NCR-A, NCR-P, and SPS regulons respond specifically to nitrogen limitation, whereas the GAAC and UPR regulons are not differentially expressed in nitrogen-limited chemostats compared with carbon, sulfur, phosphorus, leucine, and uracil growth limitation. Gene expression values in Brauer et al. (2008) were relative to a common reference taken from a glucose-limited chemostat grown at a dilution rate of 0.25 h−1. (B) A small number of nitrogen-regulated genes respond significantly to variation in nitrogen source in nitrogen-limited conditions. Regulon membership of genes is denoted by color corresponding to A (NCR-A, green; GAAC, orange; NCR-P, blue; UPR, red). GDH2 has been proposed to be a member of both NCR-A and GAAC regulons (Godard et al., 2007). Filled symbols represent significant effects as determined by permutation testing. Gene expression values are relative to gene expression in an ammonium-limited chemostat growing at a dilution rate of 0.12 h−1.
To identify genes that are differentially expressed when grown in the presence of different nitrogen sources in nitrogen-limited chemostats, we determined the global gene expression profiles of cells growing at four different growth rates in chemostats limited for six different nitrogen sources (Materials and Methods). Using the same criteria as Brauer et al. (2008; i.e., R2 > 0.7, p < 0.05, for linear regression of log2-transformed relative gene expression against growth rate), we find that 24% of transcripts (1345 of 5590) respond significantly to growth rate across different nitrogen-limited conditions compared with 27% (1474 of 5452) in Brauer et al. (2008). The small discrepancy in these results is likely due to the reduction in statistical power associated with the smaller sample size in our study (24 vs. 36 conditions), as the set of genes that respond to growth rate and the magnitude of their responses are very similar between the two studies (Supplemental Figure S3). Consistent with this finding, we previously showed that a predictive model of the “instantaneous growth rate” trained on gene expression data from diverse nutrient-limited chemostats (Brauer et al., 2008) accurately predicts growth rates in steady-state nitrogen-limited chemostats on the basis of gene expression alone (Airoldi et al., 2009). Together these results indicate that growth rate–correlated gene expression programs are highly reproducible.
To identify genes that are differentially expressed in response to variation in environmental nitrogen source, we performed an ANCOVA in which we modeled the expression of each gene as a function of nitrogen source and growth rate (Materials and Methods and Supplemental Table S2). Although the inclusion of nitrogen source in the model improves the fit of the data for more than half of the transcripts (3520 of 5590 genes at a 10% FDR) compared with a simple linear model, the nitrogen-source effect is only significant for 38 of the 5590 analyzed genes (Supplemental Table S2). Only 14 genes that belong to known nitrogen-responsive regulons exhibit a significant response to variation in nitrogen source (Figure 2B). The majority of these (10 of 14) belong to the NCR-A regulon and encode permeases or enzymes required for specific nitrogen sources. Thus, in nitrogen-limited conditions, most nitrogen-responsive gene expression is regulated in a manner independent of nitrogen source, with minimal differential expression in response to variation in the molecular form of environmental nitrogen.
Rapid physiological and transcriptional response to transient relief from nitrogen limitation
To characterize the physiological and transcriptional response to relief from nitrogen limitation, we studied cells growing in nitrogen-limited chemostats that were transiently relieved from nitrogen limitation via addition of a “pulse” of nitrogen using an experimental design previously applied to carbon-limited cultures (Kresnowati et al., 2006; Ronen and Botstein, 2006). This experimental design is similar to classic “shift-up” experiments (Kjeldgaard et al., 1958); however, our aim was to minimally and transiently perturb the steady-state condition and study the coordination of gene expression and cell growth changes in the absence of wholesale remodeling of cell physiology.
We studied the response of ammonium-limited chemostats growing at 0.12 V/h (i.e., a doubling time of 5.9 h) to a pulse of glutamine, proline, or a mixture of both. On the basis of steady-state cell density measurements, we estimate that the steady-state nitrogen concentration in chemostats growing at a dilution rate of 0.12 V/h is ∼80 μM. To study the effect of dose and nitrogen source on the kinetics and magnitude of the response, we used either a small (80 μM) or large (800 μM) nitrogen pulse of glutamine (40 or 400 μM) or proline (80 or 800 μM). We also performed a single small pulse experiment in which we added a mixture of 20 μM glutamine and 40 μM proline.
Using a mathematical model of the chemostat, we simulated the expected effect on culture density when a nitrogen-limited chemostat is provided with a pulse of nitrogen (Materials and Methods and Supplemental Methods). This model predicts a dose-dependent increase in cell density on addition of 400 µM (Figure 3A) or 40 μM (Figure 3B) glutamine. Similarly, addition of 800 µM (Figure 3C) or 80 μM (Figure 3D) proline is predicted to result in a dose-dependent increase in culture density with delayed kinetics compared with the same molar amount of nitrogen in the form of glutamine. A mixture of proline and glutamine is predicted to have a similar effect (Supplemental Figure S4). In all cases, the model predicts that the increase in culture density is followed by a gradual return to the initial steady-state density as the additional nitrogen is consumed and lost through dilution. We performed pulse experiments and monitored changes in culture density. Surprisingly, pulses of 40 μM glutamine (Figure 3B) and 800 µM (Figure 3C) and 80 μM (Figure 3D) proline failed to result in the expected increase in culture density, and, strikingly, a pulse of 400 μM glutamine resulted in a decline in culture biomass (Figure 3A). This may be the result of a pause or delay in population growth before physiological remodeling for growth in an altered environment (Kjeldgaard et al., 1958; Ludwig et al., 1977; Waldron, 1977; Kief and Warner, 1981) and the continuous dilution of the culture.
FIGURE 3:

Kinetics of growth and predicted growth rate response in cultures transiently relieved from nitrogen limitation. Ammonium-limited chemostat cultures grown at 0.12 h−1 were subjected to an instantaneous addition of (A) 400 μM glutamine, (B) 40 μM glutamine, (C) 800 μM proline, or (D) 80 μM proline. For each perturbation experiment, we modeled the change in growth rate as a continuous function of time (blue line). We measured culture density at discrete times points (blue points) and predicted the instantaneous growth rate on the basis of global gene expression (red points).
To study the transcriptional response of cells transiently relieved from nitrogen limitation, we performed time-series analysis of the transcriptome using DNA microarrays (Figure 4A). Qualitatively, many of the observed changes in gene expression were similar in response to both the addition of glutamine and the addition of proline. We observed that many of the transcripts that changed in expression also vary with growth rate in steady-state cultures. Therefore we computationally predicted the instantaneous growth rate (Brauer et al., 2008; Airoldi et al., 2009) after perturbation, using gene expression data (Materials and Methods). These predictions indicate a rapid increase in the instantaneous growth rate in response to large and small doses of both proline and glutamine (Figure 4A). Whereas the instantaneous growth rate remains high for the large doses of glutamine (Figure 3A) and proline (Figure 3C), cultures provided with a small dose of glutamine (Figure 3B) or proline (Figure 3D) return to the original steady-state instantaneous growth rates within 2 h. A nitrogen-limited chemostat culture provided with a mixture of glutamine and proline (Figure 4A) shows similar behavior (Supplemental Figure S4). The discordance between the inferred growth rate from gene expression (i.e., the instantaneous growth rate) and the measured change in culture density is consistent with gene expression reflecting the cells’ perception of the growth potential of the environment rather than the actual realized growth rate.
FIGURE 4:

Remodeling of the transcriptome in response to relief from nitrogen limitation. (A) We clustered global gene expression data for cultures limited for proline and glutamine grown at different growth rates with gene expression profiles for ammonium-limited cultures transiently perturbed by the addition of 80 and 800 μM proline or 40 and 400 μM glutamine or a mixture of 20 μM glutamine and 40 μM proline. Many transcripts that increase (red bar) or decrease (green bar) systematically with growth rate in steady-state chemostats are also altered in expression when transiently relieved from nitrogen limitation. Average response of the NCR-A, NCR-P, GAAC, SPS, and UPR regulons to a (B) large or (C) small pulse of glutamine or a (D) large or (E) small pulse of proline. Gene expression values are relative to gene expression in an ammonium-limited chemostat growing at a dilution rate of 0.12 h−1.
Because the critical cell size threshold for cell division is affected by nutrient abundance, an improvement in nitrogen status may alter the average cell size. To assess whether this occurs in response to an increase in nitrogen, we measured changes in cell size after the nitrogen pulse. We were able to detect a transient increase in the median cell size for a large (Supplemental Figure S5A) but not a small pulse of glutamine (Supplemental Figure S5B). Similarly, a large pulse of proline results in a transient increase in cell size (Supplemental Figure S5C), but a small one does not (Supplemental Figure S5D). This suggests that the critical cell size is reset in response to an instantaneous increase in nitrogen abundance.
We examined the dynamics of nitrogen-regulated gene expression in response to the transient relief from nitrogen limitation by studying previously defined regulons (Godard et al., 2007). In response to a large (Figure 4B) or small (Figure 4C) pulse of glutamine, we observed rapid and strong repression of the NCR-A and NCR-P regulons. Surprisingly, we also observed repression of the NCR-A and NCR-P regulons in response to a large (Figure 4D) or small (Figure 4E) pulse of the nonpreferred nitrogen source, proline, the magnitude and kinetics of which are less pronounced than in response to glutamine. Conversely, the GAAC and SPS regulons are induced in response to relief from nitrogen limitation. The activation of the GAAC regulon under these conditions is surprising, as it is generally believed to respond to amino acid starvation (Hinnebusch, 2005), although amino acid imbalances may also activate GAAC expression (Niederberger et al., 1981). The UPR regulon does not change dramatically in response to the addition of nitrogen. Thus, changes in nitrogen-regulated gene expression occur in response to both the preferred nitrogen source, glutamine, and the nonpreferred nitrogen source, proline, when ammonium-limited cells encounter a sudden increase in nitrogen abundance.
The RP and RiBi regulons are strongly induced in response to relief from nitrogen limitation
Although a compendium of diagnostic transcripts can be used to predict growth rate in a variety of both chemostat and nonchemostat conditions (Airoldi et al., 2009), the extent to which any individual transcript responds to growth rate may differ between dynamic conditions and steady-state conditions. To test this hypothesis, we compared the growth rate response of each transcript in steady-state nitrogen-limited chemostats in which growth rate is experimentally controlled with the response in our perturbation experiments of each transcript to the computationally estimated instantaneous growth rate. In this case, “response” is defined as the slope of the regression of gene expression against the measured or predicted growth rate (Supplemental Table S3). This approach provides a means of directly comparing gene expression differences between steady-state and dynamic conditions. We restricted this analysis to the small glutamine pulse, as this experiment exhibited the strongest transcriptional response without apparent physiological remodeling.
We found that the response of most genes to changes in steady-state growth rates compared with their response to computationally predicted growth rates in dynamic conditions is largely uncorrelated, although transcripts that exhibit a strong growth rate response (i.e., a strongly negative or strongly positive slope) do so in both dynamic and steady-state conditions (Figure 5A). Nonetheless, we identified several sets of functionally related transcripts that show coordinated but distinct behaviors with respect to growth rate in the two conditions. In particular, the response of transcripts belonging to the RP and RiBi regulons exhibited a much stronger positive response in dynamic than in steady-state conditions. Conversely, transcripts annotated as responding to stress exhibited a much stronger negative response to growth rate in steady-state than in dynamic conditions (Supplemental Figure S6). These results suggest that the transition from a nitrogen-limited to a nonlimited condition results in an increased expression of the RP and RiBi regulons relative to their expression in steady-state nitrogen-limited environments. This pulse of overproduction of transcripts may facilitate accelerated production of the corresponding protein products, as proposed for stress-responsive transcripts (Lee et al., 2011).
FIGURE 5:

Growth rate response of gene expression in steady-state and dynamic conditions. (A) We estimated the growth rate response of each transcript in steady-state and dynamic conditions by linear regression. In dynamic conditions in which nitrogen limitation is transiently relieved, the mRNAs belonging to the RiBi and RP regulons exhibit a growth rate response that exceeds that observed in steady-state chemostats. High temporal resolution shows a reciprocal relationship between expression of the RiBi and RP regulons, which increase in expression, and the NCR-A and NCR-P regulons, which decrease in expression, in response to a (B) 40 μM glutamine pulse and a (C) 80 μM proline pulse. Time points (in seconds) at which samples were obtained and analyzed are indicated on the x-axis.
To obtain a high-resolution view of mRNA abundance changes during the first 10 min after addition of nitrogen, when changes in gene expression are maximal (Figure 4), we repeated the pulse experiments and assayed global gene expression at 1–2 min intervals after the addition of 40 μM glutamine or 80 μM proline. We observed a rapid increase in expression of the RiBi and RP regulons in response to a pulse of glutamine, with a concomitant rapid decrease in expression of the NCR-A and NCR-P regulons (Figure 5B). Consistent with our initial observation, we observed a similar response to a pulse of proline (Figure 5C).
Accelerated degradation of mRNAs contributes to remodeling of the transcriptome
The majority of NCR transcripts are strongly repressed in response to a nitrogen pulse (Figure 4). If gene expression is repressed at the promoters of these genes and mRNA synthesis ceases, the decrease in mRNA abundance is expected to be a function of the degradation rate of the corresponding mRNA. Using our high-density time-series data, we estimated the rate of change in abundance for all transcripts, assuming a first-order exponential degradation model (Materials and Methods; Supplemental Table S7), which is the standard method for estimating mRNA degradation rates (Wang et al., 2002; Grigull et al., 2004). We found that in response to a glutamine pulse, 269 genes fit a first-order exponential decay model (FDR < 0.05; Supplemental Table S4), whereas 458 transcripts fit a first-order exponential decay model in response to the proline pulse (Supplemental Table S4).
We compared the half-lives of rapidly degraded transcripts after the glutamine pulse with half-life estimates in steady-state conditions determined using RATE-seq (Neymotin et al., 2014). We found that some transcripts decay significantly faster than expected, suggesting that their degradation rate is accelerated in response to the glutamine pulse (Figure 6A). Batch culture growth in proline also results in derepression of the NCR regulon (Godard et al., 2007). To test whether accelerated mRNA decay is specifically a response to the nitrogen-limited conditions of a chemostat, we added a pulse of glutamine to cells growing in batch cultures containing proline as a sole nitrogen source and measured genome-wide gene expression (Supplemental Table S7). The half-lives of transcripts that exhibit an exponential decrease is similar in chemostat and batch cultures (Supplemental Figure S7B), and many of the same transcripts show evidence of accelerated degradation rates in batch cultures (Figure 6B and Supplemental Table S4). Strikingly, the five nitrogen permease genes GAP1, DIP5, MEP2, PUT4, and DAL5 are the most rapidly cleared mRNAs in both the chemostat and batch culture experiments.
FIGURE 6:

Accelerated mRNA degradation contributes to gene expression remodeling. Upon addition of glutamine to NCR-derepressed cells, a subset of transcripts degrade more rapidly than their steady-state degradation rate both (A) in cells grown in ammonia-limited chemostats and (B) in cells growing in proline media in batch cultures. All points are genes that fit a model of exponential decrease in abundance (FDR < 0.05). Orange points are NCR genes that show significant accelerated degradation, blue points are NCR genes that are not significant, green points are non-NCR genes that show significantly accelerated degradation, and gray points are genes that are neither accelerated nor NCR. The dashed line denotes equal degradation rates in both conditions (i.e., slope equal to 1). Names of nitrogen transporter genes are displayed. We measured the transient changes in the degradation rates of (C) GAP1 and (D) DIP5 mRNA using a pulse-chase experiment. Cells were grown for 24 h in the presence of 4-thiouracil, which was chased at t = 0 min by the addition of excess uracil. At t = 13 min, we added either glutamine in water (orange) or equal volume of water (blue). We extracted and quantified the abundance of 4-thiouracil–labeled mRNA relative to a thiolated external spike-in using qPCR. We found significant acceleration of degradation for both GAP1 and DIP5 mRNAs (p < 0.001). Points are the mean of triplicate qPCR measurements, error bars are the propagated SD of transcript and spike-in measurements, and dotted lines are the log-linear model fit.
To verify that the addition of glutamine stimulates accelerated degradation of specific NCR transcripts, we performed pulse-chase experiments using the metabolic label 4-thiouracil (4-tU). After several generations of batch culture growth in proline medium in the presence of 4-tU to allow complete labeling of mRNAs, we added unlabeled uracil to the culture. We allowed the chase to occur for 13 min and then added either glutamine or water (mock) to the cells. We purified labeled transcripts and analyzed GAP1 and DIP5 mRNAs using quantitative PCR (qPCR) and normalization to external spike-ins. Consistent with our genome-wide assay, the addition of glutamine results in a clear accelerated degradation of both GAP1 mRNA (Figure 6C) and DIP5 mRNA (Figure 6D), confirming that the transition from NCR-derepressed to NCR-repressed conditions results in the accelerated degradation of some transcripts.
The NCR and RP/RiBi regulons show reciprocal abundance relationships
The rapid induction kinetics of the RP and RiBi regulons and the corresponding rapid repression of the NCR-A and NCR-P regulons in response to increases in nitrogen are consistent with reciprocal regulation of these regulons (Cardenas et al., 1999). Whereas the DNA microarrays used in our experiments provide a comparative measure of gene expression, RNA sequencing (RNA-seq) allows estimation of the fraction of the transcriptome represented by sets of coregulated mRNAs. We used RNA-seq (Supplemental Table S5) to compare the apportionment of the transcriptome in NCR-depressing conditions with the transcriptome in NCR-repressed conditions (Figure 7). In NCR-repressed conditions, the RP and RiBi regulons comprise 28.4% of the transcriptome, whereas the NCR-A and NCR-P regulons comprise just 0.9% of the transcriptome. Conversely, in NCR-derepressed conditions, the NCR-A and NCR-P regulons expand ninefold to comprise 8.8% of the transcriptome, and the RP and RiBi regulons shrink to constitute just 7.6% of the transcriptome. Thus, remodeling of gene expression in nitrogen-limited cells provided with a sudden bolus of nitrogen requires both a fourfold expansion of the RP/RiBi regulons and a ninefold reduction in the NCR regulon. We propose that the transition from NCR-derepressed to NCR-repressed conditions requires accelerated degradation of highly expressed NCR transcripts to liberate ribosomes for reallocation to RP and RiBi transcripts to facilitate rapid growth.
FIGURE 7:

Transcriptome allocation is dramatically altered in response to nitrogen availability. RP and RiBi transcripts represent 28.4% of the transcriptome and NCR transcripts comprise just 0.9% of the transcriptome in NCR-repressing conditions (growth in yeast extract/peptone/dextrose media; data from Waern and Snyder, 2013). In NCR-derepressing conditions (minimal media depleted of nitrogen), the RP and RiBi transcripts comprise 7.6% of the transcriptome and NCR mRNAs comprise 8.8%.
DISCUSSION
The molecular form of environmental nitrogen is a central determinant of cell growth rate and differential gene expression (Cooper, 1982; Magasanik and Kaiser, 2002). Although different nitrogen sources support different growth rates, we found that yeast cells convert different nitrogen sources into biomass and total cell number with similar yields. To study the effect of different nitrogen sources on gene expression in nitrogen-limiting conditions, we used chemostats (Monod, 1950; Novick and Szilard, 1950), which uniquely allow experimental control of cell growth rate independent of media composition. On the basis of our reanalysis of the data reported in Brauer et al. (2008), we find that the NCR regulon is derepressed in ammonium-limited conditions, whereas the SPS regulon is repressed. By contrast, the GAAC and UPR regulons are not differentially expressed in ammonium-limited chemostats compared with other nutrient-limited conditions.
Using chemostats limited for six different nitrogen sources grown at matched growth rates, we found that the expression of only 38 genes depends on nitrogen source. Of note, of the 75 NCR transcripts that we analyzed, only 10 were significantly affected by variation in nitrogen source in nitrogen-limiting conditions. Many of the NCR and non-NCR transcripts that respond to variation in nitrogen source in nitrogen-limiting conditions respond to one (e.g., CAR1, ARG5,6, ARG3, and CAR2 in arginine-limited conditions and PUT2, PUT4, PUT1, and GDH2 in proline-limited conditions) or two (DAL4, DAL7, DAL2, and DAL1 in urea- and allantoin-limited conditions) specific nitrogen sources and encode enzymes and transporters required for assimilation of specific nitrogen sources.
Our results support a model in which the NCR regulon is primarily activated by limitation for nitrogen, with finer-scale regulatory control exerted in response to the specific molecular form of environmental nitrogen. An important implication of this result is that the signal leading to regulation of the NCR regulon must be downstream of the environmental source of nitrogen. However, it remains to be determined how intracellular levels of nitrogen are sensed. Our results also suggest that mechanisms must exist by which a specific nitrogen source can affect the abundance of transcripts specifically required for their metabolism. One known mechanism is the direct interaction of proline with the PUT3 transcription factor (Axelrod et al., 1991; Sellick and Reece, 2003), which activates expression of transcripts required for proline metabolism. Additional feedback mechanisms must exist for controlling the expression of transcripts specific for use of arginine (e.g., CAR1, ARG5,6, ARG3, and CAR2) and allantoin (e.g., DAL4, DAL7, DAL, and DAL1), and in some cases, these have been proposed (Messenguy and Dubois, 1983). Identifying the molecular bases of these responses is important for understanding the mechanisms by which specific metabolic pathways affect gene expression regulation.
To study the dynamics with which cells respond to an instantaneous increase in environmental nitrogen, we performed transient perturbation experiments. In contrast to the response predicted by mathematical modeling of the chemostat, a sudden excess of nitrogen does not result in an increase in cell number or culture density. In the most dramatic case, a 10-fold molar increase in nitrogen in the form of glutamine led to a decrease in culture density. We suggest that this discord between model and data is due to a halt in the cell growth that is required for physiological remodeling that is not accounted for in the model. A pause in cell growth before physiological adjustment and reinitiation of growth at a new growth rate has been observed in up-shift experiments in microbes (Kjeldgaard et al., 1958; Ludwig et al., 1977; Waldron, 1977; Kief and Warner, 1981). In budding yeast, environmental nutrients and growth rate affect the critical cell size required for cell division (Johnston et al., 1979; Lorincz and Carter, 1979; Tyson et al., 1979). In the case of our large pulses, we were able to detect an increase in cell size. The remodeling of cell physiology, resetting of the critical cell size, and changes in growth rate may be interconnected phenomena (Soifer and Barkai, 2014).
Computational prediction of the instantaneous growth rate using global gene expression is consistent with a rapid and dose-dependent increase in cell growth rate when nitrogen-limited cells are provided with additional nitrogen. The rapid effect on growth rate–related gene expression is consistent with transcriptional regulation downstream of signaling pathways that transmit information about the growth potential of the extracellular environment. Previously, we reported that the Ras/PKA pathway is involved in transmitting this signal (Airoldi et al., 2009). Because nitrogen signaling may generally be regarded as being transmitted through the TORC1 pathway, our results suggest that TORC1 and Ras/PKA signaling converge on growth rate–regulated gene expression. This is consistent with the observation that TORC1 is both an upstream regulator of Ras/PKA and acts in parallel to Ras/PKA to converge on common targets (Martin et al., 2004; Soulard et al., 2010).
We developed a novel means of normalizing between different experimental conditions by comparing the growth rate response, defined as the slope of the linear regression against growth rate, of each transcript using either measured or computationally predicted growth rate. We identified several transcripts that exhibit distinct responses to growth rate in steady-state and dynamic conditions, many of which fall within distinct functional classes. Of note, the RP and RiBi regulons show an amplified response to growth rate in dynamic conditions as compared with steady-state chemostats, suggesting that when a cell transitions from a growth-limited to a growth-promoting environment, it undergoes a burst of expression in transcripts required for protein translation. The fact that increased RP and RiBi transcription inhibits passing START and committing to the cell cycle (Jorgensen et al., 2002) is consistent with our observation that cell size, but not cell number, increases in the chemostat upon addition of excess nitrogen to nitrogen-limited cells.
In parallel to the rapid induction of transcripts required for protein translation, we detect a rapid repression of NCR transcripts. Both NCR-A and NCR-P transcripts rapidly decrease in abundance in response to addition of either proline or glutamine and exhibit a dose-dependent response. The fact that NCR transcripts are repressed in response to addition of both glutamine and proline provides further support for the argument that the ultimate NCR signal must be downstream of the environmental source of nitrogen. It has been shown that TORC1-mediated regulation of one of the activators of NCR expression, GLN3, depends on intracellular glutamine (Crespo et al., 2002). Our results are not inconsistent with this model, as proline is metabolized to glutamate in the cell, which is subsequently converted to glutamine, which may elicit a TORC1-mediated response. However, intracellular glutamine does not explain all TORC1-mediated transcriptional responses (Crespo et al., 2002; Loewith and Hall, 2011). Testing the role of glutamine sensing by TORC1 under these conditions will be important for understanding the mechanism underlying coordinated regulation of transcription required for cell growth and nitrogen catabolism.
Consistent with repression of the SPS regulon in steady-state nitrogen-limiting conditions, we observed its activation upon addition of both proline and glutamine to nitrogen-limited cells, which may reflect TORC1-dependent stabilization of STP1 (Shin et al., 2009). Surprisingly, we also observed activation of the GAAC regulon. Because addition of nitrogen is expected to lead to increased TORC1 activity, we expected to observe decreased expression of GCN4 targets as a result of TORC1 phosphorylation of GCN2. The observed activation of GCN4 targets may reflect different time scales for transcriptional regulation by GCN4 regulation and translational regulation of GCN4.
Some mRNAs are rapidly degraded when cells transition from NCR-activating to NCR-repressing conditions in both chemostats and batch culture. Comparison with mRNA degradation rates suggests that the degradation of some of these transcripts is accelerated. Using in vivo metabolic labeling with 4-tU, we provide additional evidence that the addition of glutamine to nitrogen-limited cells accelerates the degradation of specific transcripts. A previous study of the transcriptional response to glucose addition in carbon-limited chemostats suggested a role for accelerated degradation of mRNAs (Kresnowati et al., 2006), and there is increasing evidence that mRNA stability plays an important role in regulating gene expression programs (Puig et al., 2005; Bennett et al., 2008; Baumgartner et al., 2011). Consistent with a posttranscriptional mechanism underlying the rapid clearing of some NCR transcripts, previous work showed that GAP1 mRNA transiently decreases in abundance during a nitrogen up-shift in the absence of URE2 (ter Schure et al., 1998), which is required for NCR repression by sequestering GLN3 in the cytoplasm. Several studies have shown that TORC1 can affect transcript stability (Albig and Decker, 2001; Munchel et al., 2011). Our results suggest that posttranscriptional regulation of mRNA stability may play an important role in remodeling gene expression in response to changes in environmental nitrogen. Transient stabilization of the RP and RiBi regulons also could contribute to their rapid increase in expression (Yin et al., 2003). Defining the role of regulated changes in mRNA stability in dynamic conditions is an important area for further study.
What is the underlying rationale for rapid induction of RP/RiBi transcripts occurring in parallel with accelerated degradation of NCR transcripts? We propose that accelerated degradation of NCR transcripts may allow for reallocation of ribosomes to transcripts required for growth and proliferation (Kief and Warner, 1981; Lee et al., 2011). Our observations are consistent with a model in which TORC1 orchestrates the balance between transcripts required for protein production and transcripts required for the acquisition and assimilation of nitrogen. When nitrogen is abundant, TORC1 activates the expression of the RP and RiBi regulons while actively repressing the NCR-A and NCR-P regulons. Conversely, when nitrogen levels are in growth-limiting concentrations, TORC1 activity decreases, leading to reduced activation of the RP and RiBi regulons and derepression of the NCR-A and NCR-P regulons. In NCR-derepressing conditions, NCR transcripts, including GAP1, MEP2, and PUT4, are the most abundant transcripts (Supplemental Table S5). When a cell encounters a sudden increase in environmental nitrogen, some highly expressed transcripts may be targeted for accelerated degradation to increase the pool of free ribosomes facilitating rapid translation of newly transcribed RiBi and RP transcripts, thereby accelerating physiological remodeling of the cell for rapid growth.
MATERIALS AND METHODS
Strains and culturing conditions
We used the prototrophic haploid strain FY4 (MATa), which is isogenic to the S288c reference strain, for all experiments. We used minimal defined media for all experiments, using a common base medium for nitrogen limitation, as described previously (Brauer et al., 2008; Boer et al., 2010). The appropriate concentrations of allantoin, glutamine, glutamate, urea, ammonium sulfate, proline, and arginine were added from 100 mM stock. Batch culture experiments were performed in 30°C shaking incubators using 100-ml cultures. Continuous culturing in chemostats using Sixfors bioreactors (Infors, Laurel, MD) was performed as described (Brauer et al., 2008; Boer et al., 2010) using a 300-ml working volume. Culture parameters were determined using either a Klett colorimeter or a Coulter counter after sonication. For perturbation studies, a single bolus of proline, glutamine, or a mix of both was added to the chemostat to a final concentration of 80 or 800 μM nitrogen.
RNA analysis
Cell samples for mRNA analysis were preserved by rapid filtration and quick freezing using liquid nitrogen. We isolated total RNA using hot acid–phenol extraction and subsequently purified RNA samples using RNeasy columns. We performed gene expression profiling using Agilent (Santa Clara, CA) 60-mer DNA microarrays and Cy3 and Cy5 incorporation as previously described (Brauer et al., 2008). We used a common reference obtained from a sample growing in an ammonium sulfate–limited chemostat at a dilution rate of 0.12 h−1 for all hybridization experiments and hybridized labeled cRNA to Agilent Yeast DNA microarrays for 20 h at 65°C. We washed arrays and scanned microarrays using an Agilent two-color scanner and extracted hybridization signals using Agilent Feature Extractor Software. Supplemental Table S6 gives the entire data set of processed log2 ratios.
Pulse chase
Cells were grown in 600 ml of minimal medium containing 800 μM proline, 500 μM uracil, and 500 μM 4-thiouracil at 30°C for 24 h. The culture was divided into two 300-ml cultures, and uracil was added to a final concentration of 2 mM. We acquired 20-ml samples after the chase using rapid filtration and flash freezing in liquid nitrogen. At 13 min after starting the chase, we added either glutamine to a final concentration of 400 μM or an equal volume of water and acquired additional samples.
After RNA extraction, samples were mixed with an in vitro–transcribed thiolated spike-in (BAC1200) at a ratio of 1 ng of spike-in to 25 μg of total RNA and reacted with EZ-Link HPDP-Biotin (ThermoFisher Scientific, Waltham, MA) at 2 mg/ml for 200 min. Reactions were cleaned up by centrifugation and ethanol precipitation and then conjugated with 180 μl of streptavidin magnetic beads (M0253L; NEB, Ipswich, MA). Labeled RNA was eluted using 5% β-mercaptoethanol.
Samples were reverse transcribed with Moloney murine leukemia virus reverse transcriptase (NEB) and random hexamer priming. We performed qPCR in technical triplicate on a LightCycler 480 (Roche, Branchburg, NJ) using the following primers: 5′-ACGGTATCAAGGGTTTGCCAAG-3′ and 5′-GCATAAATGGCAGAGTTAC-3′ for GAP1, 5′-TGGCGTACATGAATGTGTCTTCA-3′ and 5′-GGTGATCCAACTCAAGATTC-3′ for DIP5, and 5′-CTGGACGACTTCGACTACGG-3′ and 5′-ATCAGCCTTTCCTTTCGTCA-3′ for the BAC1200 spike-in. Cp values were calculated for each sample and the spike-in and log-linear regression performed using the ratio of either GAP1 mRNA or DIP5 mRNA to the spike-in in R.
Estimation of gene expression response to growth rate
We estimated the response of each gene to growth rate by performing a linear regression of gene expression against growth rate as in Brauer et al. (2008). In brief, we modeled the level of gene expression Xij for gene i in condition j growing at rate Rj using the linear model
|
|
in which αij is the intercept (i.e., average expression), βi is the slope of the regression (i.e., effect of growth rate on expression), Rj is the growth rate (i.e., the actual chemostat dilution rate), and εij is the error. To estimate the significance of the growth rate response βi, we bootstrapped the data 100,000 times, creating 100,000 artificial gene expression profiles. We fitted the linear regression in Eq. 1 to the artificial expression profiles and used the resulting distribution of growth rate coefficients estimated on these artificial expression profiles as the null distribution to determine the significance of each regression coefficient, βi, obtained for the observed expression profiles. We used a two-sided threshold of FDR-corrected p values at 0.05 to identify genes that are significant.
Estimation of gene expression response to nutrient source and growth rate using analysis of covariance
We searched for genes differing in growth rate response in at least one nutrient by performing an ANCOVA, with nutrient source as a factor and growth rate as a covariate. In brief, we model the level of gene expression Xij for gene i in array j growing at rate Rj with nutrient limitation k using the linear model
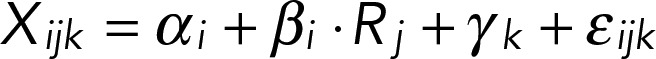 (2) (2)
|
in which αi is the intercept (i.e., average expression in the absence of growth and nutrient-specific effects), βi is the slope of the regression (i.e., effect of growth rate on expression), Rj is the actual chemostat dilution rate, γk is a nutrient-specific shift in expression due to limiting nutrient k, and εijk is the error.
To estimate the significance of the nutrient source–specific response, γk, we bootstrapped the data 100,000 times. We fitted the linear regression in Eq. 2 to the artificial expression profiles and used the resulting distribution of nutrient source–specific coefficients estimated as the null distribution to determine the significance of each regression coefficient, γk, obtained for the observed expression profiles. We used a two-sided threshold of FDR-corrected p values at 0.05 to identify genes that are significant. We then performed ANCOVA to compare the fit of the models in Eqs. 1 and 2 and confirm that indeed a nutrient source–specific effect was present for those genes that were found to have a significant response to a nutrient source using the bootstrap analysis.
Mathematical modeling of chemostat growth
We modeled the population growth rate (dx/dt) and the rate of change in the limiting nutrient concentration (ds/dt) using the coupled system of ordinary differential equations (ODEs)
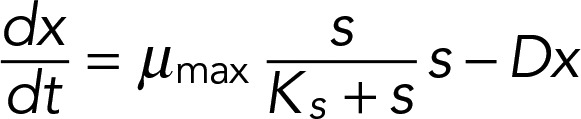 |
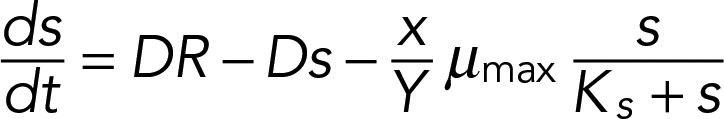 |
where the parameters are R, the concentration of the limiting nutrient in the medium, and Y, the culture yield per mole of the limiting nutrient. D is the dilution rate of the culture. Monod (1949) proposed that growth rate (μ) is related to the concentration ([s]) of the limiting nutrient with saturating kinetics as described by the relationship
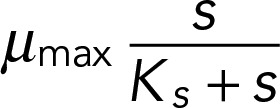 |
where Ks is the substrate concentration at half-maximal μ. ODEs were solved using XXPAUT (www.math.pitt.edu/∼bard/xpp/xpp.html). Refer to the Supplemental Methods for details of parameter estimation and modeling.
mRNA decay estimation
We estimated rates of mRNA decay for all transcripts using high–temporal resolution data. We used ratios (yt) of hybridization intensities for each transcript obtained from two-color DNA microarrays cohybridized with a common reference. Data were normalized to the initial data point (y0) and then log-transformed. We modeled the degradation rate kdeg of each gene:
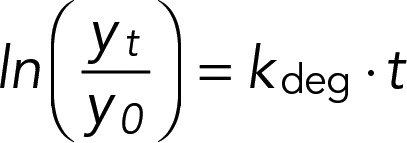 |
where t is the sampling time in minutes. Transcript half-lives were computed as ln(2)/kdeg. Accelerated degradation was assessed by fitting the model
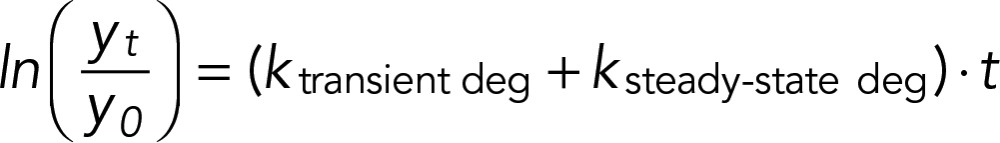 |
where ksteady-state deg is the specific degradation rate for transcript i as reported in Neymotin et al. (2014). For all linear modeling, we assessed statistical significance of coefficients using a t statistic and determined empirical p values by permuting data for each gene 1000 times. The false discovery rate was determined using the qvalue package in R.
RNA-seq analysis
We prepared RNA from replicate cultures growing in minimal medium containing glutamine 24 h after inoculation, at which point nitrogen is depleted from the media (NCR-derepressing conditions). Total RNA, fragmented to an average molecular weight of 350 nucleotides, was depleted of rRNA using RiboMinus (ThermoFisher Scientific). The efficiency of rRNA depletion was assessed with Bioanalyzer (Agilent Technologies). First-strand synthesis of RNA was performed using the Super Script III kit (ThermoFisher Scientific) and random hexamer/poly dT priming. Second-strand synthesis was performed with dUTP in place of dTTP to enable strand-specific sequencing. Samples were end repaired, A tailed, and ligated to adaptors (BIOO Scientific, Austin, TX) containing DNA Barcodes for multiplex sequencing. Adapter dimers were removed using AMPure beads (Beckman Coulter, Indianapolis, IN). Samples were treated with UNG (uracil-DNA glycosylase) and amplified using 12 cycles of PCR before sequencing. Samples were sequenced in 6-plex using an Illumina 2000 paired-end 50–base pair run.
Sequence reads were filtered for rRNA sequences by aligning to the ribosomal DNA of the yeast genome using Bowtie with default settings. All remaining reads were then aligned to the rest of the yeast genome using Bowtie2 and TopHat. The combined BAM files were filtered to remove PCR duplicates and alignment quality scores (MAPQ) of <20. We calculated FPKMs (fragments per kilobase of transcript per million mapped reads) for each gene using Cufflinks (Trapnell et al., 2010).
Definition of regulons
We used nitrogen-regulated gene sets defined in Godard et al. (2007) and RP and RiBi membership defined in Jorgensen (2004).
Data availability
DNA microarray data are available through gene expression omnibus (GEO) GSE57293. RNA-seq data are available through sequence read archive (SRA) PRJNA246199.
Supplementary Material
Acknowledgments
We thank members of the Gresham and Airoldi labs, Birgitte Regenberg, Arthur Kruckeberg, and David Botstein for helpful comments. We thank Alexandra Ward for technical assistance. This work was supported by grants from the National Science Foundation (MCB-1244219 to D.G.) and the National Institutes of Health (R01-GM107466 to D.G., R01-GM096193 to E.M.A., and RO1-GM046406 to D.B.), a DuPont Young Professor Award (to D.G.), and an Alfred P. Sloan Research Fellowship (to E.M.A).
Abbreviations used:
- GAAC
general amino acid control
- NCR
nitrogen catabolite repression
- RiBi
ribosome biogenesis
- RP
ribosomal protein
- SPS
SSY1-PTR3-SSY5
- TORC1
TOR complex 1
- UPR
unfolded protein response.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E14-05-1013) on March 3, 2016.
REFERENCES
- Airoldi EM, Huttenhower C, Gresham D, Lu C, Caudy AA, Dunham MJ, Broach JR, Botstein D, Troyanskaya OG. Predicting cellular growth from gene expression signatures. PLoS Comput Biol. 2009;5:e1000257. doi: 10.1371/journal.pcbi.1000257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albig ARA, Decker CJC. The target of rapamycin signaling pathway regulates mRNA turnover in the yeast Saccharomyces cerevisiae. Mol Biol Cell. 2001;12:3428–3438. doi: 10.1091/mbc.12.11.3428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Axelrod JD, Majors J, Brandriss MC. Proline-independent binding of PUT3 transcriptional activator protein detected by footprinting in vivo. Mol Cell Biol. 1991;11:564–567. doi: 10.1128/mcb.11.1.564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumgartner BL, Bennett MR, Ferry M, Johnson TL, Tsimring LS, Hasty J. Antagonistic gene transcripts regulate adaptation to new growth environments. Proc Natl Acad Sci USA. 2011;108:21087–21092. doi: 10.1073/pnas.1111408109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck T, Hall MN. The TOR signalling pathway controls nuclear localization of nutrient-regulated transcription factors. Nature. 1999;402:689–692. doi: 10.1038/45287. [DOI] [PubMed] [Google Scholar]
- Bennett MR, Pang WL, Ostroff NA, Baumgartner BL, Nayak S, Tsimring LS, Hasty J. Metabolic gene regulation in a dynamically changing environment. Nature. 2008;454:1119–1122. doi: 10.1038/nature07211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boer VM, Crutchfield CA, Bradley PH, Botstein D, Rabinowitz JD. Growth-limiting intracellular metabolites in yeast growing under diverse nutrient limitations. Mol Biol Cell. 2010;21:198–211. doi: 10.1091/mbc.E09-07-0597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boer VM, de Winde JH, Pronk JT, Piper MDW. The genome-wide transcriptional responses of Saccharomyces cerevisiae grown on glucose in aerobic chemostat cultures limited for carbon, nitrogen, phosphorus, or sulfur. J Biol Chem. 2003;278:3265–3274. doi: 10.1074/jbc.M209759200. [DOI] [PubMed] [Google Scholar]
- Brauer MJ, Huttenhower C, Airoldi EM, Rosenstein R, Matese JC, Gresham D, Boer VM, Troyanskaya OG, Botstein D. Coordination of growth rate, cell cycle, stress response, and metabolic activity in yeast. Mol Biol Cell. 2008;19:352–367. doi: 10.1091/mbc.E07-08-0779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardenas ME, Cutler NS, Lorenz MC, Di Como CJ, Heitman J. The TOR signaling cascade regulates gene expression in response to nutrients. Genes Dev. 1999;13:3271–3279. doi: 10.1101/gad.13.24.3271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castrillo JI, Zeef LA, Hoyle DC, Zhang Z, Hayes A, Gardner DCJ, Cornell MJ, Petty J, Hakes L, Wardleworth L, et al. Growth control of the eukaryote cell: a systems biology study in yeast. J Biol. 2007;6:4. doi: 10.1186/jbiol54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper TG. Nitrogen metabolism in Saccharomyces cerevisiae. In: Strathern JN, editor. The Molecular Biology of the Yeast Saccharomyces: Metabolism and Gene Expression. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1982. pp. 39–99. [Google Scholar]
- Cooper TG. Transmitting the signal of excess nitrogen in Saccharomyces cerevisiae from the Tor proteins to the GATA factors: connecting the dots. FEMS Microbiol Rev. 2002;26:223–238. doi: 10.1111/j.1574-6976.2002.tb00612.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crespo JL, Powers T, Fowler B, Hall MN. The TOR-controlled transcription activators GLN3, RTG1, and RTG3 are regulated in response to intracellular levels of glutamine. Proc Natl Acad Sci USA. 2002;99:6784–6789. doi: 10.1073/pnas.102687599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godard P, Urrestarazu A, Vissers S, Kontos K, Bontempi G, van Helden J, André B. Effect of 21 different nitrogen sources on global gene expression in the yeast Saccharomyces cerevisiae. Mol Cell Biol. 2007;27:3065–3086. doi: 10.1128/MCB.01084-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigull J, Mnaimneh S, Pootoolal J, Robinson MD, Hughes TR. Genome-wide analysis of mRNA stability using transcription inhibitors and microarrays reveals posttranscriptional control of ribosome biogenesis factors. Mol Cell Biol. 2004;24:5534–5547. doi: 10.1128/MCB.24.12.5534-5547.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall MN, Raff MC, Thomas G. Cell Growth: Control of Cell Size. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 2004. [Google Scholar]
- Hinnebusch AG. Translational regulation of GCN4 and the general amino acid control of yeast. Annu Rev Microbiol. 2005;59:407–450. doi: 10.1146/annurev.micro.59.031805.133833. [DOI] [PubMed] [Google Scholar]
- Ingraham JL, Maaløe O, Neidhardt FC. Growth of the Bacterial Cell. Sunderland, MA: Sinauer; 1983. [Google Scholar]
- Ishii N, Nakahigashi K, Baba T, Robert M, Soga T, Kanai A, Hirasawa T, Naba M, Hirai K, Hoque A, et al. Multiple high-throughput analyses monitor the response of E. coli to perturbations. Science. 2007;316:593–597. doi: 10.1126/science.1132067. [DOI] [PubMed] [Google Scholar]
- Johnston GC, Ehrhardt CW, Lorincz A, Carter BL. Regulation of cell size in the yeast Saccharomyces cerevisiae. J Bacteriol. 1979;137:1–5. doi: 10.1128/jb.137.1.1-5.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen P. A dynamic transcriptional network communicates growth potential to ribosome synthesis and critical cell size. Genes Dev. 2004;18:2491–2505. doi: 10.1101/gad.1228804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen P, Nishikawa JL, Breitkreutz B-J, Tyers M. Systematic identification of pathways that couple cell growth and division in yeast. Science. 2002;297:395–400. doi: 10.1126/science.1070850. [DOI] [PubMed] [Google Scholar]
- Kief DR, Warner JR. Coordinate control of syntheses of ribosomal ribonucleic acid and ribosomal proteins during nutritional shift-up in Saccharomyces cerevisiae. Mol Cell Biol. 1981;1:1007–1015. doi: 10.1128/mcb.1.11.1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kjeldgaard NO, Maaloe O, Schaechter M. The transition between different physiological states during balanced growth of Salmonella typhimurium. J Gen Microbiol. 1958;19:607–616. doi: 10.1099/00221287-19-3-607. [DOI] [PubMed] [Google Scholar]
- Klumpp S, Zhang Z, Hwa T. Growth rate-dependent global effects on gene expression in bacteria. Cell. 2009;139:1366–1375. doi: 10.1016/j.cell.2009.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kresnowati MTAP, van Winden WA, Almering MJH, Pierick ten A, Ras C, Knijnenburg TA, Daran-Lapujade P, Pronk JT, Heijnen JJ, Daran JM. When transcriptome meets metabolome: fast cellular responses of yeast to sudden relief of glucose limitation. Mol Syst Biol. 2006;2:49. doi: 10.1038/msb4100083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubitschek HE. Introduction to Research with Continuous Cultures. Englewood Cliffs, NJ: Prentice Hall; 1970. [Google Scholar]
- Lee MV, Topper SE, Hubler SL, Hose J, Wenger CD, Coon JJ, Gasch AP. A dynamic model of proteome changes reveals new roles for transcript alteration in yeast. Mol Syst Biol. 2011;7:514. doi: 10.1038/msb.2011.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loewith R, Hall MN. Target of rapamycin (TOR) in nutrient signaling and growth Control. Genetics. 2011;189:1177–1201. doi: 10.1534/genetics.111.133363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorincz A, Carter BLA. Control of cell size at bud initiation in Saccharomyces cerevisiae. J Gen Microbiol. 1979;113:287–295. [Google Scholar]
- Ludwig JR, II, Oliver SG, McLaughlin CS. The regulation of RNA synthesis in yeast II: amino acids shift-up experiments. Mol Gen Genet. 1977;158:117–122. doi: 10.1007/BF00268303. [DOI] [PubMed] [Google Scholar]
- Magasanik B, Kaiser CA. Nitrogen regulation in Saccharomyces cerevisiae. Gene. 2002;290:1–18. doi: 10.1016/s0378-1119(02)00558-9. [DOI] [PubMed] [Google Scholar]
- Martin D, Soulard A, Hall M. TOR regulates ribosomal protein gene expression via PKA and the forkhead transcription factor FHL1. Cell. 2004;119:969–979. doi: 10.1016/j.cell.2004.11.047. [DOI] [PubMed] [Google Scholar]
- Messenguy F, Dubois E. Participation of transcriptional and post-transcriptional regulatory mechanisms in the control of arginine metabolism in yeast. Mol Gen Genet. 1983;189:148–156. doi: 10.1007/BF00326068. [DOI] [PubMed] [Google Scholar]
- Monod J. The growth of bacterial cultures. Annu Rev Microbiol. 1949;3:371–394. [Google Scholar]
- Monod J. La technique de culture continue: théorie et applications. Ann Inst Pasteur. 1950:390–410. [Google Scholar]
- Munchel SE, Shultzaberger RK, Takizawa N, Weis K. Dynamic profiling of mRNA turnover reveals gene-specific and system-wide regulation of mRNA decay. Mol Biol Cell. 2011;22:2787–2795. doi: 10.1091/mbc.E11-01-0028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neymotin B, Athanasiadou R, Gresham D. Determination of in vivo RNA kinetics using RATE-seq. RNA. 2014;20:1645–1652. doi: 10.1261/rna.045104.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niederberger P, Miozzari G, Hütter R. Biological role of the general control of amino acid biosynthesis in Saccharomyces cerevisiae. Mol Cell Biol. 1981;1:584–593. doi: 10.1128/mcb.1.7.584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novick A, Szilard L. Description of the chemostat. Science. 1950;112:715–716. doi: 10.1126/science.112.2920.715. [DOI] [PubMed] [Google Scholar]
- Puig S, Askeland E, Thiele DJ. Coordinated remodeling of cellular metabolism during iron deficiency through targeted mRNA degradation. Cell. 2005;120:99–110. doi: 10.1016/j.cell.2004.11.032. [DOI] [PubMed] [Google Scholar]
- Regenberg B, Grotkjaer T, Winther O, Fausbøll A, Akesson M, Bro C, Hansen LK, Brunak S, Nielsen J. Growth-rate regulated genes have profound impact on interpretation of transcriptome profiling in Saccharomyces cerevisiae. Genome Biol. 2006;7:R107. doi: 10.1186/gb-2006-7-11-r107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronen M, Botstein D. Transcriptional response of steady-state yeast cultures to transient perturbations in carbon source. Proc Natl Acad Sci USA. 2006;103:389–394. doi: 10.1073/pnas.0509978103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saldanha AJ, Brauer MJ, Botstein D. Nutritional homeostasis in batch and steady-state culture of yeast. Mol Biol Cell. 2004;15:4089–4104. doi: 10.1091/mbc.E04-04-0306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaechter M, Maaloe O, Kjeldgaard NO. Dependency on medium and temperature of cell size and chemical composition during balanced grown of Salmonella typhimurium. J Gen Microbiol. 1958;19:592–606. doi: 10.1099/00221287-19-3-592. [DOI] [PubMed] [Google Scholar]
- Scott M, Gunderson CW, Mateescu EM, Zhang Z, Hwa T. Interdependence of cell growth and gene expression: origins and consequences. Science. 2010;330:1099–1102. doi: 10.1126/science.1192588. [DOI] [PubMed] [Google Scholar]
- Scott M, Klumpp S, Mateescu EM, Hwa T. Emergence of robust growth laws from optimal regulation of ribosome synthesis. Mol Syst Biol. 2014;10:747–747. doi: 10.15252/msb.20145379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellick CA, Reece RJ. Modulation of transcription factor function by an amino acid: activation of Put3p by proline. EMBO J. 2003;22:5147–5153. doi: 10.1093/emboj/cdg480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin C-S, Kim SY, Huh W-K. TORC1 controls degradation of the transcription factor Stp1, a key effector of the SPS amino-acid-sensing pathway in Saccharomyces cerevisiae. J Cell Sci. 2009;122:2089–2099. doi: 10.1242/jcs.047191. [DOI] [PubMed] [Google Scholar]
- Soifer I, Barkai N. Systematic identification of cell size regulators in budding yeast. Mol Syst Biol. 2014;10:761–761. doi: 10.15252/msb.20145345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soulard A, Cremonesi A, Moes S, Schütz F, Jenö P, Hall MN. The rapamycin-sensitive phosphoproteome reveals that TOR controls protein kinase A toward some but not all substrates. Mol Biol Cell. 2010;21:3475–3486. doi: 10.1091/mbc.E10-03-0182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ter Schure EG, Silljé HH, Vermeulen EE, Kalhorn JW, Verkleij AJ, Boonstra J, Verrips CT. Repression of nitrogen catabolic genes by ammonia and glutamine in nitrogen-limited continuous cultures of Saccharomyces cerevisiae. Microbiology. 1998;144:1451–1462. doi: 10.1099/00221287-144-5-1451. [DOI] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28:516–520. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyson CB, Lord PG, Wheals AE. Dependency of size of Saccharomyces cerevisiae cells on growth rate. J Bacteriol. 1979;138:92–98. doi: 10.1128/jb.138.1.92-98.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urban J, Soulard A, Huber A, Lippman S, Mukhopadhyay D, Deloche O, Wanke V, Anrather D, Ammerer G, Riezman H, et al. Sch9 is a major target of TORC1 in Saccharomyces cerevisiae. Mol Cell. 2007;26:663–674. doi: 10.1016/j.molcel.2007.04.020. [DOI] [PubMed] [Google Scholar]
- Usaite R, Patil KR, Grotkjaer T, Nielsen J, Regenberg B. Global transcriptional and physiological responses of Saccharomyces cerevisiae to ammonium, L-alanine, or L-glutamine limitation. Appl Environ Microbiol. 2006;72:6194–6203. doi: 10.1128/AEM.00548-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waern K, Snyder M. Extensive transcript diversity and novel upstream open reading frame regulation in yeast. G3 (Bethesda) 2013;3:343–352. doi: 10.1534/g3.112.003640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldron C. Synthesis of ribosomal and transfer ribonucleic acids in yeast during a nutritional shift-up. J Gen Microbiol. 1977;98:215–221. doi: 10.1099/00221287-98-1-215. [DOI] [PubMed] [Google Scholar]
- Wang Y, Liu CL, Storey JD, Tibshirani RJ, Herschlag D, Brown PO. Precision and functional specificity in mRNA decay. Proc Natl Acad Sci USA. 2002;99:5860–5865. doi: 10.1073/pnas.092538799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Z, Wilson S, Hauser NC, Tournu H, Hoheisel JD, Brown AJP. Glucose triggers different global responses in yeast, depending on the strength of the signal, and transiently stabilizes ribosomal protein mRNAs. Mol Microbiol. 2003;48:713–724. doi: 10.1046/j.1365-2958.2003.03478.x. [DOI] [PubMed] [Google Scholar]
- Ziv N, Siegal ML, Gresham D. Genetic and nongenetic determinants of cell growth variation assessed by high-throughput microscopy. Mol Biol Evol. 2013;30:2568–2578. doi: 10.1093/molbev/mst138. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
DNA microarray data are available through gene expression omnibus (GEO) GSE57293. RNA-seq data are available through sequence read archive (SRA) PRJNA246199.