

Abstract
Fish and human cytochrome P450 (P450) 17A1 catalyze both steroid 17α-hydroxylation and 17α,20-lyase reactions. Fish P450 17A2 catalyzes only 17α-hydroxylation. Both enzymes are microsomal-type P450s, integral membrane proteins that bind to the membrane through their N-terminal hydrophobic segment, the signal anchor sequence. The presence of this N-terminal region renders expression of full-length proteins impossible or challenging. For some proteins, variable truncation of the signal anchor sequence precludes expression or results in poor expression levels. To crystallize P450 17A1 and 17A2 in order to gain insight into their different activities, we used an alternative N-terminal sequence to boost expression together with in situ proteolysis. Key features of our approach to identify crystallizable P450 fragments were the use of an N-terminal leader sequence, a screen composed of 12 proteases to establish optimal cleavage, variations of protease concentration in combination with an SDS-PAGE assay, and analysis of the resulting fragments using Edman sequencing. Described in this unit are protocols for vector preparation, expression, purification, and in situ proteolytic crystallization of two membrane-bound P450 proteins.
INTRODUCTION
Cytochrome P450 enzymes (P450s) play crucial roles in the metabolism of endogenous and exogenous compounds (Ortiz de Montellano, 2005). Microsomal P450s also participate in the detoxification of xenobiotics and the synthesis of steroid hormones (Guengerich, 2005; Miller and Auchus, 2011). The hydrophobic N-terminal leader sequence of microsomal P450s that includes a membrane-spanning helix targets them to the endoplasmic reticulum, where the catalytic domain of about 460 amino acids is lodged on the cytoplasmic side of the membrane (Johnson and Stout, 2013). Hydrophobic patches on helix A and the connector between the F/F’ and G helices provide additional anchoring points on the membrane surface (Figure 1). Like many other membrane proteins, membrane-bound P450s remain challenging targets for recombinant expression and crystallization. Thus, Escherichia coli (E. coli) that continues to be the most commonly used organism for over-expression of recombinant proteins, including P450s (Guengerich and Martin, 2006), may not be capable of providing the folding machinery and specific lipid environment for production of a membrane protein in milligram amounts, needed for biophysical and structural investigations. Even with pure protein available, the presence of flexible hydrophobic N- and/or C-terminal tails in P450s can frequently hamper crystallization efforts, and then requires a time consuming search for alternative protein constructs that can be expressed in high yields and are suitable for production of diffraction-quality crystals. Our experiments suggest that truncation of the N-terminal membrane anchor sequence will not affect P450 function and, combined with addition of a soluble N-terminal linker, will increase protein expression yields and solubility. Moreover, co-expression of chaperon EL/ES assists in protein folding. Compared to this approach, the use of eukaryotic expression systems such as Sf9 insect cells is expensive, time consuming and often suboptimal in terms of yields. However, P450 enzymes expressed in E. coli lack post-translational modifications such as phosphorylation, glycosylation, ubiquitination or nitration that have been observed in various eukaryotic P450 subfamilies (Lamb and Waterman, 2013).
Figure 1.

Tertiary structure diagram of human P450 17A1 (PBD ID code 3RUK; http://www.rcsb.org) with the ribbon colored in the rainbow spectrum, from N-terminus (blue) to C-terminus (red). Amino acid side chains in hydrophobic regions of the protein associated with the membrane (horizontal gray bar) are depicted in ball and stick mode. The P450 enzyme is located on the cytoplasmic side of the bilayer (Monk et al., 2014). The image was generated with the program UCSF Chimera (Pettersen et al., 2004).
To date, 57 human P450s have been identified and among these at least 14 serve important roles in steroid metabolism (Guengerich, 2005; Miller and Auchus, 2011). For example, human P450 17A1 is primarily expressed in adrenal and testicular tissues and catalyzes two-step oxidation reactions of progesterone and pregnenolone. In the first step, 17α-hydroxylation of these substrates results in 17α-hydroxy-progesterone and 17α-hydroxy-pregnenolone, respectively. The 17α,20-lyase reaction in the second step subsequently produces androstenedione and dehydroepiandrosterone, respectively. Interestingly, the reactions catalyzed by P450 17A1 are dependent on the cellular location of the enzyme. Thus, in the gonads 17A1 converts the 21-carbon steroid to the 19-carbon androgen in a 3-step hydroxylation reaction. Conversely, in the adrenal cortex the enzyme stops after the initial hydroxylation reaction (Lamb and Waterman, 2013).
For catalytic activity, P450 enzymes require electrons that are provided by reduced nucleotides NADPH and NADH and redox partner proteins that shuttle the electrons to the iron center: Fe-containing ferredoxin (Fdx) and flavin adenine dinucleotide (FAD)-containing ferredoxin reductase (FDR) (prokaryotic P450s); flavin mononucleotide (FMN)/FAD-containing NADPH cytochrome P450 oxidoreductase (CPR) (eukaryotic P450s); and adrenodoxin (ADX) and adrenodoxin reductase (ADR) (mitochondrial P450s) (Lamb and Waterman, 2013). In some cases, cytochrome b5 can provide the second electron (Johnson and Stout, 2013).
Prostate cancer cells proliferate in response to androgen steroids, and inhibition of the 17α,20-lyase reaction only is a new strategy to prevent androgen synthesis and treat lethal metastatic, castration-resistant prostate cancer without interfering with mineralocorticoid and glucocorticoid synthesis. The drug abiraterone that inhibits both enzymatic functions of human P450 17A1 is used in the clinic for treatment of metastatic castration-resistant prostate cancer (DeVore and Scott, 2012). Teleost fish feature two P450 17A enzymes; like human P450 17A1, fish P450 17A1 catalyzes both steroid 17α-hydroxylation and 17α,20-lyase reactions. However, P450 17A2 only catalyzes the 17α-hydroxylation. We analyzed the substrate binding properties, enzymatic behavior and three-dimensional structures of P450 17A1 and 17A2 from zebrafish in order to gain insight into the biochemical basis of the 17α,20-lyase reaction, and to potentially identify differences in the active sites between the enzymes that could explain the absence of the lyase reaction in 17A2 (Pallan, Nagy et al., 2015). Both enzymes are microsomal-type P450s, integral membrane proteins that bind to the membrane through their N-terminal hydrophobic segment. Extensive attempts to crystallize the two proteins failed, despite testing a variety of constructs with portions of the N-terminal regions truncated and/or alternative leader sequences (Pallan, Nagy et al., 2015), mutation of N-terminal amino acids, and screening a large number of crystallization conditions
The present protocol describes an approach that allowed us to successfully grow diffraction-quality crystals for both P450 17A1 and 17A2 from zebrafish. In the first step, we produced the two membrane proteins in E. coli both in good quality and quantity by truncating the N-terminal hydrophobic sequence, and adding a leader sequence, in addition to co-expressing the GroEL/ES chaperonin to assist P450 protein expression and folding (Figure 2a,b). Secondly, we screened twelve different proteases in regard to their cleavage patterns with P450 17A1 and 17A2, by varying protease concentration in combination with a gel electrophoretic assay and analyzing the resulting fragments using Edman sequencing. Compared to the time-consuming preparation of discrete expression systems for proteins with systematically shortened N-terminal regions, in situ proteolysis allows for potentially rapid identification of crystallizable fragments. Thirdly, the 17A1 and 17A2 proteins featuring an N-terminal leader sequence conducive for expression (Richardson et al., 1995), in conjunction with the optimal protease for generating N-terminally truncated proteins, were used for in situ proteolysis in the crystallization and subsequent structure determination.
Figure 2.

Generation of alternative P450 constructs for improving expression, solubility, stability and crystal formation, using bovine P450 21A2 as an example (Zhao et al., 2012). (a) Wild type sequence of bovine P450 21A2. (b) A soluble fragment was obtained by replacing the hydrophobic N-terminal 29 amino acids in bovine P450 21A2 by the shorter MAKKTSSKGK leader sequence (from the N-terminus of P450 2C3). Residues that were mutated individually are highlighted in red in the wild type (Thr-241 and Leu-442) and alternative protein constructs (Arg-222 and Ala-423, respectively). (c) Identification of candidate surface residues for point mutation (T241R and L442A) by aligning the sequences of P450 21A2 from selected eukaryotic species, combined with (d) hydrophilicity analyses of wild type (with alternative leader; c3B21) and mutated protein sequences (c3B21RA). The alternative protein construct shown in panel b was used for determining the crystal structure of bovine P450 21A2 (Zhao et al., 2012).
Strategic Planning
EXPERIMENTAL DESIGN
Expression system
E. coli remains a commonly used organism for expression of membrane proteins as it is relatively inexpensive and numerous proteins can be produced in recombinant form easily and efficiently. BL21 cells and derivatives thereof offer multiple choices such as different cell culture temperatures (14 °C to 37 °C) to optimize expression of a wide range of target proteins, including membrane or toxic proteins and those encoded by rare codons. To increase target protein expression and augment folding, a GroEL/ES chaperonin expression vector is co-transformed into E. coli BL21 (DE3) cells.
Protein construct
Eukaryotic membrane-bound P450s have a hydrophobic N-terminal anchor region that serves to attach the protein to the membrane. N-Terminal truncation of P450s can affect both activity (e.g. by limiting proximity between the P450 catalytic domain and the reductase that are both anchored to the membrane) and expression levels. Thus, in our experience protein constructs with truncated N-terminal portions fail to express or the expression levels are poor. Replacing the missing hydrophobic region by a soluble linker can sometimes rescue protein expression in E. coli. In our case we introduced the P450 2C3 N-terminal sequence that is known to improve expression (Richardson et al., 1995; Zhao et al., 2012) (Figure 2a,b). Alternative leader sequences include a histidine tag at the N-terminal end that enables both increased protein expression and protein purification by Ni-NTA agarose affinity chromatography. However, we introduced a hexa-histidine tag encoded by 18 nucleotides at the C-terminal end. We relied on the pET17 expression vector for producing both the P450 17A1 and 17A2 proteins of zebrafish. Other pET expression vectors or pET-derived vectors include pAT, pBG and pSV. With some vectors, it is possible to introduce a specific protease cleavage site between protein and tag or the leader to help cleave off the latter. If, after truncating the N- or C-terminal hydrophobic tails, and replacing the former with a leader sequence and co-expression of GroES/EL chaperonin, protein expression and/or solubility could not be improved, we used the Protean software package (https://www.dnastar.com/t-protean.aspx) to analyze the protein sequence and, based on a sequence alignment, picked hydrophobic amino acid(s) that map to the surface of the protein and mutated them to less hydrophobic or hydrophilic amino acid(s) as identified in the same protein subfamily or in other species in the alignment (Figure 2). In an alternative mutation experiment, we produced the P450 17A1 C57R mutant in order to test whether trypsin in combination with this single arginine mutation would promote crystal growth, because in situ proteolysis with trypsin and cleavage at an N-terminal arginine had previously produced diffraction-quality crystals of P450 17A2 (Pallan, Nagy et al., 2015).
The zebrafish P450 17A1 and 17A2 enzymes were expressed at levels of between 500 and 1100 nmole/liter of culture (Figure 3) and used for both biochemical experiments and crystallization (Pallan, Nagy et al., 2015).
Figure 3.
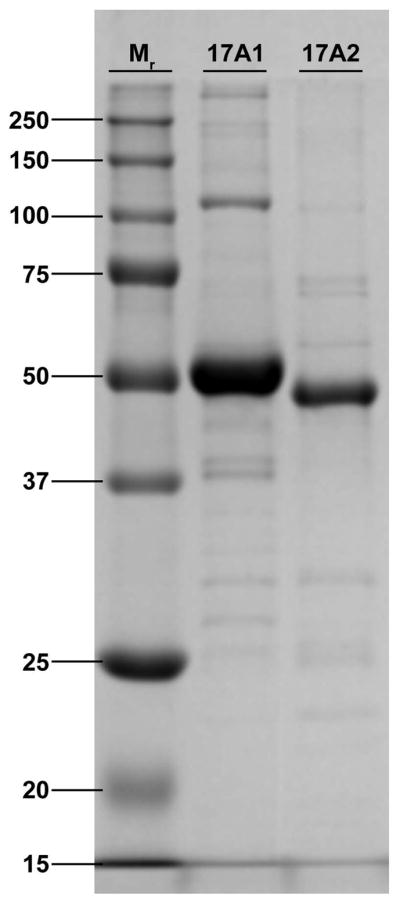
SDS-PAGE of expressed and purified P450 17A1 and 17A2 proteins from zebrafish.
Detergent screen
Not every membrane-bound P450 protein requires the use of detergent for crystallization. Thus, we relied on 5-cyclohexyl-1-penthyl-β-D-maltoside to crystallize bovine P450 21A2 (Zhao et al., 2012), but no detergent was used to produce crystals of human P450 21A2 (Pallan, Wang et al., 2015). In cases where the protein cannot be concentrated to 20 mg/ml at 50 mM potassium phosphate, pH 7.0–7.4, 5–10% glycerol, 50–100 mM NaCl, and 1mM DTT, like with zebrafish P450 17A1 and 17A2, a detergent that helps solubilize the membrane protein needs to be identified. A good starting point is the use of a screening kit such as the Detergent Screen HT offered by Hampton Research.
In situ proteolysis and crystallization
Following prescreening of 12 proteases, optimal cleavage conditions and protease concentrations were determined. For the crystallization experiments, we used a mosquito nanodrop setting robot (Molecular Dimensions) in combination with 96-well plates. For each well in the plate, two droplets containing the protein with or without addition of the protease were set up and nearly 300 different crystallization conditions screened. After several days, crystals were observed only in drops that were treated with the respective proteases. Initial diffraction tests indicated that these diffracted to resolutions of between 2.8 and 3 Å. Edman sequencing revealed that the N-terminal residue of P450 17A2 in the crystals was Ser-52, consistent with trypsin cleaving C-terminal to Arg-51 and in accord with the first residue visible in the electron density maps being Leu-54. In the case of P450 17A1 treatment with subtilisin and analysis of the proteolytic products with SDS-PAGE indicated a mixture of fragments. Edman sequencing of the major product placed the N-terminus at Gly-37. However, the crystal contained a shorter fragment as indicated by Ser-83 being the first residue visible in the electron density maps.
BASIC PROTOCOL: Vector Preparation, Expression, Purification, and in situ Proteolytic Crystallization of Membrane-Bound P450 Proteins
This protocol provides instructions for the generation of expression plasmids for P450 proteins with alternate N-terminal sequences, their expression of in E. coli, purification of the protein by nickel affinity, ion exchange, and size exclusion chromatography, detergent screening, in situ proteolytic crystallization experiments, and analysis of the protein in crystals using N-terminal Edman sequencing. The experimenter may also consult existing protocols on the preparation of plasmids, expression in E. coli, protein purification and crystallization.
Materials
KHPO4 (Research Products International Corp. cat. no. 41300-500)
K2HPO4 (Research Products International Corp. cat. no. 41300-500)
NaCl (Research Products International Corp. cat. no. 7647-14-5)
Glycerol (Research Products International Corp. cat. no. G22020-4.0)
Imidazole (Sigma, cat. no. I2399-500G)
Dithiothreitol (DTT) (Research Products International Corp., D11000-5.0)
EDTA (Research Products International Corp. cat. no. D11000-5.0)
Methanol (Fisher Scientific, cat. no. A433P-4)
Acetic acid, glacial (Fisher Scientific, cat. no. A38-212) CAUTION Corrosive, avoid inhalation, exposure to skin and eyes.
Phenylmethanesulfonyl fluoride (PMSF) (Sigma, cat. no. P7626-25G) CAUTION Corrosive, avoid inhalation, contacts with skin and eyes.
3-Cyclohexylamino-1-propanesulfonic acid (CAPS) (Research Products International Corp., cat no. c30060)
Tris base (Research Products International Corp., cat. no. T60040)
Coomassie Brilliant Blue R 250 (MP Biomedical. cat. no. 04821616)
Plasmid vectors: pET 17b expression vectors (Novagen)
E. coli DH5 cells for cloning (Vanderbilt University Core Lab)
BL21 (DE3) Gold (or plysS) for overexpressing protein (Invitrogen)
Bacto Agar (Becton, Dickinson and Company, cat. no. 214010)
Antibiotics: e.g., Kanamycin sulfate (Research Products International Corp., cat. no. K22000-5.0); Ampicillin sodium salt (Research Products International Corp., cat. no. A40040-5.0)
LB Broth Miller (EMD, cat. no. 1.10258.5007)
Terrific Broth, Modified (Research Products International Corp., cat. no. T15000-500.0)
IPTG (Research Products International Corp., cat. no. I56000-5.0)
L-Arabinose (Gold Bio Technology Inc., CAS No. 87-72-9)
δ-Aminolevulinic acid hydrochloride (ALA)(CHEM-IMPEX International, cat. no. 01433)
For SDS-PAGE: mini-protean TGX precast 4–20% (w/v) Tris-glycine gel, (BIO-RAD, cat. no 601-1105)
Invitrolon PVDF filter paper sandwich, 0.45-μm pore size (Invitrogen, cat. no. LC2005)
-
Detergents:
Tween 20 (Anatrace, cat. no. T1003)
C12E9 Polyoxyethylene(9)dodecyl Ether • Thesit • Polydocanol • α-Dodecyl-w-Hydroxy-Poly(oxy-1,2-Ethanediyl) (Anatrace, cat. no. Apo-12950X1ml)
Sodium cholate (Sigma cat. no. C1254)
Detergent Screen HT (Hampton research, cat. no. HR2-406)
Ni-NTA Agarose resin (Qiagen, cat. no. 30250)
Q sepharose fast flow (GE Healthcare, cat. no. 17-0510-01)
S sepharose fast flow (GE Healthcare, cat. no. 17-0720-01)
Superdex 75 10/30 GL (GE Healthcare, cat. no. 17-5174-01)
pregnenolone (Steraloids), 17 α-OH pregnenolone (Steraloids), Abiraterone (Selleckchem, Houston, TX).
Laemmli Sample buffer for SDS-PAGE (Bio-Rad, cat. no. 161-0737)
PhastGel Blue R pre-measured tablets for SDS-PAGE gel staining (Sigma-Aldrich B4921)
Amicon ultra centrifugal filters, 30K (Merck Millipore Ltd, cat. no. UFC 903024)
Proti-Ace Kit (Hampton Research, cat. no. HR2-429)
Proti-Ace 2 Kit (Hampton Research, cat. no. HR2-432)
-
Crystallization screens:
Index kit (Hampton Research, cat. no. HR2-144),
Crystal screen (Hampton Research, cat. no. HR2-110),
Crystal screen 2 (Hampton Research, cat. no. HR2-112),
PEG/Ion screen (Hampton Research, cat. no. HR2-126)
MRC 2 well crystallization plates (Hampton Research, cat no. HR3-082)
CrystalQuick 96 well Greiner 609191 (Hampton Research, cat. no. HR3-192)
ClearSeal Film (Hampton Research, cat. no. HR4-521)
Seed bead (Hampton Research, cat. no. 2-320)
Equipment
Ultracentrifuge Optima L-80 (Beckman Instruments Inc., cat. no. 355884)
Beckman GS6 KR centrifuge (Beckman Instrument Inc., cat. no. 362134)
Leica MZ28 stereomicroscope (Leica Microsystems)
Mosquito crystal liquid handler (TTP labtech Inc., cat. no. Mosquito)
Rock Imager (Formulatrix cat. no., rock imager 1000)
Spectrophotometer (Shimadzu Scientific Inc., cat. no. UV-2401PC)
FPLC (GE Amersham Biosciences, cat. no. 11001479 AKTAbasic UPC10 incl. Frac-920)
REAGENTS AND SOLUTIONS
LB broth
Add 25 g of LB Broth Miller to 1 liter of water and autoclave for 25 min. Store at room temperature (25 °C) until use.
LB agar plates
Prepare LB medium as above and add 15g/L of agar before autoclaving, cool to 50°C after autoclaving and add appropriate antibiotic. Dispense into sterile Petri dishes (10 cm diameter) and allow to set at room temperature. Store plates at 4 °C.
TB broth
47g, add Terrific Broth and 4 ml of glycerol to 1 liter of water, autoclave for 25 min. Store at room temperature until use.
ALA stock solution
Dissolve ALA into water to 1 M and store at −20 °C.
PMSF stock
Dissolve PMSF in DMSF to 0.1 M and store at −20 °C
Buffers for chromatography
Buffer A: cell lysis and Ni-NTA column equilibration and washing. Mix 50 mM potassium phosphate (pH 7.4), 300–500 mM NaCl, 0.1 mM DTT, 0.1 mM EDTA, 20% (v/v) glycerol, 1%(v/v) Tween-20, 1% (w/v) sodium cholate. Buffer B: Ni-NTA wash buffer. Mix 50 mM potassium phosphate (pH 7.4), 0.1 mM DTT, 0.1 mM EDTA, 20%(v/v) glycerol, 1%(v/v) Tween-20, 1% (w/v) sodium cholate. Buffer C: Q and S sepharose fast flow column. . Mix 50 mM potassium phosphate (pH7.4), 0.1 mM DTT, 0.1 mM EDTA, 0.005% (v/v) C12E9. Buffer D: Size exclusion chromatography. Buffer C, 100 mM NaCl.
TES buffer
For lysozyme treatment of cells. 100 mM Tris-HCl pH 8.0, 1 mM EDTA, 500 mM sucrose.
SDS-PAGE gel running buffer
Dissolve 1 g of SDS, 3 g of Tris base and 14.4 g of glycine in 600 ml of water. Add water to a final volume of 1 liter. Store at room temperature.
2 × SDS protein-loading dye
Mix 950μl of Laemmli sample buffer (Bio-Rad 161-0737) with 50 μl of β-mercaptoethanol before use.
SDS Gel stain
Make 0.2% w/v stock solution as manufacturer recommends: 1 tablet in 80 ml of ultra pure water, add 120 ml of methanol. Store at 4 °C.
SDS Gel destain
Mix 120 ml of methanol, 120 ml of glacial acetic acid and 460 ml of water. Store at room temperature.
PVDF electroblotting buffer
CAPS buffer (10 mM CAPS, 10% methanol 0.1% (w/v) SDS, pH 11): dissolve 2.21 g CAPS in 500 ml deionized H2O; add 100 ml of methanol and 10 ml of 10% SDS; add deionized H2O to 1 L. Adjust pH to 11 with NaOH. CRITICAL STEP The SDS concentration used in CAPS buffer depends on the size of the target protein.
PROCEDURE
Generating the modified expression vector
-
1
Design and order primers for PCR amplification of the P450 17A1 and P450 17A2 DNAs The 3’-primer contains the codons of the NdeI site, MAKKTSSKGK and codons for five amino acids of the truncated N-terminal region of the target proteins. The 5’-primer contains the codons of the HindIII site, the region encoding six histidines and codons for five C-terminal amino acids of the target proteins. Use RNA extracted from zebrafish as template with the Qiagen One Step RT-PCR kit and follow the manufacturer’s instructions to amplify the genes. Alternatively, order the N-terminal truncated gene with the NdeI site and MAKKTSSKSK in the protein’s N-terminal region and the HindIII site, stop codon and six histidines in the protein’s C-terminal region.
-
2
Clone the modified genes into the pET17b vector using the NdeI and HindIII sites and confirm them by sequencing: In the zebrafish P450 17A1 open reading frame, the region encoding the N-terminal transmembrane helix (residues 1–26) was replaced by DNA coding for MAKKTSSKGK (P450 2C3 N-terminal region), and the 3’-end was extended by 18 nucleotides encoding six histidines. For zebrafish P450 17A2, the region encoding the N-terminal transmembrane helix (residues 1–25) was replaced by DNA coding for MAKKTSSKGK (P450 2C3 N-terminal region) and the 3’-end was extended by 18 nucleotides encoding six histidines. Both modified cDNAs were inserted into the pET17b vector.
TROUBLESHOOTING
Expression in E. coli
-
3
Co-transform E. coli BL21 (DE3)Gold or BL21(DE)3 PlysS cells by heat shock according to the manufacturer’s protocol with the vectors containing the target gene and pGro12 ES/EL gene. Plate the transformed cells onto LB agar plates with the appropriate antibiotic selection. Grow at 37 °C overnight.
-
4
Pick colonies and inoculate 20 ml of LB medium with the appropriate antibiotics in a flask. Culture cells in an incubator/shaker overnight at 37 °C and 225 r.p.m.
-
5
1:100 inoculate each 250ml of TB/flask x6 with the overnight growth culture and add the appropriate antibiotics. Grow the cultures in an incubator shaker at 37 °C and using 175–225 r.p.m. After 3.5–4 hours (OD600 reaches 1.0–1.2), apply 1 mM ALA to help heme production and 2 mg/ml arabinose to induce chaperones. After another hour induce target protein expression by adding IPTG to a final concentration of 1 mM. Continue to grow the cultures at 26 °C and at 100–120 r.p.m. for 36 hrs.
CRITICAL STEP Getting high expression of target protein is one of the critical steps for getting enough purified protein to screen crystallization conditions. For overnight cultures at 37 °C, the optimal time is less than 17 hours. If more time is needed, it will be better to culture at 32 °C. Freshly transformed clones usually result in higher expression. The culturing time after IPTG induction is dependent on the individual protein; it may be necessary to determine the best time course for a particular target protein.
We normally use 1mM ALA to help E.coli produce more heme and the concentration from 0 to 2 mM. For example, 2 mM ALA affords better expression of a fusion protein that consists of two proteins that both require heme. Regarding the arabinose concentration: if the experiment shows a large amount of chaperone expression – in any case more than needed by the target protein - decrease the arabinose concentration or induce chaperone and target protein expression at the same time and drop the culture temperature.
-
6
Transfer the culture to centrifuge tubes and harvest the cells by centrifuging for 25 min at 3000 r.p.m. and 4 °C. Discard the supernatant and resuspend the pellet in 80–100 ml of 100 mM Tris buffer pH 8.0 or TES buffer pH8.0 and addition of lysozyme to 1 mg/ml, keep the sample at 4 °C and stir for 25 min. Collect cells by centrifugation at 3000 r.p.m. and 4 °C for 25 min. Discard the supernatant and store cells at −80 °C overnight. PAUSE POINT The cells can be frozen at this point and stored at −80 °C. Overnight storage is recommended as freezing and thawing will help cells lysis, but tolerance for storage should be determined for each target protein. Lysozyme treatment is also important for cell lysis.
TROUBLESHOOTING
Purification of target protein
All steps should be carried out on ice or at 4 °C. All buffers should be kept at 4 °C. Since our target proteins are colored, no FPLC is needed. The following protocol is based on using our own packed columns and the gravity method except for the size exclusion column.
-
7
Nickel affinity purification
Add 200–300 ml of buffer A to thaw and lyse the cells, then aliquot the sample to 50 ml tubes and sonicate for 30” x3 on ice for each tube to break cells and DNA. Centrifuge the sample at 35k r.p.m. and 4 °C for 40–50 min. Collect all supernatant.
Prepare the appropriate amount (here we use 15–20 ml) of Ni-NTA agarose resin according to the manufacturer’s instructions. Equilibrate the column with buffer A. Load the supernatant collected onto the column, collect and save the flow-through containing the unbound material. Measure the CO spectrum for P450 protein, otherwise take a 10–20 μl sample of the flow-through for analysis by SDS-PAGE and store at 4 °C. The flow-through should not contain or contain only a small amount of the target protein.
-
Wash the column with buffer A until the signal of the eluent at 280 nm returns nearly to baseline and then wash with 50–100 ml of buffer A with 10–20 mM imidazole. Check flow through P450 CO spectrum or take a 10 μl sample of the wash out for SDS-PAGE analysis and store at 4 °C. Wash the column with 50–100 ml of buffer A and 50 ml of buffer B until the flow through OD at 280 nm returns to nearly baseline. Elute the protein with buffer B with 200–300 mM imidazole and collect the peak(s). Take a 2–10 μl sample for analysis by SDS-PAGE and store at 4 °C. Flash freeze sample in liquid nitrogen and transfer to −80 °C.
Optional: the column can be washed with up to 50 mM imidazole wash buffer, dependent on how tight the target protein binds to Ni-NTA or how large an amount of target protein is bound to the column. Some target protein may be sacrificed in exchange for obtaining purer protein.
TROUBLESHOOTING
Ion exchange chromatography
-
8
Before ion exchange chromatography, determine the size, pI and the charges etc. of the target protein using the Protean software.
Make “Q” and “SP” columns by adding 6–10 ml of sepharose into the columns, and washing them with 50 ml of deionized water and then 20 ml of buffer B.
-
1:1 dilute the eluent from the Ni-NTA column with buffer C, load onto and pass the “Q” sepharose fast flow column with a flow rate of 1 ml/min and then load the sample which passes the “Q” onto the “SP” sepharose fast flow column. Wash with 50 ml of buffer C with 50 mM NaCl and 50 ml of buffer C with 100 mM NaCl until OD 280nm returns to base line. Elute the sample with buffer C with 250–300 mM NaCl. Take a 1–5 μl sample of the elution for SDS-PAGE analysis and store at 4 °C. If a size exclusion column is needed, concentrate the protein sample to 1–2 ml. Flash freeze the sample in liquid nitrogen and transfer to −80 °C.
CAUTION Use freshly made protein for crystallization as soon as possible. The tolerance for storage should be determined for each target protein.
Note: passing the target protein through the same charge column is important; proteins that carry opposite charge can bind to the column, while others that are nearly neutral in the buffer have the opportunity to bind the ion exchange column they first encounter. TROUBLESHOOTING
-
9
Detergent screen:
-
Produce detergent free protein. Detergents can be eliminated during column purification by reducing their concentration stepwise during washing and simultaneously increasing the NaCl concentration from 50 mM to elution concentration. This can be done while washing the SP column and eluting the protein without detergent.
CAUTION Try to avoid concentrating the membrane protein. If it has to be done at all, watch carefully whether there is precipitation while concentrating the protein.
Make 10X CMC detergent stock. Add 10X CMC detergent to the protein sample (2–10mg/ml) for a final concentration of 1X CMC and leave the sample at 20 °C. Check if any precipitation occurs and analyze the P450 CO spectrum each day for seven days with a detergent free protein control. Select the sample with the highest ratio of P450 to P420 peak. Alternatively, perform a detergent screen using commercial screening kits. Detergent C12E9 was chosen for zebrafish P450 17A1 and 17A2 crystallization.
-
Size exclusion chromatography
-
10
Equilibrate the column with buffer D, inject 0.5 ml of sample and run it with buffer D at a flow rate at 0.5 ml/min. Collect 0.5–1 ml fractions of the eluent while monitoring at 280 nm. To determine which peak on the chromatogram contains the target protein, check the P450 peak of the fractions collected or run them on a 4–20% (w/v) tris-glycine gel or a native gel and stain with Coomassie (Figure 3). If only a single peak is obtained and the protein is sufficiently pure for crystallization after the ion exchange column, no size exclusion column in needed.
Preparation of P450 substrates and inhibitor complexes for crystallization
-
11
Measure the concentration of target protein; in a 1:2–10 molar ratio (1:10 for abiraterone inhibitor and 1:2 – 1:5 for substrates), add either 100 mM progesterone dissolved in ethanol or 10 mM abiraterone dissolved in DMSO, mix well and remove any aggregated protein or particulate matter by centrifugation in a micro centrifuge for 10 min at 13000 r.p.m. and 4 °C. Concentrate the protein-substrate or protein-inhibitor complexes to a concentration of 30 mg/ml. Aliquot the protein to the volume needed for individual crystallization experiments, in our case 22μl/plate (200 nl x 96 wells). Flash freeze in liquid nitrogen and transfer to −80 °C.
In situ proteolysis for crystallization of zebrafish P450 17A1 and 17A2 proteins
-
12
Make 1 mg/ml protease stock solution as per the manufacturer’s suggestion. Prescreen the target protein to protease ratio that yields the largest, most stable domain that will be used for subsequent crystallization by mixing protease and target protein at 1:100, 1:1000, and 1:2000 ratios (w/w), and incubate the mixtures at room temperature for 30 min, add PMSF to a 1 mM final concentration. Add SDS-PAGE sample buffer and boil for 2 minutes, load on a 4–20% Mini-PROTEAN TGX Precast Gel, run the gel at 30–40 mA until the blue dye migrates to the bottom of the gel. Stain the gel with 10 ml of SDS gel stain solution by heating in a microwave oven for 15 seconds and keeping at room temperature for another 2 min. Destain gel with SDS gel destain solution until the background is clear.
A 1:1000 protease to target protein ratio was found to be optimal in our case for the crystallization experiments (Figures 4, 5).
CAUTION If the protease has no specific cleavage site within the protein sequence, the ratios among protein fragments observed in the SDS-PAGE gel may not be representative of those formed in the crystallization drop. Ratios vary with temperature, screening reagent condition and the time of exposure of the protein to the particular protease. The size of the protein in crystals formed in different screen conditions may be different.
Figure 4.

SDS-PAGE (4–20%) gel depicting zebrafish P450 17A1 cleavage with 12 proteases, using various protein to protease ratios: Subtilisin, lane 1 1:2000, lane 2 1:1000, lane 3 1:100; papain, lane 4 1:2000, lane 5 1:1000, lane 6 1:100; trypsin, lane 7 1:2000, lane 8 1:1000, lane 9 1:100; α-chymotrypsin, lane 10 1:2000, lane 11:1000, lane 12 1:100; endoproteinase Glu-C, lane 13 1:2000, lane 14 1:1000, lane 15 1:100; elastase, lane 16 1:2000, lane 17 1:1000, lane 18 1:100; pepsin: lane 19 1:2000, lane 20 1:1000, lane 21 1:100; protease K, lane 22 1:2000, lane 23 1:1000, lane 24 1:1000; thermolysin, lane 25 1:2000, lane 26 1:1000, lane 27 1:100; actimase, lane 28 1:2000, lane 29 1:1000, lane 30 1:100; bromelain, lane 31 1:2000, lane 32 1:1000, lane 33 1:100; clostripain, lane 34 1:2000, lane 35 1:1000, and lane 36 1:100. M represents a molecular marker lane.
Figure 5.

SDS-PAGE (4–20%) gel depicting zebrafish P450 17A2 cleavage with trypsin or α-chymotrypsin using various protein to protease ratios: α-chymotrypsin, lane 1 1:0, lane 2 1:100, lane 3 1:1000, lane 4 1:10000, lane 5 1:100000; and trypsin, lane 6 1:100, lane 7 1:1000, lane 8 1:10000, and lane 9 1:100000.
Crystallization experiments
-
13
At room temperature, dispense 60 μl of a commercially available crystallization screen (conditions 1–96) into reservoirs 1–96 of a 96-well flat-bottomed polypropylene plate. Prior to setting up crystallization droplets, add subtilisin (trypsin) to zebrafish P450 17A1 (zP450 17A2) inhibitor (or substrate) complex with the protease to P450s ratio (w/w) being 1:1000. Remove 10 μl of the mixture and incubate at room temperature for 30 min, then add PMSF to a 1 mM final concentration, boil for 2 min and store at −20 °C for subsequent N-terminal sequence analysis (step 13). Using a Mosquito crystal liquid handler, set sitting drops, two drops per well, one by mixing 200 nl mother liquor with 200 nl protein substrate or inhibitor complex, the other by mixing 200 nl of protein substrate or inhibitor complex and protease mixture with 200 nl of a given screening condition. Incubate the 96-well plates in a Rock Imager at 21 °C and capture images on days 1, 3, 5, 7, 14, 21, 30 and once a month after day 30. Crystals of P450 17A2 with abiraterone or progesterone appeared within 3–4 days. Their size was around 0.2–0.4 mm (Figure 6a). Crystals of P450 17A1 with abiraterone appeared within 5–7 days and, as the crystals were smaller than 0.05 mm, their size was increased by a round of microseeding.
Microseeding is carried out as follows: Pick 3–5 crystals (if they can be scooped up with a nylon loop), place in a microcentrifuge tube with 20–30 μl of mother liquor and a bead, or use 5–10 μl of mother liquor to wash the well that contains microcrystals, and put the mixture into a microcentrifuge tube with 40 μl of mother liquor and a bead. Vortex the tube for 1–2 min. Dilute the mixture with mother liquor at ratios of 1:400, 1:800, 1:1600, and 1:3200, and use these diluted mixtures to seed drops by mixing 200 nl of protein inhibitor complex and protease mixture with 200 nl of the seed solution. Crystals of P450 17A1:abiraterone complex (0.15–0.3 mm size) appeared within 3–10 days (Figure 6b).
Figure 6.

Crystals of zebrafish P450 (a) 17A2 and (b) 17A1, obtained by in situ proteolysis using trypsin and subtilisin, respectively. The modified zebrafish P450 17A1 and P450 17A2 proteins were expressed using plasmids based on the pET17b vector. The protease to target protein ratio for in situ proteolytic crystallization was 1:1000 and the maximum size of crystals was ca. 0.25 mm.
N-terminal Edman sequencing
-
14
Dilute 50-fold the samples that were treated with protease before making crystallization setups, then add the same amount of SDS-gel sample buffer, boil for 2 min and load 20 μl to each lane of a mini-protean TGX precast 12% (w/v) tris-glycine gel. Run the gel at 30–40 mA until the blue dye reaches the bottom of the gel.
Soak PVDF in methanol and then equilibrate with CAPS buffer. Rinse the gel with deionized water and balance with CAPS buffer.
Assemble gel PVDF sandwiches according to the instructions: gel holder-fiber pad-filter paper-gel-PVDF membrane-filter paper-fiber pad-gel holder. Put the sandwiches in the tank filled with CAPS buffer with the gel on the cathode side. Run at 30V/100mM, for 16 hrs (overnight) at 4 °C or in the cold room.
Stain the PVDF blot with 10 ml of coomassie blue R-250 stain solution until the protein bands show and destain the blot until the background is light. Rinse the blot with deionized water completely. Cut out the target bands and dry them at room temperature. The cut out bands are now ready for sequencing.
N-terminal Edman sequencing analysis is carried out by the Synthesis and Sequence Facility at the John Hopkins University, School of Medicine, Baltimore, MD. (http://biolchem.bs.jhmi.edu/tssf/Pages/default.aspx). TROUBLESHOOTING
COMMENTARY
Critical Parameters and Troubleshooting
Advice regarding critical parameters and troubleshooting is summarized in Table 1.
Table 1.
Critical parameters and troubleshooting
| Step | Problem | Possible reason | Solution |
|---|---|---|---|
| 1–2 | No colonies. | Wrong antibiotic or the concentration of the antibiotic is too high. | Switch to the correct antibiotic. Lower the concentration of the antibiotic. |
| Competent cells are not viable. | Use fresh competent cells. | ||
| Insufficient competent cells or plasmids. | Use a larger amount of competent cells or plasmids. | ||
| Culturing time at 37°C is too short. | Culture the plate at 37°C for up to 20 hours. | ||
| 3–6 | Very low or no protein expression. | Competent cells are not viable or glycerol stock was used for an overnight culture. | Use healthy competent cells, and make a fresh transformation. |
| Used wrong medium for overnight culture | Use LB medium. | ||
| Overnight culture at 37°C is too long | Culture at 37° C for less than 17 hours. | ||
| Insufficient expression time. | Determine the best time course. | ||
| Expression temperature is incorrect. | Determine the best temperature course. | ||
| Target protein is toxic. | Switch to competent cells that tolerate toxic proteins. | ||
| The target protein gene codons are not optimal for E.coli. | Switch to BL21Codonplus expression cells. | ||
| The protein is expressed in inclusion bodies. | Switch to expression competent cell line that allows expression of protein at low temperature, such as ArcticExpression competent cells. Add an N-terminal or C-terminal soluble fragment. Mutate hydrophobic amino acid(s) on the surface of the protein. See Experimental Design. |
||
| 7 | The protein does not bind to Ni resin. | DTT or EDTA concentration is too high. | Lower the concentration. |
| Affinity tag is sequestered by detergent micelle or is buried inside the protein. | Lower the detergent concentration or add the linker between the protein and the affinity tag. | ||
| Too much imidazole in the lysis buffer. | Lower imidazole concentration to 2–5mM. | ||
| The protein from Ni resin is heavily contaminated. | The proteins from host cell bind to Ni resin non-specifically. | Add to 5–10 mM imidazole and 100 mM glycine to lysis buffer. Increase imidazole to 20–50 mM in wash buffer. |
|
| 8 | Protein does not bind to ion exchange column. | The protein carries the same charge as the resin. | Check the pI and the charge of protein in the buffer. |
| Ionic strength of eluent is too high. | Lower the ionic strength. | ||
| Protein is denatured or has precipitated. | Keep or add detergent to the protein solution. After loading, immediately wash with buffer containing 50–100 mM NaCl. | ||
| 14 | No protein bands on either the SDS gel or PVDF. | Not enough protein loaded onto the gel. | Load more protein. |
| Protein is smaller than 15kDa which passes PDVF. | Remove SDS from blotting buffer or increase MeOH concentration. | ||
| Protein band cannot be transferred to PVDF membrane. | Protein is bigger than 80kDa. | Add 0.1–0.2% SDS or omit MeOH from blotting buffer. |
Background Information
Comparison with Previous Applications of Limited Proteolysis in Macromolecular Crystallization
An initial report on the use of in situ proteolysis with either trypsin or chymotrypsin involving 55 bacterial and 14 human proteins that proved resistant to crystallization found a 15% success rate, including the steps from pure protein to structure determination (Dong et al., 2007). With an expanded set of 270 soluble proteins and testing six proteases (trypsin, chymotrypsin, elastin, papain, subtilisin and V8 protease), the success rate was ca. 13% (Wernimont and Edwards, 2009). Both studies were primarily concerned with increasing the number of crystallizable protein fragments as part of a structural genomics effort. Several other preliminary protein crystallization notes and publications involving structure determination of proteins or domains thereof reported the use of in situ proteolysis (Gaur et al., 2004; Johnson et al., 2006; Taneja et al., 2006; Forsgren et al., 2009; Little et al., 2012; Kobayashi et al., 2014; Civril & Hopfner, 2014). At least in two cases, the importance of proteolysis for growing diffraction-quality crystals was a serendipitous discovery in that the solution was infected with a fungus that secreted the protease (Mandel et al., 2006; Bai et al., 2007). Instead of screening crystallization conditions directly by in situ proteolysis, it may be advisable to apply limited proteolysis of a protein prior to crystallization in order to test the suitability of the protease (e.g. (Forsgren et al., 2009)). It is possible that in situ proteolysis will produce a fragment enabling successful crystallization of a protein, but subsequent interference of the protease with crystal growth may hamper crystal quality. In such a case, the cleavage site can be established using SDS-PAGE and mass spectrometry or other means of sequencing and the construct then reengineered for optimal crystal quality (Little et al., 2012). Although only one or at best a few proteases were tested in the context of crystallization in most cases, the use of in situ proteolysis with a larger number of proteins in structural genomics projects (Dong et al., 2007; Wernimont and Edwards, 2009) points to some proteases that seem to be particularly useful for paving the road to diffraction-quality crystals. Thus, trypsin, chymotrypsin and subtilisin appear to have been used successfully more often than other proteases. However, while one may want to try these three first, it remains a good idea to test as many proteases as possible to increase the likelihood of obtaining diffraction-quality crystals.
Although in situ proteolysis to promote crystallization has been used in quite a few studies, no experimental protocol covering in detail individual steps, including the screening of proteases, establishing optimal concentrations for their use in situ, and establishing the cleavage site, has been published to date. The present protocol seeks to fill that void and provide biochemists and structural biologists with guidelines based on the successful crystallization of two membrane-bound P450 enzymes using in situ proteolysis that can be applied to any protein or protein domain resisting crystallization. In our efforts to crystallize the P450 17A1 and 17A2 enzymes from zebrafish in complex with substrate and/or inhibitor, we screened an increased set of proteases (12) compared with previous reports. Moreover, we combined efforts to optimize expression using alterative leader sequences at the N-terminal end with the identification of fragments by in situ proteolysis that yield to crystallization. To establish optimal conditions for in situ proteolytic crystallization, we tested a wide range of concentrations and time ranges. As well, we used a combination of SDS-PAGE and Edman sequencing to rapidly and reliably identify fragment size and cleavage sites.
Although the expanded set of proteases tested undoubtedly constitutes an advantage over published accounts of the use of in situ proteolysis to promote crystallization and relying on one or a few proteases (it is possible that the authors may not have disclosed how many were tested or not mentioned those that proved to be unhelpful), it is likely that inclusion of even more proteases may proved beneficial for the crystallization success rate. We relied on commercially available protease kits (Proti Ace and Proti-Ace 2; Hampton Research Inc., Aliso Viejo, CA; https://hamptonresearch.com). However, many more proteases than those included in the kits can be purchased individually from diverse vendors.
In a further extension of the in situ proteolysis approach to macromolecular crystallization, we tested whether mutation to arginine of a residue in the N-terminal region of P450 17A1 in combination with trypsin would promote crystallization. Trypsin cleaves C-terminal to Arg or Lys (Olson et al., 2004), and we had established that trypsin-catalyzed cleavage at the corresponding arginine in the N-terminal region of P450 17A2 promoted crystallization. Although the mutation in the N-terminal sequence of P450 17A1 ultimately proved not to be conducive to the production of diffraction-quality crystals of this enzyme, variations in the length or amino acid composition of flexible N- and C-terminal tails or internal loop regions could turn out to be fruitful for promoting crystal growth.
Anticipated Results
The in situ proteolytic crystallization approach is expected to be beneficial for proteins with hydrophobic (including membrane-spanning domains such as those found in microsomal cytochrome P450 enzymes) and/or flexible N-terminal regions in general that are targeted for analysis by single crystal X-ray diffraction or small angle X-ray scattering (SAXS). For the latter application it is important to identify protein domains or fragments that lack flexible N- or C-terminal regions that interfere with the conformational analysis of the proteins and their complexes in solution. Unlike the P450 17A1 and 17A2 proteins from zebrafish for which in situ proteolysis proved key to successful crystal structure determination (Pallan, Nagy et al., 2015), the human P450 17A1 protein could be crystallized by somewhat shortening the N-terminal leader sequence and the first residue visualized in the electron density was Leu-31 (DeVore and Scott, 2012). In the crystal lattice hydrophobic N-termini of human P450 17A1 mediate dimer formation, whereby β1 strands are part of an extended β-sheet (Figure 7). Dimer formation by human P450 17A1 persists in solution, but unlike for crystal lattice formation, where associations among neighboring molecules are a prerequisite, oligomerization hampers solution studies such as SAXS and complicates conformational analysis of proteins and their complexes (Putnam et al., 2007). Conversely, the N-terminally truncated forms of zebrafish P450 17A1 and 17A2 do not dimerize in solution, rendering them suitable for scattering experiments in principle. Beyond the use for structural or biophysical applications, purified proteins after undergoing proteolytic cleavage can also be employed in functional studies and the technique is therefore of interest to structural biologists, biophysicists and enzyme chemists alike.
Figure 7.

Dimerization of catalytic domains under formation of an antiparallel β-sheet, mediated by N-terminal hydrophobic tails in the crystal structure of human P450 17A1 in complex with abiraterone (DeVore and Scott, 2012).
Time Considerations
Steps 1–2, generating the expression vector: 1–2 weeks
Steps 3–6, expressing the target protein: 4–5 days
Steps 7–9, purifying the target protein: 2–3 days (using your own columns; 1–2 days with prepacked columns)
Step 7, nickel affinity purification: 1 day
Step 8, ion exchange chromatography: 0.5–1 day
Step 9, Detergent screen: 7 days Step 10, size exclusion chromatography: 2 hours
Step 11, preparations for crystallization experiments: 2–5 hours
Step 12, in situ proteolysis: 1 day
Step 13, protein crystallization: 3–15 days
Step 14, N-terminal Edman sequencing: 1–3 days
Acknowledgments
Supported by US NIH grant R01 GM103937. We would like to thank Drs. F. P. Guengerich, M. R. Waterman and P. S. Pallan for valuable advice and helpful discussions, and Dr. N. Kagawa for an initial draft of Figure 2.
Literature Cited
- Bai Y, Auperin TC, Tong L. The use of in situ proteolysis in the crystallization of murine CstF-77. Acta Cryst F. 2007;63:135–138. doi: 10.1107/S1744309107002904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Civril F, Hopfner KP. Crystallization of mouse RIG-I ATPase domain: in situ proteolysis. Methods Mol Biol. 2014;1169:27–35. doi: 10.1007/978-1-4939-0882-0_3. [DOI] [PubMed] [Google Scholar]
- DeVore NM, Scott EE. Structures of cytochrome P450 17A1 with prostate cancer drugs abiraterone and TOK-001. Nature. 2012;482:116–119. doi: 10.1038/nature10743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong A, Xu X, Edwards AM. In situ proteolysis for protein crystallization and structure determination. Nat Methods. 2007;4:1019–1021. doi: 10.1038/nmeth1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaur RK, Kupper MB, Fischer R, Hoffmann KMV. Preliminary X-ray analysis of a human VH fragment at 1.8 Å resolution. Acta Cryst D. 2004;60:965–967. doi: 10.1107/S0907444904004834. [DOI] [PubMed] [Google Scholar]
- Guengerich FP. Human cytochrome P450 enzymes. In: Ortiz de Montellano PR, editor. Cytochrome P450: Structure, Mechanism, and Biochemistry. 3. Kluwer Academic/Plenum Press; New York: 2005. pp. 377–353. [Google Scholar]
- Guengerich FP, Martin MV. Purification of cytochromes P450: products of bacterial recombinant expression systems. Methods Mol Biol. 2006;320:31–37. doi: 10.1385/1-59259-998-2:31. [DOI] [PubMed] [Google Scholar]
- Forsgren N, Lamont RJ, Persson K. A crystallizable form of the Streptococcus gordonii surface antigen SspB C-domain obtained by limited proteolysis. Acta Cryst F. 2009;65:712–714. doi: 10.1107/S1744309109021046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson S, Roversi P, Espina M, Deane JE, Birket S, Picking WD, Blocker A, Picking WL, Lea SM. Expression, limited proteolysis and preliminary crystallographic analysis of IpaD, a component of the Shigella flexneri type III secretion system. Acta Cryst F. 2006;62:865–868. doi: 10.1107/S1744309106027047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson EF, Stout CD. Structural diversity of eukaryotic membrane cytochrome P450s. J Biol Chem. 2013;288:17082–17090. doi: 10.1074/jbc.R113.452805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi K, Suzuki T, Dohmae N, Ishitani R, Nureki O. Crystallization and preliminary X-ray crystallographic analysis of YfcM: an important factor for EF-P hydroxylation. Acta Cryst F. 2014;70:1236–1239. doi: 10.1107/S2053230X14015726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb DC, Waterman MR. Unusual properties of the cytochrome P450 superfamily. Phil Trans R Soc B. 2013;368:20120434. doi: 10.1098/rstb.2012.0434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little DJ, Whitney JC, Robinson H, Yip P, Nitz M, Howell PL. Combining in situ proteolysis and mass spectrometry to crystallize Escherichia coli PgaB. Acta Cryst F. 2012;68:842–845. doi: 10.1107/S1744309112022075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandel CR, Gebauer D, Zhang H, Tong L. A serendipitous discovery that in situ proteolysis is essential for the crystallization of yeast CPSF-100 (Ydh1p) Acta Cryst F. 2006;62:1041–1045. doi: 10.1107/S1744309106038152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller WL, Auchus RJ. The molecular biology, biochemistry, and physiology of human steroidogenesis and its disorders. Endocrin Rev. 2011;32:81–151. doi: 10.1210/er.2010-0013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monk BC, Tomasiak TM, Keniya MV, Huschmann FU, Tyndall JDA, O’Connell JD, III, Cannon RD, McDonald JG, Rodriguez A, Finer-Moore JS, Stroud RM. Architecture of a single membrane spanning cytochrome P450 suggests constraints that orient the catalytic domain relative to a bilayer. Proc Natl Acad Sci USA. 2014;111:3865–3870. doi: 10.1073/pnas.1324245111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen JV, Ong SE, Mann M. Trypsin cleaves exclusively C-terminal to arginine and lysine residues. Mol Cell Proteomics. 2004;3:608–614. doi: 10.1074/mcp.T400003-MCP200. [DOI] [PubMed] [Google Scholar]
- Ortiz de Montellano PR, editor. Cytochrome P450: Structure, Mechanism, and Biochemistry. 3. KluwerAcademic/Plenum Publishers; New York: 2005. [Google Scholar]
- Pallan PS, Nagy LD, Lei L, Gonzalez E, Kramlinger VM, Azumaya CM, Wawrzak Z, Waterman M, Guengerich FP, Egli M. Structural and kinetic basis of steroid 17α,20-lyase activity in teleost fish cytochrome P450 17A1 and its absence in cytochrome P450 17A2. J Biol Chem. 2015;290:3248–3268. doi: 10.1074/jbc.M114.627265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pallan PS, Wang C, Lei L, Yoshimoto FK, Auchus RJ, Waterman MR, Guengerich FP, Egli M. Human cytochrome P450 21A2, the major steroid 21-hydroxylase: structure of the enzyme-progesterone substrate complex and rate-limiting C-H bond cleavage. J Biol Chem. 2015;290:13128–13143. doi: 10.1074/jbc.M115.646307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putnam CD, Hammel M, Hura GL, Tainer JA. X-ray solution scattering (SAXS) combined with crystallography and computation: defining accurate macromolecular structures, conformations and assemblies in solution. Q Rev Biophys. 2007;40:191–285. doi: 10.1017/S0033583507004635. [DOI] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera - a visualization system for exploratory research and analysis. J Comp Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Richardson TH, Jung F, Griffin KJ, Wester M, Raucy JL, Kemper B, Bornheim LM, Hassett C, Omiecinski CJ, Johnson EF. A universal approach to the expression of human and rabbit cytochrome P450s of 2C subfamily in Escherichia coli. Arch Bioch Biophys. 1995;323:87–96. doi: 10.1006/abbi.1995.0013. [DOI] [PubMed] [Google Scholar]
- Taneja B, Patel A, Slesarev A, Mondragon A. Structure of the N-terminal fragment of topoisomerase V reveals a new family of topoisomerases. EMBO J. 2006;25:398–408. doi: 10.1038/sj.emboj.7600922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wernimont A, Edwards A. In situ proteolysis to generate crystals for structure determination: an update. PloS One. 2009;4:e5094. doi: 10.1371/journal.pone.0005094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B, Lei L, Kagawa N, Sundaramoorthy M, Banerjee S, Nagy LD, Guengerich FP, Waterman MR. Three-dimensional structure of steroid 21-hydroxylase (cytochrome P450 21A2) with two substrates reveals locations of disease-associated variants. J Biol Chem. 2012;287:10613–10622. doi: 10.1074/jbc.M111.323501. [DOI] [PMC free article] [PubMed] [Google Scholar]