

SUMMARY
Pancreatic β cells undergo postnatal maturation to achieve maximal glucose-responsive insulin secretion, an energy intensive process. We identify estrogen-related receptor γ (ERRγ) expression as a hallmark of adult, but not neonatal β cells. Postnatal induction of ERRγ drives a transcriptional network activating mitochondrial oxidative phosphorylation, the electron transport chain, and ATP production needed to drive glucose-responsive insulin secretion. Mice deficient in β cell-specific ERRγ expression are glucose intolerant and fail to secrete insulin in response to a glucose challenge. Notably, forced expression of ERRγ in iPSC-derived β-like cells enables glucose-responsive secretion of human insulin in vitro, obviating the need for in vivo ‘maturation’ to achieve functionality. Moreover, these cells rapidly rescue diabetes when transplanted into β cell-deficient mice. These results identify a key role for ERRγ in β cell metabolic maturation, and offer a reproducible, quantifiable and scalable approach for in vitro generation of functional human β cell therapeutics.
Graphical abstract
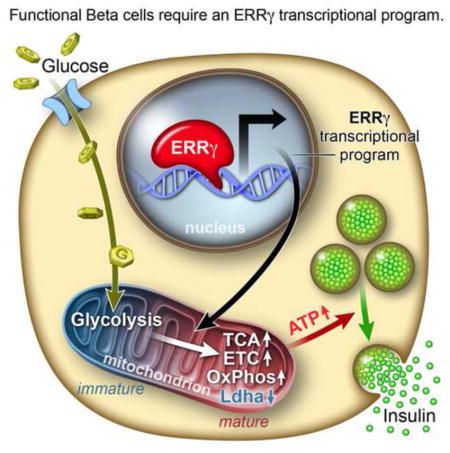
INTRODUCTION
In healthy individuals, pancreatic β cells secrete insulin in response to nutrients such as glucose, amino acids and free fatty acids to regulate blood glucose levels and lipid metabolism (Prentki et al., 2013), while β cell failure or impaired function is associated with both type 1 and type 2 diabetes (Ashcroft and Rorsman, 2012). The continuing challenges in treating diabetes placed the development of transplantable β cells as one of the central goals of stem cell therapy. While human embryonic and pluripotent stem cells (hESCs and hPSCs) offer this potential, it has been difficult to realize this therapeutic promise. The recent descriptions of ES-derived β-like cells reflect both the progress and ongoing challenges in achieving functionally mature β cells in vitro (Pagliuca et al., 2014; Rezania et al., 2014). Interestingly, transplantation of these pre-functional cells into mice results in a progressive in vivo maturation, presumably facilitated by the complex internal milieu reflecting a process that may be too complex to achieve in cell culture.
β cells are known to facilitate glucose-stimulated insulin secretion (GSIS) through increased mitochondrial oxidative ATP production (Prentki et al., 2013; Supale et al., 2012). While the cellular components required for GSIS are well established, the key transcriptional networks that regulate β cell metabolism and insulin secretion remain poorly understood.
Nuclear receptors are ligand–dependent transcription factors that play central roles in controlling development, growth and metabolism (Evans and Mangelsdorf, 2014). Estrogen related receptors are orphan nuclear receptors represented by three paralogs in mammals, ERRα (NR3B1, Esrra), ERRβ (NR3B2, Esrrb) and ERRγ (NR3B3, Esrrg). ERRβ is known to play an essential role in embryonic stem cell maintenance (Feng et al., 2009; Festuccia et al., 2012) while ERRα and ERRγ regulate oxidative pathways such as the tricarboxylic acid (TCA) cycle, the electron transport complex (ETC), and oxidative phosphorylation (OXPHOS). Notably, genetic studies in mice have revealed distinct functions for ERRα and ERRγ (Alaynick et al., 2007; Dufour et al., 2007; Luo et al., 2003). While whole body ERRα knockout (ERRαKO) mice are developmentally normal, they are lean and resistant to high fat diet-induced obesity (Luo et al., 2003). In contrast, whole body ERRγ knockout (ERRγKO) mice have significant developmental abnormalities and marked postnatal lethality attributed to defects in energy utilization, severely limiting their experimental utility (Alaynick et al., 2007).
Here we report that ERRγ is a master regulator of β cell maturation in vivo and that this function can be recapitulated in the in vitro setting. Accordingly, mice lacking ERRγ specifically in pancreatic β cells (βERRγKO mice) are glucose intolerant due to impaired insulin secretion. Reciprocally, in vitro expression of ERRγ in human iPSC-derived β-like cells yields functional and transplantable glucose-responsive cells capable of restoring glucose homeostasis in type 1 diabetic mice. These studies reveal that functional, transplantable β cells can be generated and quantified in vitro without the need for kidney capsule maturation in patients.
RESULTS
Postnatal islets acquire oxidative features
β cells are known to functionally mature postnatally, including acquiring the ability to robustly secrete insulin in response to high glucose (Bliss and Sharp, 1992; Grill et al., 1981; Lavine et al., 1971). Consistent with an immature phenotype, islets isolated from 2 week-old neonatal mice are unable to secrete insulin appropriately in response to a high glucose challenge (Figure 1A). To identify critical pathways required for glucose-stimulated insulin secretion (GSIS), we compared the transcriptomes of isolated islets during postnatal maturation. Consistent with β cell terminal differentiation being essentially completed postnatally, and the fact that adult β cells are formed by self-duplication rather than stem-cell differentiation (Dor et al., 2004), the expression of genes known to regulate pancreatic endocrine development in rodents and humans are largely unchanged (Conrad et al., 2014) (Figure S1A, Table S1A). While increased Pdx1, MafA and Nkx6.1, and decreased MafB expression are observed with maturation, in agreement with these genes being required for adult β cell function (Conrad et al., 2014), the expression of the majority of transcription factors implicated in β cell development is largely unchanged (Figure S1A, Table S1A). Functional analyses of the transcriptomes revealed that genes involved in cell proliferation are down-regulated during islet maturation, including known positive β cell proliferation regulators Pdgfrα, Pdgfrβ, Pdgfβ and Fgfr1, in line with the observed decline in clonal β cell expansion in adult islets (Figure 1B, Figure S1B, S1C and Table S1B) (Chen et al., 2011; Dor et al., 2004; Teta et al., 2005). In contrast, up-regulated genes are associated with metabolic pathways, particularly glucose metabolism and ATP biosynthesis pathways, including electron transport chain (ETC), Oxidative Phosphorylation (OxPhos) and ion channel-related exocytosis. Given the high mitochondrial activity of adult β cells, we hypothesized that this induction of metabolic genes may be critical for acquiring proper GSIS function during maturation. Consistent with this hypothesis, mitochondrial genes known to regulate GSIS, including malate dehydrogenase (Mdh1), pyruvate carboxylase (Pcx), and components of OxPhos including Cox6a2, Ndufa4 and Ndufs2, are more highly expressed, while lactate dehydrogenase (Ldha), a suppressor of GSIS, has reduced expression in adult compared to neonatal β cells (Xu et al., 2008) (Zhao and Rutter, 1998) (Figure 1C, 1F and Table S1B). These results suggest that a key metabolic transition may occur in islet β cells during postnatal functional maturation. Of particular note, expression of Errγ, a known mitochondrial gene regulator, is progressively induced during islet maturation (Figure 1C and 1D). This induction of Errγ expression is similarly observed in primary β cells isolated from mouse insulin-GFP (MIP-GFP) mice, where Errγ expression is ~5 fold higher in adult compared to neonatal β cells (Figure S1D–F). These findings, combined with the predominant expression of Errγ in endocrine islets compared to exocrine cells, as evidenced by the positive staining of islets in ERRγ-LacZ knock-in mice (Alaynick et al., 2007) and the enrichment of Errγ expression in islets compared to whole pancreas (Figure 1E and Figure S1G), are suggestive of a specific role for Errγ in orchestrating a metabolic transition in endocrine cells required for GSIS.
Figure 1. Islets acquire oxidative features postnatally.

(A) Glucose-stimulated insulin secretion from freshly isolated mouse neonatal (< 14 days) and adult (> 12 weeks) islets after sequential perfusion with 3mM and 20mM glucose for 30 min (reported as % insulin content, 10 islets per assay, n=6). (B–C) Heatmaps of transcriptional changes in islets during postnatal maturation. Row Z-score of down-regulated (B), and up-regulated (C) genes. (D) Relative Errγ expression in isolated islets measured by qPCR. (E) Relative Errγ expression in murine 12 wk old heart, liver, white adipose (WAT), pancreas and islets (n=4). X-gal staining indicating ERRγ expression in islets from 12 wk old ERRγ knock-in mice (top panel). (F) Relative expression of Ldha and selected mitochondrial metabolic genes during postnatal islet maturation, as measured by qPCR. (n=3). Data represent the mean ±s.e.m. *p<0.01 Student’s unpaired t-test. See also Figure S1.
ERRγ is required for glucose-stimulated insulin secretion
To investigate the proposed role of ERRγ in the functional maturation of pancreatic β cells, we generated β cell-specific Errγ knockout (βERRγKO) mice by crossing Errγfl/fl mice with rat insulin 2 promoter (RIP)-Cre mice. βERRγKO mice are born at the expected Mendelian frequency and exhibit normal body weights, ad lib fed glucose levels, and life expectancies (Figure S2A–C). The RIP-Cre recombinase selectively decreased Errγ expression by 80% in βERRγKO compared to wild-type Errγfl/fl islets without significantly affecting hypothalamic Errγ expression, in agreement with recent similar reports (Tang et al., 2013) (Figure S2A). However, βERRγKO mice were glucose intolerant compared to Errγfl/fl (WT) and RIP-Cre (WT(RIP-Cre)) cohorts, as determined by glucose tolerance tests (GTTs) (Figure 2A, and Figure S2D, S2E). While no significant differences in insulin sensitivity are seen, βERRγKO mice fail to increase insulin secretion in response to a glucose challenge (Figure 2C, 2D). Notably, this βERRγKO phenotype is exaggerated by metabolic stress after weaning. βERRγKO mice fed a high fat-high sucrose diet for 4 weeks from 4 weeks of age displayed more pronounced glucose intolerance and defects in insulin secretion, without any significant change in insulin sensitivity (Figure 2B, C, E).
Figure 2. ERRγ expression in β cells is required for glucose homeostasis.

(A–B) Intra-peritoneal glucose tolerance test (IP-GTT, 2g/kg, 8 wk old mice) of ERRγfl/fl (WT n=8), βERRγKO (n=6), and RIP-Cre (WT (RIP-Cre), n= 3) mice on a normal chow diet (NCD) (A), and a high fat diet (HFD, 4 weeks of HFD after weaning) (B). (C) Intra-peritoneal insulin tolerance test (IP-ITT) of WT and βERRγKO mice on NCD (9 wk old, WT n=4, βERRγKO n=5) and HFD (5 wk HFD after weaning; WT n=8, βERRγKO n=8). (D–E) Serum insulin level during IP-GTT in 8 wk old mice under NCD (WT n=4, βERRγKO n=12, WT(RIP-Cre) n=5) (D), and after 4 wks HFD (WT n=6, βERRγKO n=6, and WT(RIP-Cre), n=6) (E). Bar graphs indicate relative area under the curve (AUC). (F) Relative Errγ and Insulin 2 (Ins2) expression, (G) ex vivo glucose-stimulated (20mM) or KCl-stimulated (20mM) insulin secretion, and (H) oxygen consumption rate (OCR) in adenoviral-EGFP (Ad-Control) and adenoviral-Cre (Ad-ERRγKO) infected ERRγfl/fl islets from 12 wk old mice. Data represent the mean ±s.e.m. *p<0.05 Student’s unpaired t-test. See also Figures S2 and S3.
The inability of βERRγKO mice to secrete insulin in response to a glucose challenge is phenocopied in both inducible β cell-specific deletion (βERRγKO ER+Tam) and pancreatic-specific ERRγKO (PERRγKO) mouse models. βERRγKO ER mice treated with tamoxifen (7 days sequential i.p. injection) show a 75% reduction in islet Errγ expression and exhibit glucose intolerance similar to that observed in the developmental βERRγKO mice (Figure S2F, S2G). Furthermore, mice lacking Errγ in the entire pancreas generated by crossing Errγfl/fl mice with PDX1-Cre mice (PERRγKO mice), display impaired glucose tolerance and reduced insulin secretion compared to WT mice (Figure S2H–S2K). Collectively, these results suggest that islet Errγ expression is essential for proper GSIS function and whole body glucose homeostasis when challenged with elevated blood glucose levels.
Morphologically, islets isolated from βERRγKO mice maintained on a normal chow diet are indistinguishable from control islets, based on hematoxylin and eosin (H&E) staining and immunohistochemical analysis (Figure S3A top panels). However, when stressed by a high fat diet (HFD), significant increases in β cell mass, insulin content, and islet size are seen in βERRγKO compared to control islets (Figure S3A–S3G). These changes suggest that the glucose-intolerant phenotype of βERRγKO mice is due to a defect in GSIS rather than β cell mass reduction. To explore this notion, we evaluated the effect of transient Errγ deletion ex vivo on GSIS. Adenoviral-induced Cre-recombination in Errγfl/fl (Ad-ERRγKO) islets reduces Errγ expression by ~75% compared to control adenovirus EGFP-Errγfl/fl (Ad-Control) islets without affecting insulin2 expression (Figure 2F). Notably, the ability of Ad-ERRγKO islets to secrete insulin in response to a glucose challenge is almost totally abrogated ex vivo, while the sensitivity to KCl is largely intact (Figure 2G). Furthermore, loss of Errγ selectively in β cells (βERRγKO mice) or the entire pancreas (PERRγKO mice) reduces islet insulin secretion in response to nutrients such as glucose, L-leucine and L-glutamine (Figure S3H and S3I, respectively). As ERRγ regulates mitochondrial oxidative phosphorylation and metabolism in heart (Alaynick et al., 2007; Dufour et al., 2007), skeletal muscle (Narkar et al., 2011) and brain (Pei et al., 2015), we asked whether ERRγ is required for mitochondrial function in islets. Ad-Control islets respond robustly with a 2-fold increase in their oxygen consumption rate (OCR) when stimulated with 20mM glucose. In contrast, Ad-ERRγKO islets fail to increase their OCR in response to the glucose challenge (Figure 2H). Consistent with these results, Errγ knockdown in the rat clonal β cell line INS-1 similarly reduces OCR, cellular ATP production and GSIS function in response to a glucose challenge (Figure S3J–S3M). These results suggest that ERRγ regulation of β cell energy metabolism is required for GSIS.
ERRγ is required for β cell metabolic maturation
As mitochondrial function and morphology are tightly correlated (Anello et al., 2005; Hackenbrock, 1966), we investigated whether structural changes were detectable in βERRγKO β cell mitochondria. Electron microscopy revealed that the insulin and proinsulin granules, and the overall mitochondrial number are not affected by Errγ deletion (Figure 3A, 3B). However, mitochondrial swelling leading to a significant increase in mitochondrial volume and disrupted cristae structure are seen in the Errγ-deficient β cells, hallmarks of functionally defective mitochondria in β cells (Figure 3A, 3C).
Figure 3. ERRγ is required for functional maturation of β cells.

(A) Electron microscopy images showing mitochondrial morphology, (B) mitochondria number, and (C) mitochondrial volume in β cells from ERRγfl/fl (WT) and βERRγKO islets (n=8, 12 wk old mice). (D) Functional annotation of dysregulated gene categories in βERRγKO islets identified by Gene Ontology (GO). (E) Relative expression of metabolic genes in βERRγKO (left panel) and pancreatic ERRγKO (ERRγfl/fl x PDX1-Cre, PERRγKO, right panel) islets, as measured by qPCR. (F) Heatmap of gene expression levels (Row Z-score) during functional maturation of islets compared to βERRγKO islets. (G) Heatmap of expression changes in selected metabolic and secretion/exocytosis pathway genes in βERRγKO islets. Data represent the mean ±s.e.m. *p<0.05 Student’s unpaired t-test. See also Figure S4.
To understand the molecular role of ERRγ in β cell function, we determined the transcriptional consequences of Errγ deletion. In the developmentally-deleted βERRγKO islets, RNA-Seq revealed that the expression of 4189 genes was altered, with almost equal numbers of genes down- and up-regulated (2008 and 2181 genes, respectively; false discovery rate [FDR]< 0.01, fold change [FC]> 1.5). A similar comparison in the transiently deleted Ad-ERRγKO islets by microarray analysis identified 2205 genes with altered expression, again with similar numbers of genes down- and up-regulated (1207 and 998 genes, respectively; FDR< 0.01, FC > 1.25). As defects in GSIS are observed ex vivo in both βERRγKO islets (Figure S3H) and Ad-ERRγKO islets (Figure 2G), we performed Gene Ontology (GO) analysis on the common, differentially expressed genes (232 down- and 239 up-regulated genes) to identify global cellular processes affected by Errγ deletion (Figure S4A, Table S1C). Consistent with the diabetic phenotype, ERRγ-regulated genes are associated with processes critical for β cell function including ATP biosynthesis, cation transport, oxidative phosphorylation, electron transport and secretion (Figure 3D). Furthermore, motif analyses of promoter regions identified ERR response elements (ERREs) in more than half of these differentially expressed genes (62.1% of down- and 64.6% of up-regulated genes) suggesting direct regulation by ERRγ (Figure S4B). In support of this notion, conventional ChIP assays performed in the mouse insulinoma cell line MIN-6 confirmed the direct binding of ERRγ to the promoter regions of Atp2a2 and Mdh1 (Figure S4C). The expression changes of selected genes relevant to metabolic pathways (Mdh1, Cox6a2, Atp2a2, Ndufs2 and Atp6v0a2) were confirmed by qPCR analysis in both βERRγKO and PERRγKO islets (Figure 3E, left and right panels, respectively). These results suggest that ERRγ is a global regulator of ATP biosynthesis and metabolic genes in islet β cells.
To further clarify the role of ERRγ in the functional maturation of β cells, we compared the postnatal transcriptional changes in islets with those induced by ERRγ deletion. Notably, loss of ERRγ abrogates a large number of the transcriptional changes associated with postnatal β cell maturation (Figure 3F, S4D). Specifically, the changes in expression of 74 genes up-regulated, and 35 genes down-regulated during maturation are lost in βERRγKO islets (Figure 3F). These dysregulated genes include genes involved in energy production (ATP biosynthesis, oxidative phosphorylation and ETC) and secretory/exocytosis pathways (Figure 3G). Collectively, these results suggest that ERRγ is not only required to maintain mitochondrial function in functionally mature β cells, but directly orchestrates many of the transcriptional changes that drive the postnatal maturation of these cells.
ERRγ drives the maturation of synthetic β-like cells
Generation of transplantable human β cells from pluripotent stem cells is a major goal of stem cell therapeutics. However, current iPSC-derived β-like cells resemble fetal cells in their inability to secrete insulin in response to a glucose challenge (Hrvatin et al., 2014; Pagliuca and Melton, 2013). Based on our proposed regulatory role for ERRγ in enhancing oxidative metabolism during β cell maturation, and the enrichment of ERRγ expression in adult compared to fetal human β cells (GSE56130), we asked whether overexpression of ERRγ could drive a metabolic maturation and thereby GSIS capability in human iPSC-derived β-like cells. To address this question, we initially optimized the differentiation protocol for producing insulin-positive β-like cells from human iPSCs, utilizing a human insulin promoter driven-GFP reporter for screening and isolation (Blum et al., 2012; D’Amour et al., 2006; Kroon et al., 2008; Kunisada et al., 2012; Schulz et al., 2012; Sneddon et al., 2012; Xie et al., 2013). In our optimized protocol, insulin-positive cells are generated from iPSCs, derived from a human endothelial cell line HUVEC, 18–21 days after initiation of differentiation (Figure 4A–4C). Expression profiling during iPSC differentiation confirmed the generation of β-like insulin-producing cells (defined as iβL cells). Specifically, expression of the pluripotent marker NANOG is lost upon initiation of differentiation, while the terminal β cell differentiation marker, insulin, appears 18–21 days after initiation of differentiation. The definitive endoderm marker SOX17, and the pancreatic progenitor marker HNF1β are transiently increased around day 5, and days 7–15 respectively (Figure S5A). Furthermore, the expression of additional β cell markers including PDX1, MAFA, NKX6-1, NEUROD1, GCK and VAMP2 are strongly induced at day 21 (Figure S5A). This optimized differentiation protocol reproducibly generates insulin-positive, PDX1-positive, glucagon-negative β-like cells with significant expression of NKX6-1 and MAFA (defined here as iβL cells, Figures S5A–S5G). Moreover, while iβL cells fail to secrete c-peptide in response to a glucose challenge, they are responsive to direct cellular depolarization-mediated insulin secretion by KCl (Figure S5H).
Figure 4. ERRγ promotes maturation of human iPSC-derived β-like cells.

(A) Schematic protocol for iPSC-derived β-like cell generation (ED, endoderm; PP, pancreatic progenitors; iβL, iPSC-derived β-like cells; iβeta, ERRγ-expressing iPSC-derived β-like cells). Growth factors and small molecules were added at each stage. Vitamin C, GABA and 13.6 mM glucose were in the base media for all stages. (B) Relative expression of human insulin during iPSC differentiation (C) Human insulin reporter-driven GFP expression (left panel) and phase contrast image (right) of day 22 iβL cells. (D) Intracellular (left) and extracellular (right) c-peptide concentrations of iβL cells after adenoviral infection: Ad-EGFP infected, iβLGFP cells, open bars; Ad-ERRγ infected, iβeta cells, red bars. (E) Induced c-peptide secretion in iβL cells, iβLGFP cells, iβeta cells, and human islets. (F) Functional annotation of upregulated gene categories in iβeta cells identified by Gene Ontology (GO). (G) Heatmap of expression changes in known β cell maker genes (left) and metabolic genes (right) in iβLGFP and iβeta cells (log2 ratio relative to undifferentiated iPSC). Data represent the mean ±s.e.m. *p<0.05 Student’s unpaired t-test. See also Figure S5.
Given the role of ERRγ in endogenous β cell metabolic maturation, we asked whether overexpression of ERRγ could rescue GSIS function in iPSC-derived β-like cells. To address this question, we infected day 22–25 iPSC-derived β-like cells with an adenoviral ERRγ (Ad-ERRγ) or control (Ad-GFP) vector and performed gene expression and functional analyses at days 25–30. Given the widespread use of insulin in culture media, we used human c-peptide levels as a surrogate measure of β-like cell-derived insulin. We found that Ad-ERRγ infection successfully restores ERRγ expression in iPSC-derived β-like cells, but does not significantly affect their intracellular c-peptide content (Figure 4D left panel, and Figure S5I). Encouragingly, Ad-ERRγ infection does significantly increase the c-peptide concentration in the culture media (Figure 4D, right panel). We next examined the GSIS ability of Ad-ERRγ infected iPSC-derived β-like cells. Control Ad-EGFP infected iPSC-derived β-like cells (defined as iβLGFP cells) are not able to secrete c-peptide in response to a glucose challenge. Remarkably, Ad-ERRγ infected iPSC-derived β-like cells (defined as iβeta cells) demonstrate enhanced c-peptide secretion in response to a glucose challenge, similar to that recorded for isolated human islets, in the absence of an increase in maximum exocytosis-secretion capacity measured by KCl stimulated insulin secretion (Figure 4E and Figure S5J). Transcriptomic analyses identified increases in iβeta cells of genes associated with the generation of precursor metabolites, oxidation reduction, electron transport chain (ETC), oxidative phosphorylation and mitochondrial organization, and decreases in genes associated with cell cycle (Figure 4F, 4G, Table S2). Interestingly, directed ERRγ expression alters the expression profile of metabolic pathway genes in iβeta cells towards that seen in human islets, without significant increases in the expression of KATP channel/secretion pathway or β cell lineage-specific genes (Figure 5A–C, Figure S5K and S6A). Furthermore, ERRγ overexpression improves the cristae structure, as well as the respiration function of mitochondria in iβeta cells (Figure 5D,E). These results describe the generation of a synthetic glucose-responsive β-like cell from iPSCs, and support our hypothesis that ERRγ-regulated mitochondrial metabolic pathways are required for GSIS.
Figure 5. ERRγ regulates oxidative capacity through metabolic gene expression inβ-like cells.

(A–C) Relative expression of metabolic (A), KATP channel related (B) and β cell lineage genes (C) in iβLGFP cells, iβeta cells and human islets, as determined by qPCR (n=3). (D) Electron microscopy images showing mitochondrial morphology (left panel) and crystallized insulin granules (right panels) in iβLGFP and iβeta cells, respectively. (E) Oxidative capacity of iβLGFP and iβeta cells, as measured by oxygen consumption rate (OCR). *p<0.05 Student’s unpaired t-test.
The ultimate goal is to produce transplantable β cells capable of restoring glucose homeostasis in the setting of diabetes. To determine whether our iβeta cells function in vivo, we utilized a Streptozocin (STZ)-induced diabetic NOD-SCID mouse model. Blood glucose levels of NOD-SCID mice treated with STZ (180 mg/kg i.p. injection) were monitored daily to confirm hyperglycemia (blood glucose levels >300mg/dL). 12 days after STZ injection, 10 million iPSC-derived β-like cells infected with Ad-ERRγ (iβeta cells) or Ad-GFP (iβLGFP cells) were transplanted into the kidney capsule (Figure S6B). Human islets and mouse islets were similarly transplanted as positive controls. Remarkably, the blood glucose levels of mice that received the iβeta cells began to normalize within days of transplantation, similar to those receiving functional human or mouse islets (Figure 5A,B). Furthermore, transplanted iβeta cells controlled blood glucose levels in recipient mice chronically (56 days, Figure 5B and S6C) and insulin-positive cells were detected in the kidney 2 months after transplantation (Figure S7). Importantly, in approximately 50% of chronically treated iβeta cell recipient mice, blood glucose levels are restored to non-diabetic levels (<250mg/dL) concomitant with a glucose-responsive phenotype, as demonstrated by glucose tolerance tests, similar to mice that received human islet transplantation (Figure 5C). Additionally, pronounced improvements in the circadian regulation of glucose and lipid metabolism, which is tightly controlled by insulin, are observed in the chronically treated iβeta cell recipient mice with normalized blood glucose levels (increased night time respiratory exchange ratio (RER) consistent with improved glucose utilization), and are accompanied by increased night time activity (Figure 5D). These results demonstrate that increased oxidative mitochondrial metabolism driven by ERRγ expression may be exploited to potentiate GSIS function in human iPSC-derived β-like-cells.
DISCUSSION
Fetal development occurs under conditions of low oxygen tension and steady maternal glucose such that physiologic systems including the pancreas are functionally poised in the prenatal state. Postnatally, oxidative metabolism becomes dominant and intermittent feeding exposes the pancreas to dramatic changes in blood glucose. In particular, weaning has been reported to trigger a β cell maturation step characterized by enhanced glucose-stimulated oxidative phosphorylation and insulin secretion (Jacovetti et al., 2015; Stolovich-Rain et al., 2015). We demonstrate a direct linkage between the expression of the orphan nuclear receptor ERRγ and pre- and postnatal β cell metabolism. Based on selective expression in adult, but not fetal/neonatal β cells, our findings suggest that ERRγ is a key driver of the oxidative metabolic gene network in mature β cells, and that its postnatal induction orchestrates the metabolic maturation of β cells. Based on these findings, we posited that reversing the deficiency of ERRγ expression in iPSC-derived β-like cells may improve their ability to secrete insulin in response to a glucose challenge. Consistent with this notion, we show that targeted ERRγ expression in iPSC-derived β-like (iβeta) cells triggers a metabolic transformation that facilitates glucose-sensitive insulin secretion (GSIS). While significant variability and heterogeneity is observed in the human iβeta preparations (which we attribute to technical differences in ERRγ expression and differentiation conditions, e.g. Figure S5D and Table S6) transplantation of these cells into the kidney capsule not only restores glucose homeostasis in STZ-induced Type I diabetic mice, but re-establishes circadian rhythmicity to metabolic substrate usage, a process regulated by β cells.
As a known activator of mitochondrial function including oxidative metabolism, this dependence on ERRγ expression suggests that very high cellular energy levels are needed to achieve and maintain glucose responsiveness (Dhawan et al., 2015; Jacovetti et al., 2015; Stolovich-Rain et al., 2015). Remarkably, as a single gene, ERRγ appears sufficient to induce a gene network necessary to overcome this metabolic roadblock in iPSC-derived β-like cells. In contrast, β cell-specific ERRγ-deficient mice are glucose intolerant and fail to appropriately secrete insulin in response to a glucose challenge. Thus, genome-orchestrated metabolic maturation seems to be a critical step in both the metabolic maturation of endogenous β cells in vivo and engineered β-like cells in vitro.
Poor glucose management is associated with long-term diabetic consequences including diabetic retinopathy, nephropathy and neuropathy. While long-acting insulin formulations and programmable delivery pumps provide durable therapeutic utility, they fail to fully replicate the glucose-responsiveness of pancreatic β cells. Human islet transplantations offer superior glucose management, but require immuno-suppressive drug regimens and are limited by the availability and viability of the transplanted cells. Though insulin independence can be achieved via islet transplantation, more than 50% of allotransplanted patients and virtually all autotransplanted patients are back on insulin therapy after 5 years. In both situations, transplantation of a larger mass of islets may alleviate some of the limitations. Patient-specific iPSC-derived β cells produced in unlimited supplies and potentially encapsulated in the newly developed alginate hydrogels (Vegas et al., 2016a; Vegas et al., 2016b) could resolve many of these concerns and is one of the major goals of stem cell replacement therapy.
Despite recent advances, including generating functional β-like cells from hESCs (Pagliuca et al., 2014; Rezania et al., 2014) and the in vivo functional maturation of in vitro differentiated pancreatic progenitor cells (Kroon et al., 2008), the underlying mechanisms of β cell functional maturation remain poorly understood. Whereas dynamic chromatin remodeling (Xie et al., 2013) and sympathetic innervation stimuli (Borden et al., 2013) are implicated, our finding that ERRγ coordinates a transcriptional program regulating increased oxidative metabolism provides novel mechanistic insight into the functional limitations preventing β cell maturation (Figure 7). In support of these observations, genetic and epidemiology studies have independently implicated ERRγ as a risk factor in the development of diabetes (Chuang et al., 2008; Rampersaud et al., 2007; Tennessen and Thummel, 2011), though its functional relevance was not understood.
Figure 7. ERRγ is required for the functional maturation of β cells and iβeta cells.

Schematic describing the role of ERRγ in regulating an oxidative switch necessary for glucose stimulated insulin secretion in β and iβeta cells.
Our work suggests that the single gene (ERRγ) can drive metabolic maturation in vitro. This has practical advantages for dissecting the cellular events underlying this postnatal maturation process. Hopefully, the pivotal role of ERRγ in generating glucose-responsive β cells will accelerate the development of patient-specific glucose-sensitive insulin-producing β cells by overcoming a long standing conceptual and molecular roadblock.
Experimental Procedures
Generation and maintenance of mouse lines
β cell-specific ERRγ-knockout mice (βERRγKO) were generated by crossing ERRγfl/fl (generously provided by Johan Auwerx) and RIP-Cre (B6N.Cg-Tg(Ins2-cre)25Mgn/J) mice on a pure C57BL/6J genetic background. Tamoxifen-inducible β cell-specific ERRγ-knockout mice were generated by crossing ERRγ fl/fl and RIP-CreER (STOCK Tg (Ins2-cre/Esr1)1Dam/J) mice. Pancreas-specific ERRγ-knockout mice (PERRγKO) were generated by crossing ERRγ fl/fl and PDX1-Cre (B6.FVB-Tg(PDX1-cre)6Tuv/J). Insulin promoter GFP (MIP-GFP) mice (Tg(Ins1-EGFP)1Hara) and all above Cre lines were purchased from Jackson Laboratory. The ERRγ-LacZ knock-in mice were described previously (Alaynick et al., 2007). Animals were maintained in a specific pathogen-free animal facility (SPF) on a 12-hour light-dark cycle at an ambient temperature of 23°C. Water and food were provided ad lib. All animal experiments used age-matched male mice. All procedures involving animals were performed in accordance with protocols approved by the IACUC and Animal Resources Department (ARD) of the Salk Institute for Biological Studies. Details on Tamoxifen-inducible mice are described in Supplemental Information.
Intra-peritoneal glucose (IP-GTT) or insulin (IP-ITT) tolerance tests
IP-GTTs were performed on overnight fasted mice. Blood glucose values were assessed before and at 15, 30, 60 and 120 min after intra-peritoneal administration of 2g/kg of glucose using glucose PILOT. Serum insulin levels were assessed before and at 5, 15 and 30 min after the intra-peritoneal administration of glucose using a Rat/mouse Insulin ELISA kit (Millipore). IP-ITT assays were performed on mice after a 6 hour fast with an injection of 0.75 U/ kg of insulin (Humalin R, Eli Lilly).
Isolation of pancreatic islets
Mouse pancreatic islets were isolated as previously described(Yoshihara et al., 2010) with slight modification. Briefly, 0.5mg/ml collagenase P (Roche REF11213873001, diluted in HBSS buffer, Gibco, 14170–112) was injected through the common bile duct, and the perfused pancreas dissected and incubated at 37°C for 21 min. Digested exocrine cells and intact islets were separated via centrifugation (900g for 15 min) over Histopaque-1077 (Sigma, H8889), and intact islets were manually selected. Human islets were provided by the Integrated Islets Distribution Program (IIDP) under an approved protocol. Additional information on human islets is provided in Table S3.
Insulin secretion assay
Insulin release from intact islets was monitored using batch incubation methods (Yoshihara et al., 2010). Overnight-cultured isolated pancreatic islets (RPMI-1640 supplemented with 10% (v/v) fetal bovine serum and 1% (v/v) Antibiotic-Antimycotic (Gibco) were pre-cultured at 37°C for 30 min (Krebs-Ringer bicarbonate buffer (KRBB) containing 129.4 mM NaCl, 3.7 mM KCl, 2.7 mM CaCl2, 1.3 mM KH2PO4, 1.3 mM MgSO4, 24.8 mM NaHCO3 (equilibrated with 5% CO2, 95% O2, pH7.4), 10mM HEPES and 0.2% (v/v) BSA (fraction V, Sigma) (KRBH) with 3 mM glucose). Pancreatic islets were then incubated in KRBH buffer (500 μl/10 islets) with 3 mM or 20 mM glucose to determine insulin secretion levels. After 30 min, islets were pelleted by centrifugation and insulin levels determined by ELISA (Rat/mouse Insulin ELISA KIT (Millipore, EZRMI-13K) for mouse and human islets, respectively). For hiPSC-derived cells, cells (1×106 cells/well in 24 well) were pre-cultured in 3mM glucose KRBH buffer (500 μl/well). The cells were then incubated in KRBH buffer (200 μl/well) with 3mM or 20mM glucose to determine c-peptide secretion levels as an indicator of insulin secretion levels. After 30 min, the cells were pelleted by centrifugation and c-peptide levels were determined in the supernatant using a human c-peptide ELISA KIT (Millipore, EZHCP-20K). Details for rat INS-1 cell culture, transfection and insulin secretion assay are in Supplemental Information.
Chromatin immunoprecipitation
Chromatin was prepared from mouse insulinoma, MIN-6 cells. Briefly, MIN-6 cells were cross-linked with 1% formaldehyde for 10 min, followed by the addition of glycine at 125 mM. Chromatin was sheared by enzymes (CHIP IT Express Kit, Active Motif, 53009) and immuno-precipitated with 2μg anti-H3, control mouse IgG, or anti-ERRγ antibodies. ChIP-qPCR primers are listed in Table S4.
Generation of Reporter hiPSC
Human induced pluripotent stem cells (hiPSCs) derived from HUVECs were obtained from the Stem Cell Core (Salk Institute) and maintained on matrigel (BD)-coated dishes in complete mTeSR1 Media (Ludwig et al., 2006). HiPSCs were infected with a human insulin GFP reporter (System Biosciences, SR10028PA-1; hINS-GFP-Zeo) and insulin producing cells selected with Zeocin. To generate hiPSCs stably expressing a human insulin reporter, a human insulin-GFP-EF1a-neo reporter was cloned into the 307bp human insulin promoter from pGZ-hInsulin vector (SR10028, System Biosciences), replacing the Nanog promoter in pGF-Nanog-EF1a-neo (System Biosciences, TR019PA1) with the insulin promoter (hINS-GFP-EF1a-Neo). A human Pdx1 GFP reporter lentivirus (System Biosciences, SR10039PA-1) was used to mark pancreatic specification. HiPSCs infected with fresh virus using Spinfection (800g, 1 hour) and hINS-GFP-EF1a-Neo were selected using G418 (100μg/ml for 1 week from 3 days after Spinfection, Corning, 30–234-Cl).
Differentiation of hiPSC to insulin-producing glucose-responsive cells
Reporter hiPSCs were cultured to 90% confluency in complete mTeSR Media, at which time the media was changed to 100ng/ml Activin (R&D Systems, 338-AC), 25ng/ml Wnt3a (R&D Systems, 5036-WN) in differentiation media (800ml DMEM/F12, 13.28g BSA, 10ml Glutamax, 560mg NaHCO3, 330mg thiamine, 100mg reduced glutathione, 3300 mg Vitamin C, 14μg Selenium, 10ml NEAA, 2ml Trace Element B, 1ml Trace Element C, 7μl β-ME, 2ml DLC, 2ml GABA, 2ml LiCl, 129.7μg pipecolic acid, Insulin 2mg up to 1000ml) for 2 days and then 100ng/ml Activin in differentiation media for another 2 days (Stage 1). Subsequently, media was replaced with differentiation media with 1μM dorsomorphin (Biovision, 1656–5), 2μM Retinoic Acid (Sigma, R2625–1G), 10μM SB431542 (Sigma, S4317–5MG) and 1% B27 supplement (Gibco, 17504-044) for 7 days (Stage 2). Media was then replaced with differentiation media with 10μM Forskolin (Sigma, F6886-25MG), 10 μM dexamethasone (Sigma, D4902-100MG), 10μM Alk5 inhibitor II (Calbiochem, 616452 or Enzo, ALX-270-445), 10mM Nicotinamide (Sigma, 72340-100G) and 1% B27 supplement with or without 1μM 3,3′,5-Triiodo-L-thyronine sodium salt (T3, Sigma, T6397-100MG) for 10 days (stage 3). Media was replaced every day (stage 1, Endoderm differentiation), every day or every other day (stage 2, Pancreatic specification) and every other day (stage 3, β-like cells). Cells were split once during differentiation. At day 5, cells were treated with Accutase for 5 min at 37°C. Released single cells were rinsed with PBS and spun at 1000 r.p.m for 5 min. The resulting cell pellet was resuspended in stage 2 media with or without 10μM Y-27632 (Reagents Direct, 53-B85) and a single cell suspension seeded at a 1:3 dilution on matrigel-coated plates. At days 22–25, the expression of human insulin genes and GFP were confirmed regularly by qPCR and fluorescence microscopy. Positive cells were used in subsequent experiments. EGFP-adenovirus (Ad-GFP) or human ERRγ adenovirus (Ad-ERRγ) were diluted in RPMI-1640 with 2% FCS, and 1×108 – 5×108 pfu/ml (MOI 100–500) used to infect β-like cells for 2 hrs. Media was changed to differentiation media containing 10μM Forskolin, 10μM dexamethasone, 10μM Alk5 inhibitor II, 10mM Nicotinamide and 1% B27 supplement with or without 1μM T3 for 3–5 days, then GFP-expressing β-like cells (iβLGFP cells) and ERRγ-expressing β-like cells (iβeta cells) were analyzed for RNA-Seq, EM, Seahorse and transplantation studies. Additional information for the differentiation protocol and small molecules screened or used for above protocol is listed in Table S5. During the differentiation, cells were maintained on matrigel (BD)-coated dishes. Raw data of c-peptide secretion assay results from these cells are shown in Table S6.
Virus production
Lentiviruses were produced using 2nd generation or 3rd generation lentiviral systems in HEK293T cell line. Adenovirus EGFP and Cre were purchased from Illinois University and Adenovirus ERRγ was purchased from Welgen, Inc.
NOD-SCID mice transplantation study
Immunodeficient NOD-SCID mice (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ) were purchased from Jackson Laboratory and bred and maintained in autoclaved cages in a SPF facility at the Salk Institute. Mice were rendered diabetic by a single i.p. high dose of streptozotocin (STZ; 180mg/kg, S0130–500MG) injection. 1 week after STZ injection, mice with blood glucose levels higher than 400mg/dL were used as recipients for transplantation analyses.
Human and mouse islets (200–500 islets or 500–1000 IEQ for mouse islets, 500–1000 islets or 1000–2000 IEQ for human islets per animal) or human iPSC-derived insulin-producing cells (iβLGPF or iβeta cells; 10 million cells per animal) were resuspended in 200ul RPMI-1640 media, loaded into laboratory tubing (SiLastic, 508–004) and centrifuged (400g for 1–2 min) to generate cell clusters in the center of the tubing. Cell clusters were transplanted (approximately 30–50ul) under kidney capsules in 8–16 week old STZ-injected diabetic mice. Ketamine (80mg/kg) and Xylazine (10mg/kg) were used as surgical anaesthetics and mice were placed on 37°C heat pads to recover.
Metabolic cage analyses
Metabolic cage analyses were conducted with a Comprehensive Lab Animal Monitoring System (Columbus Instruments). CO2 production, O2 consumption, Respiratory Exchange Rate (RER) and ambulatory counts by x-peak were determined for 5 consecutive days and nights, with at least 24 hour adaptation before data recording.
OCR and ECAR measurements
Oxygen consumption rates (OCRs) and extracellular acidification rates (ECARs) were recorded in 96-well plates using an XF96 seahorse (Seahorse Biosciences). Briefly, 70 isolated islets/well were pre-cultured with 3mM glucose XF DMEM media (pH7.4) for 1 hour prior to transfer to matrigel-coated XF96 cell culture plates and subsequent culture in 3mM glucose XF DMEM media (pH7.4). OCRs (reported as % change compared to 3mM glucose) were recorded during the incremental addition of glucose, up to a final concentration of 20mM glucose. Insulin-positive β-like cells, sorted by flow cytometry, were cultured for 3 days on matrigel-coated XF96 cell culture plates (1×105 cells/ well) prior to infection with adenoviral EGFP or ERRγ vectors. Infected cells were pre-cultured in XF DMEM media (pH 7.4) with 3mM glucose for 1 hour, then the media was changed to XF DMEM media (pH 7.4) with 20mM glucose, 1mM sodium pyruvate, and appropriate mitochondrial stress reagents (oligomycin, Fccp, Rotenone and Antimycin A), as instructed in the Mitostress Kit (Seahorse Biosciences).
Statistical methods
Results were expressed as the mean ± standard error of the mean (s.e.m.). Statistical comparisons were made using Student’s t-test. Statistically significant differences are indicated as *P<0.05.
Histology and Electron Microscopy
Details of methods for H&E staining, Immunohistochemistry, LacZ staining and electron microscopy are described in Supplemental Information.
Expression Analyses
Details of methods for quantitative RT-PCR, microarray, RNA-Seq library generation and high-throughput sequencing and analyses are described in Supplemental Information.
Supplementary Material
Figure 6. iβeta cells restore glucose homeostasis in diabetic mice.

(A) Acute effects on ad lib fed blood glucose levels in STZ-induced hyperglycemic NOD-SCID mice after mock transplantation (n=3), transplantation of iβLGFP cells (n=8), iβeta cells (n=7) and mouse islets (n=5). (B) Chronic effects on ad lib fed blood glucose levels of mock transplantation (n=3), transplantation of iβLGFP cells (n=14/12; 2 mice died 2 wks after transplantation), iβeta cells (n=13), mouse islets (n=5) and human islets (n=2). (C) Human c-peptide levels before and 15 minutes after a glucose challenge (2g/kg) in mice 2 months after the indicated transplantation (c-peptide ELISA limit of detection indicated by dotted line). (D) Oxygen consumption (VO2), carbon dioxide production (VCO2), Respiratory Exchange Ratio (RER), and ambulatory motion of STZ treated NOD-SCID mice 56 days after mock (n=4) or iβeta cell transplantation (mice with blood glucose levels <250mg/dL, n=4). See also Figures S6 and S7.
Acknowledgments
We thank J. Alvarez, S. Kaufman, B. Collins, M. He and H. Juguilon for technical assistance, and L. Ong and C. Brondos for administrative assistance. E.Y. was supported by Uehara Memorial Foundation, Japan Society for the Promotion of Science (JSPS) and Kanae Fundation for the promotion of medical science. Z.W. is supported by a CIRM training grant. R.M.E. is an Investigator of the Howard Hughes Medical Institute (HHMI) at the Salk Institute and March of Dimes Chair in Molecular and Developmental Biology, and is supported by National Institutes of Health (NIH) grants (DK057978, DK090962, HL088093, HL105278 and ES010337), the Glenn Foundation for Medical Research, the Leona M. and Harry B. Helmsley Charitable Trust (#2012-PG-MED002), Ipsen/Biomeasure, California Institute for Regenerative Medicine (CIRM), The Ellison Medical Foundation, and by a gift from Steven and Lisa Altman. C.L. and M.D. are funded by grants from the National Health and Medical Research Council of Australia Project Grants 512354, 632886 and 1043199.
Footnotes
AUTHOR CONTRIBUTIONS
E.Y., R.T.Y., A.R.A., M.D. and R.M.E. designed, supervised research and wrote the manuscript. E.Y., Z.W., C.S.L., S.F., M.A., Y.K., T.T., Y.D., R.T.Y., C.L. performed experiments. E.Y., R.T.Y., A.R.A., M.D. and R.M.E. analyzed data.
ONLINE CONTENT
Supplemental Methods, along with any additional Extended Data display items and Source Data, are available in the online version of the paper; references unique to these sections appear only in the online paper. The microarray data is deposited in the NCBI Gene Expression Omnibus and accessible through GEO Series accession number GSE56080. RNA-Seq data can be accessed on the NCBI Sequence Read Archive under the accessions SRP048600 and SRP048605.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alaynick WA, Kondo RP, Xie W, He W, Dufour CR, Downes M, Jonker JW, Giles W, Naviaux RK, Giguere V, et al. ERRgamma directs and maintains the transition to oxidative metabolism in the postnatal heart. Cell metabolism. 2007;6:13–24. doi: 10.1016/j.cmet.2007.06.007. [DOI] [PubMed] [Google Scholar]
- Anello M, Lupi R, Spampinato D, Piro S, Masini M, Boggi U, Del Prato S, Rabuazzo AM, Purrello F, Marchetti P. Functional and morphological alterations of mitochondria in pancreatic beta cells from type 2 diabetic patients. Diabetologia. 2005;48:282–289. doi: 10.1007/s00125-004-1627-9. [DOI] [PubMed] [Google Scholar]
- Ashcroft FM, Rorsman P. Diabetes mellitus and the beta cell: the last ten years. Cell. 2012;148:1160–1171. doi: 10.1016/j.cell.2012.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bliss CR, Sharp GW. Glucose-induced insulin release in islets of young rats: time-dependent potentiation and effects of 2-bromostearate. The American journal of physiology. 1992;263:E890–896. doi: 10.1152/ajpendo.1992.263.5.E890. [DOI] [PubMed] [Google Scholar]
- Blum B, Hrvatin SS, Schuetz C, Bonal C, Rezania A, Melton DA. Functional beta-cell maturation is marked by an increased glucose threshold and by expression of urocortin 3. Nature biotechnology. 2012;30:261–264. doi: 10.1038/nbt.2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borden P, Houtz J, Leach SD, Kuruvilla R. Sympathetic innervation during development is necessary for pancreatic islet architecture and functional maturation. Cell reports. 2013;4:287–301. doi: 10.1016/j.celrep.2013.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Gu X, Liu Y, Wang J, Wirt SE, Bottino R, Schorle H, Sage J, Kim SK. PDGF signalling controls age-dependent proliferation in pancreatic beta-cells. Nature. 2011;478:349–355. doi: 10.1038/nature10502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang JC, Cha JY, Garmey JC, Mirmira RG, Repa JJ. Research resource: nuclear hormone receptor expression in the endocrine pancreas. Molecular endocrinology. 2008;22:2353–2363. doi: 10.1210/me.2007-0568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad E, Stein R, Hunter CS. Revealing transcription factors during human pancreatic beta cell development. Trends in endocrinology and metabolism: TEM. 2014 doi: 10.1016/j.tem.2014.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Amour KA, Bang AG, Eliazer S, Kelly OG, Agulnick AD, Smart NG, Moorman MA, Kroon E, Carpenter MK, Baetge EE. Production of pancreatic hormone-expressing endocrine cells from human embryonic stem cells. Nature biotechnology. 2006;24:1392–1401. doi: 10.1038/nbt1259. [DOI] [PubMed] [Google Scholar]
- Dhawan S, Tschen SI, Zeng C, Guo T, Hebrok M, Matveyenko A, Bhushan A. DNA methylation directs functional maturation of pancreatic beta cells. The Journal of clinical investigation. 2015;125:2851–2860. doi: 10.1172/JCI79956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dor Y, Brown J, Martinez OI, Melton DA. Adult pancreatic beta-cells are formed by self-duplication rather than stem-cell differentiation. Nature. 2004;429:41–46. doi: 10.1038/nature02520. [DOI] [PubMed] [Google Scholar]
- Dufour CR, Wilson BJ, Huss JM, Kelly DP, Alaynick WA, Downes M, Evans RM, Blanchette M, Giguere V. Genome-wide orchestration of cardiac functions by the orphan nuclear receptors ERRalpha and gamma. Cell metabolism. 2007;5:345–356. doi: 10.1016/j.cmet.2007.03.007. [DOI] [PubMed] [Google Scholar]
- Evans RM, Mangelsdorf DJ. Nuclear Receptors, RXR, and the Big Bang. Cell. 2014;157:255–266. doi: 10.1016/j.cell.2014.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng B, Jiang J, Kraus P, Ng JH, Heng JC, Chan YS, Yaw LP, Zhang W, Loh YH, Han J, et al. Reprogramming of fibroblasts into induced pluripotent stem cells with orphan nuclear receptor Esrrb. Nature cell biology. 2009;11:197–203. doi: 10.1038/ncb1827. [DOI] [PubMed] [Google Scholar]
- Festuccia N, Osorno R, Halbritter F, Karwacki-Neisius V, Navarro P, Colby D, Wong F, Yates A, Tomlinson SR, Chambers I. Esrrb is a direct Nanog target gene that can substitute for Nanog function in pluripotent cells. Cell stem cell. 2012;11:477–490. doi: 10.1016/j.stem.2012.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grill V, Lake W, Freinkel N. Generalized diminution in the response to nutrients as insulin-releasing agents during the early neonatal period in the rat. Diabetes. 1981;30:56–63. doi: 10.2337/diab.30.1.56. [DOI] [PubMed] [Google Scholar]
- Hackenbrock CR. Ultrastructural bases for metabolically linked mechanical activity in mitochondria. I. Reversible ultrastructural changes with change in metabolic steady state in isolated liver mitochondria. The Journal of cell biology. 1966;30:269–297. doi: 10.1083/jcb.30.2.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hrvatin S, O’Donnell CW, Deng F, Millman JR, Pagliuca FW, Diiorio P, Rezania A, Gifford DK, Melton DA. Differentiated human stem cells resemble fetal, not adult, beta cells. Proceedings of the National Academy of Sciences of the United States of America. 2014 doi: 10.1073/pnas.1400709111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacovetti C, Matkovich SJ, Rodriguez-Trejo A, Guay C, Regazzi R. Postnatal beta-cell maturation is associated with islet-specific microRNA changes induced by nutrient shifts at weaning. Nature communications. 2015;6:8084. doi: 10.1038/ncomms9084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroon E, Martinson LA, Kadoya K, Bang AG, Kelly OG, Eliazer S, Young H, Richardson M, Smart NG, Cunningham J, et al. Pancreatic endoderm derived from human embryonic stem cells generates glucose-responsive insulin-secreting cells in vivo. Nature biotechnology. 2008;26:443–452. doi: 10.1038/nbt1393. [DOI] [PubMed] [Google Scholar]
- Kunisada Y, Tsubooka-Yamazoe N, Shoji M, Hosoya M. Small molecules induce efficient differentiation into insulin-producing cells from human induced pluripotent stem cells. Stem cell research. 2012;8:274–284. doi: 10.1016/j.scr.2011.10.002. [DOI] [PubMed] [Google Scholar]
- Lavine RL, Chick WL, Like AA, Makdisi TW. Glucose tolerance and insulin secretion in neonatal and adult mice. Diabetes. 1971;20:134–139. doi: 10.2337/diab.20.3.134. [DOI] [PubMed] [Google Scholar]
- Ludwig TE, Bergendahl V, Levenstein ME, Yu J, Probasco MD, Thomson JA. Feeder-independent culture of human embryonic stem cells. Nature methods. 2006;3:637–646. doi: 10.1038/nmeth902. [DOI] [PubMed] [Google Scholar]
- Luo J, Sladek R, Carrier J, Bader JA, Richard D, Giguere V. Reduced fat mass in mice lacking orphan nuclear receptor estrogen-related receptor alpha. Molecular and cellular biology. 2003;23:7947–7956. doi: 10.1128/MCB.23.22.7947-7956.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narkar VA, Fan W, Downes M, Yu RT, Jonker JW, Alaynick WA, Banayo E, Karunasiri MS, Lorca S, Evans RM. Exercise and PGC-1alpha-independent synchronization of type I muscle metabolism and vasculature by ERRgamma. Cell metabolism. 2011;13:283–293. doi: 10.1016/j.cmet.2011.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagliuca FW, Melton DA. How to make a functional beta-cell. Development. 2013;140:2472–2483. doi: 10.1242/dev.093187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagliuca FW, Millman JR, Gurtler M, Segel M, Van Dervort A, Ryu JH, Peterson QP, Greiner D, Melton DA. Generation of Functional Human Pancreatic beta Cells In Vitro. Cell. 2014;159:428–439. doi: 10.1016/j.cell.2014.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei L, Mu Y, Leblanc M, Alaynick W, Barish GD, Pankratz M, Tseng TW, Kaufman S, Liddle C, Yu RT, et al. Dependence of hippocampal function on ERRgamma-regulated mitochondrial metabolism. Cell metabolism. 2015;21:628–636. doi: 10.1016/j.cmet.2015.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prentki M, Matschinsky FM, Madiraju SR. Metabolic signaling in fuel-induced insulin secretion. Cell metabolism. 2013;18:162–185. doi: 10.1016/j.cmet.2013.05.018. [DOI] [PubMed] [Google Scholar]
- Rampersaud E, Damcott CM, Fu M, Shen H, McArdle P, Shi X, Shelton J, Yin J, Chang YP, Ott SH, et al. Identification of novel candidate genes for type 2 diabetes from a genome-wide association scan in the Old Order Amish: evidence for replication from diabetes-related quantitative traits and from independent populations. Diabetes. 2007;56:3053–3062. doi: 10.2337/db07-0457. [DOI] [PubMed] [Google Scholar]
- Rezania A, Bruin JE, Arora P, Rubin A, Batushansky I, Asadi A, O’Dwyer S, Quiskamp N, Mojibian M, Albrecht T, et al. Reversal of diabetes with insulin-producing cells derived in vitro from human pluripotent stem cells. Nature biotechnology. 2014 doi: 10.1038/nbt.3033. [DOI] [PubMed] [Google Scholar]
- Schulz TC, Young HY, Agulnick AD, Babin MJ, Baetge EE, Bang AG, Bhoumik A, Cepa I, Cesario RM, Haakmeester C, et al. A scalable system for production of functional pancreatic progenitors from human embryonic stem cells. PloS one. 2012;7:e37004. doi: 10.1371/journal.pone.0037004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sneddon JB, Borowiak M, Melton DA. Self-renewal of embryonic-stem-cell-derived progenitors by organ-matched mesenchyme. Nature. 2012;491:765–768. doi: 10.1038/nature11463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolovich-Rain M, Enk J, Vikesa J, Nielsen FC, Saada A, Glaser B, Dor Y. Weaning Triggers a Maturation Step of Pancreatic beta Cells. Developmental cell. 2015 doi: 10.1016/j.devcel.2015.01.002. [DOI] [PubMed] [Google Scholar]
- Supale S, Li N, Brun T, Maechler P. Mitochondrial dysfunction in pancreatic beta cells. Trends in endocrinology and metabolism: TEM. 2012;23:477–487. doi: 10.1016/j.tem.2012.06.002. [DOI] [PubMed] [Google Scholar]
- Tang T, Abbott MJ, Ahmadian M, Lopes AB, Wang Y, Sul HS. Desnutrin/ATGL activates PPARdelta to promote mitochondrial function for insulin secretion in islet beta cells. Cell metabolism. 2013;18:883–895. doi: 10.1016/j.cmet.2013.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tennessen JM, Thummel CS. Coordinating growth and maturation - insights from Drosophila. Current biology : CB. 2011;21:R750–757. doi: 10.1016/j.cub.2011.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teta M, Long SY, Wartschow LM, Rankin MM, Kushner JA. Very slow turnover of beta-cells in aged adult mice. Diabetes. 2005;54:2557–2567. doi: 10.2337/diabetes.54.9.2557. [DOI] [PubMed] [Google Scholar]
- Vegas AJ, Veiseh O, Doloff JC, Ma M, Tam HH, Bratlie K, Li J, Bader AR, Langan E, Olejnik K, et al. Combinatorial hydrogel library enables identification of materials that mitigate the foreign body response in primates. Nature biotechnology. 2016a doi: 10.1038/nbt.3462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vegas AJ, Veiseh O, Gurtler M, Millman JR, Pagliuca FW, Bader AR, Doloff JC, Li J, Chen M, Olejnik K, et al. Long-term glycemic control using polymer-encapsulated human stem cell-derived beta cells in immune-competent mice. Nature medicine. 2016b doi: 10.1038/nm.4030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie R, Everett LJ, Lim HW, Patel NA, Schug J, Kroon E, Kelly OG, Wang A, D’Amour KA, Robins AJ, et al. Dynamic chromatin remodeling mediated by polycomb proteins orchestrates pancreatic differentiation of human embryonic stem cells. Cell stem cell. 2013;12:224–237. doi: 10.1016/j.stem.2012.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Han J, Long YS, Epstein PN, Liu YQ. The role of pyruvate carboxylase in insulin secretion and proliferation in rat pancreatic beta cells. Diabetologia. 2008;51:2022–2030. doi: 10.1007/s00125-008-1130-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshihara E, Fujimoto S, Inagaki N, Okawa K, Masaki S, Yodoi J, Masutani H. Disruption of TBP-2 ameliorates insulin sensitivity and secretion without affecting obesity. Nature communications. 2010;1:127. doi: 10.1038/ncomms1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C, Rutter GA. Overexpression of lactate dehydrogenase A attenuates glucose-induced insulin secretion in stable MIN-6 beta-cell lines. FEBS letters. 1998;430:213–216. doi: 10.1016/s0014-5793(98)00600-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.