

ABSTRACT
Tissue microenvironment adjusts biological properties of different cells by modulating signaling pathways and cell to cell interactions. This study showed that epithelial–mesenchymal transition (EMT)/ mesenchymal–epithelial transition (MET) can be modulated by altering culture conditions. HPV E6/E7‐transfected immortalized oral keratinocytes (IHOK) cultured in different media displayed reversible EMT/MET accompanied by changes in cell phenotype, proliferation, gene expression at transcriptional, and translational level, and migratory and invasive activities. Cholera toxin, a major supplement to culture medium, was responsible for inducing the morphological and biological changes of IHOK. Cholera toxin per se induced EMT by triggering the secretion of interleukin 6 (IL‐6) from IHOK. We found IL‐6 to be a central molecule that modulates the reversibility of EMT based not only on the mRNA level but also on the level of secretion. Taken together, our results demonstrate that IL‐6, a cytokine whose transcription is activated by alterations in culture conditions, is a key molecule for regulating reversible EMT/MET. This study will contribute to understand one way of cellular adjustment for surviving in unfamiliar conditions. J. Cell. Biochem. 116: 2552–2562, 2015. © 2015 The Authors. Journal of Cellular Biochemistry Published by Wiley Periodicals, Inc.
Keywords: IL‐6, CULTURE CONDITION, EPITHELIAL MESENCHYMAL TRANSITION (EMT), MESENCHYMAL EPITHELIAL TRANSITION (MET), CHOLERA TOXIN
Microenvironment is a crucial factor that regulates cellular growth and differentiation by modulating complex signaling pathways and intercellular interactions in different cell types [Abbott et al., 2008]. All viable organisms persistently respond to an altered environment to maintain homeostasis, and epithelial cells actively interact with their microenvironment to facilitate normal tissue homeostasis [Cousin et al., 2009]. Likewise, molecular crosstalk occurs between cancer cells and their microenvironment, which actively participates in cancer initiation and progression [Ghotra et al., 2009]. Culture medium represents in vitro microenvironment, and different culture conditions have triggered diverse phenotypic alterations of cells [Yu et al., 2009]. For human cancer cells, biochemical, and morphological changes have been induced by various culture conditions [Terasaki et al., 1986; Winkenwerder et al., 2003].
Epithelial–mesenchymal transition (EMT) is defined as a phenomenon in which epithelial cells are transformed into motile mesenchymal cells through a process known as mesenchymal metaplasia or the bidirectional differentiation of undifferentiated stem cells [Battifora, 1976]. EMT is a well‐known physiologic process in development [Lee et al., 2006]. Moreover, EMT plays a central role for invasion and metastasis of cancer cells [Niu et al., 2012]. Similarly, mesenchymal–epithelial transition (MET) occurs as a physiological process in embryonic development [Vainio and Lin, 2002], and MET enables migratory cancer cells to colonize and survive at remote sites for successful completion of metastasis [Brabletz et al., 2005]. EMT occurs at an early stage of carcinogenesis for migration and invasion, while MET occurs at a later stage for stabilization of invasive or migratory cells [Chaffer et al., 2006]. In this sense, EMT and MET are transient processes that occur sequentially to facilitate the formation of metastatic cancer [Brabletz et al., 2004].
Upon conducting an experiment in which culture medium was altered to assess the consequent changes in cultured cells, we made an interesting discovery: EMT/MET occurred reversibly in immortalized oral keratinocytes (IHOK) upon changing the culture medium in which the cells were grown. As EMT/MET is a coupled physiologic process that plays a pivotal role in development and cancer progression, the finding that EMT/MET can be reversibly induced by modifying microenvironment implies that developmental abnormalities or cancer progression can be controlled by making alterations to the environment. Hence, we attempted to search for key components and molecules in culture conditions that modulate EMT/MET. To achieve this goal, we closely examined reversible EMT that occurred in human papilloma virus (HPV) E6/E7‐transfected IHOK when they were grown in different culture media.
Because cholera toxin stimulates the growth of mammary cells, it has been used as a culture supplement for epithelial biology and carcinogenesis studies [Stampfer, 1982]. Cholera toxin affects G‐protein through the stimulation of adenylate cyclase, which results in an increased level of intracellular cAMP [Evans et al., 1987]. In turn, cAMP regulates IL‐6 as a transcription factor in a cell type‐specific manner [Hershko et al., 2002]. IL‐6, a multifunctional cytokine, has shown diverse effects depending on cell types. That is, IL‐6 induced differentiation of neuronal or neuroglial cells in malignant glioma cells [Kimura et al., 2001; Shu et al., 2011], while IL‐6 promoted cancer progression by inducing EMT in human breast or prostate cancer cell lines [Sullivan et al., 2009; Rojas et al., 2011].
From this study, we found that cholera toxin triggered the up‐regulated secretion of IL‐6, an E‐cadherin repressor. In turn, IL‐6 was responsible for the induction of reversible EMT, representing an adaptive process for IHOK cells to adjust to an altered culture condition. This study will contribute to understand one way of cellular adjustment for surviving in unfamiliar conditions, opening up the possibility of controlling incurable human diseases through modulation of tissue microenvironment.
MATERIALS AND METHODS
REAGENTS AND ANTIBODIES
Antibodies against E‐cadherin, snail, p21, CDK4, vimentin, and cyclin D1 were obtained from Cell Signaling Technology (Danvers, MA). Antibodies against p27 and CDK2 and Alexa Fluor 488‐conjugated anti‐rabbit IgG secondary antibody were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Anti‐actin, FITC monoclonal anti‐pan cytokeratin conjugate, and monoclonal anti‐vimentin cy3 conjugate were purchased from Sigma (St. Louis, MO). Type I collagen was purchased from Nitta Gelatin Inc (Osaka, Japan).
CELL CULTURE
IHOK cell line was established and reported previously [Lee et al., 2005]. To analyze medium‐dependent phenotypic changes, two kinds of keratinocyte culture media were used. IHOK cells were maintained in keratinocyte growth medium (KGM, Lonza, MD) with supplementary bullet kit (Lonza), which were then named as IHOK‐KGM. As an alternative culture medium, EF‐medium was used to culture IHOK, and the cells were named as IHOK‐EF. After IHOK‐EF cells were cultured for 120 days in EF‐medium, they were cultured again in KGM medium for 120 days, and the cell line was subsequently named as IHOK‐EFKGM. IHOK‐EF and IHOK‐EFKGM required 120 days for adjustment before the aforementioned culture method could be applied. When IHOK cells started to proliferate without floating, 1 × 106 cells were plated and then transferred to a new dish once in 3–4 days. The medium was changed every other day. Lovo colorectal cancer cell line, MCF‐7 human breast cancer cell line, and HT1080 human fibrosarcoma cells were obtained from American Type Culture Collection (ATCC, Manassas, VA). Lovo cells were maintained in minimum essential medium (MEM, gibco, Carlsbad, CA). MCF‐7 and HT1080 cells were maintained in Dulbecco's Modified Eagles Medium (DMEM, Gibco BRL). YD‐38 cells, an oral squamous cell carcinoma cell line established in our laboratory, were maintained in EF‐medium. MCF‐7 and Lovo cells with 1 × 106 cells/8 mL were seeded in 100 mm dishes. YD‐38 and HT1080 cells with 5 × 105 cells/8 mL were seeded in 100 mm dishes, respectively. Each medium was changed every other day, and the cells were transferred to a new dish once in 3 days. For the control of mesenchymal cells, normal oral gingival fibroblasts were used, which were derived from mucosal disease‐free patients who underwent wisdom tooth extraction. Informed consent was obtained from the patients for this study (IRB‐2‐2009‐0002). Gingival fibroblasts were selected by a versene solution (0.2% EDTA in PBS) supplemented with 0.04% glucose from the explanted tissues, and the selected cells were maintained in F‐medium. The detailed composition of each medium was presented in Supplementary Table S1.
IMMUNOBLOTTING
When the cells were grown overnight to 80% confluency, the cells (3 × 106 cells) were washed with cold PBS and lysed in Cell Lysis Buffer (Cell Signaling Technology). After lysis, protein complexes were boiled for 5 min in sodium dodecyl sulfate (SDS) sample buffer. Thirty microgram protein per sample was loaded on gel and separated by 10% SDS‐PAGE. Then, proteins were transferred to polyvinylidene fluoride membranes, and the membranes were blocked in 5% milk in Tris‐buffered saline containing tween 20 and then immunoblotted with appropriate antibodies.
CELL PROLIFERATION ASSAY
Proliferation of IHOKs was determined directly by counting cells. In brief, IHOKs cells (2 × 103) were seeded in 6‐well plates. The number of cells per well was measured in a time‐dependent manner. We detached the cells at indicated time points and centrifuged the cells with culture media. Supernatant medium was discarded, and the cells were resuspended in a fresh medium. Ten microliter of of culture medium‐cell mixture was placed in hematocytometer, and the number of cells was counted using microscope. In the proliferation assay conducted for the normalization of invasion assay, 2 × 104 IHOK cells were seeded in 6‐well plates.
IMMUNOFLUORESCENCE
IHOKs were seeded in a 4‐well chamber slide. Cells were fixed in 4% formaldehyde in PBS for 15 min, permeabilized in 0.5% triton X‐100 solution, and rinsed three times in PBS for 5 min each. Then cells were labeled with antibodies against E‐cadherin or snail (1:100), followed by incubation with Alexa Fluor 488‐conjugated anti‐rabbit IgG secondary antibodies. For cytokeratin and vimentin staining, cells were incubated with FITC monoclonal anti‐pan cytokeratin conjugate (1:200) and monoclonal anti‐vimentin cy3 conjugate (1:200), respectively. To detect DNA, cells were counterstained with DAPI, and the slides were mounted with mounting medium. Fluorescent images were captured and analyzed using LSM 510 laser scanning confocal microscope and software with 40× lens (Carl Zeiss Inc., Oberkochen, Germany).
TRANSWELL MIGRATION ASSAY
IHOKs were plated and cultured to 70% confluency. To evaluate the migratory activity of each cell line, we used each cell line's own culture medium to fill the lower chamber, for the migratory potential was altered by changing the medium. In brief, 2 × 104 cells/well were resuspended either in F‐medium without FBS or in KGM without the supplementary bullet kit. Then, the cells were seeded in the upper wells of 24‐well transwell chambers (Coster, Cambridge, MA) for 48 hr. The cells' own media were placed in the lower wells, and the cells migrated to the lower wells. After 48 hr incubation in 5%CO2 incubator, cells were fixed in 10% formalin solution and stained in 0.025% crystal violet (Junsei, Japan). Then, the number of migratory cells was counted in five fields at 200× magnification. The mean for each chamber was calculated and was normalized by counting the number of IHOK cells that were separately incubated in the same condition.
TRANSWELL INVASION ASSAY
The transwell invasion assay was carried out by coating the upper chamber with Type I collagen, and the rest of the procedure was same as the procedure for transwell migration assay explained above. The data were normalized by counting the number of IHOK cells that were separately incubated in the same condition.
GELATIN ZYMOGRAPHY
Gelatinolytic activities of MMP‐2 and MMP‐9 were analyzed by gelatin zymography. IHOKs were seeded in 100 mm dish and incubated for 16 hr. Cells were then incubated in proper media for 48 hr. The conditioned media were centrifuged and quantified for protein content. The samples were loaded on a 10% SDS polyacrylamide gel copolymerized with 0.2% gelatin. After electrophoresis, the gel was renaturated in zymogram renaturation buffer (Bio–Red Laboratories) at room temperature for 2 hr, and then incubated in zymogram development buffer (Bio–Red Laboratories) for 16 hr at 37°C. The gels were stained with Coomassie blue and then destained.
IHOKs XENOGRAFT IN ZEBRAFISH AND MICROSCOPIC OBSERVATION
Zebrafish were maintained in accordance with the standard guidelines [Nüsslein‐Volhard and Dahm, 2002]. Care and treatment of zebrafish were conducted in accordance with the guidelines established by the Animal Care and Ethics Committee of the Gwangju Institute of Science and Technology, Republic of Korea. After staining of cells, zebrafish embryos were de‐chorionized using micro‐forceps, anesthetized with 0.0016% tricaine, and positioned on their right side on a wet 1.0% agarose pad. IHOKs were detached from culture dishes using 0.05% trypsin‐EDTA and washed twice with PBS at room temperature. Cells were stained with 2 µg/ml DiI diluted in PBS and washed four times. 1 × 102 cells / ml were suspended in 10% FBS, which were then injected into the center of yolk sac using an injector (PV820 pneumatic picopump, World Precision Instruments, FL) equipped with borosilicate glass capillaries (World Precision Instruments, FL). The number of embryos exhibiting cell dissemination from the injection site was counted by inverted microscopy (Leica DMIRB microscope 090‐132.101, JH Technologies, Boston, MA), and the embryos were imaged at 4 days post injection (DPI) by upright microscopy (Leica DMRBE microscope 301‐371.011, JH Technologies).
MICROARRAY DATA ANALYSIS
IHOK‐KGM, IHOK‐EF, and IHOK‐EFKGM were cultured to 70% confluency. Total RNA was isolated using RNeasy kit (Qiagen) according to the manufacturer's instructions. Each total RNA sample (200 ng) was labeled and amplified using Low Input Quick Amp labeling kit (Agilent technologies, CA). The Cy3‐labeled aRNAs were resuspended in 50 µl of hybridization solution (Agilent technologies). After labeled aRNAs were placed on Agilent Sure Print G3 Human GE 8 × 60K array (Agilent technologies) and covered by a Gasket 8‐plex slide (Agilent technologies), the arrays were analyzed using an Agilent scanner with associated software. Gene expression levels were calculated with Feature Extraction v10.7.3.1 (Agilent technologies). Relative signal intensity for each gene was generated using the Robust Multi‐Array Average algorithm. The data were processed based on the median polish normalization method using Gene Spring GX 7.3.1 (Agilent technologies). The normalized and log‐transformed intensity values were then analyzed using Gene Spring GX 7.3.1 (Agilent technologies).
QUANTITATIVE REAL‐TIME PCR
Total RNA was extracted using RNeasy kit (Qiagen), and cDNA was synthesized from 1 µg of the total RNA by using Transcriptor First Strand cDNA synthesis Kit (Roche) according to the manufacturer' protocol. Real‐time PCR was carried out using SYBER green I Master (Roche applied science, Switzerland), and the results were analyzed using the Light Cycler 480 software. The primers used for real‐time PCR were presented in Supplementary Table 2.
ENZYME LINKED IMMUNOSORBENT ASSAY (ELISA)
Each IHOK cell line was cultured with proper culture medium for 48 hr in a 100 mm dish. Their conditioned media were harvested for the analysis of IL‐6 secretion. Concentration of IL‐6 was measured according to the manufacturer's protocol using IL‐6 ELISA kit (R&D, Minneapolis, MN).
STATISTICAL ANALYSIS
To determine if the difference between control and experimental groups is significant in western blot, invasion assay, transwell migration assay, RT‐PCR, and Zymography, Mann‐Whitney U test (SPSS Inc, IBM, Chicago) was used. P < 0.05 was considered to be statistically significant.
RESULTS
CHANGING THE CULTURE MEDIUM INDUCED PHENOTYPE CHANGES IN IHOK CELLS
When oral keratinocytes were immortalized by HPV E6/E7 transfection, KGM culture medium was used. Therefore, IHOK cells were maintained in KGM (IHOK‐KGM), and they showed typical epithelial cobblestone‐like morphology regardless of culture periods (Fig. 1Aa−d). When IHOK‐KGM cells were cultured in a different medium (EF‐medium) for 60 days, the cells lost cell to cell contacts, scattered, and exhibited elongated shape. After additional 60 days, the cells were completely changed to long spindle‐like, fibroblastic morphology (IHOK‐EF) (Fig. 1Ah). To examine whether this change was reversible, IHOK‐EF cells were moved back to KGM medium (IHOK‐EFKGM). For 30 days, IHOK‐EFKGM cells maintained its spindle‐like shape. But IHOK‐EFKGM cells were morphologically recovered to polygonal epithelial shape, the shape of IHOK‐KGM cells, in 120 days (Fig. 1Al).
Figure 1.
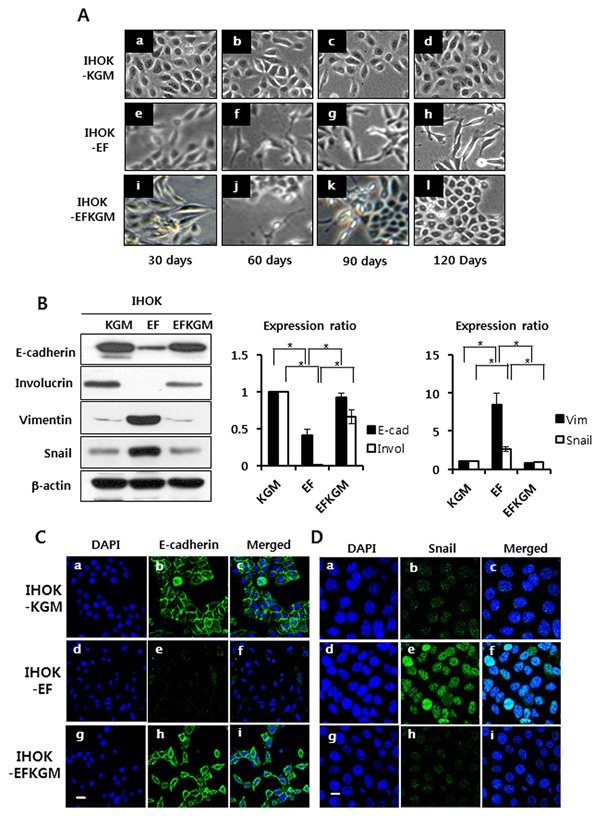
Modulation of IHOK phenotype by culture conditions. (A) a‐d, IHOK‐KGM was maintained in KGM medium for 120 days, which was used as a control IHOK cell line. e‐h, IHOK‐KGM was cultured in EF‐medium for 120 days (IHOK‐EF). i‐l, IHOK‐EF was re‐cultured in KGM medium, which was the original medium for the control cells, for 120 days (IHOK‐EFKGM). (B) Western blot was performed to measure the protein expression levels of E‐cadherin and involucrin, the epithelial markers, and vimentin and snail, the mesenchymal markers. (C), (D) Cells were labeled with antibody against E‐cadherin (Bar indicates 20μm.) or antibody against snail (Bar indicates 10 μm.) (1:100), followed by incubation with Alexa Fluor 488‐conjugated anti‐rabbit IgG secondary antibody.
To investigate whether spindle‐shaped cells of IHOK‐EF went through EMT, EMT marker proteins were examined. Expression of E‐cadherin, an epithelial differentiation marker, was reduced by 58% in IHOK‐EF compared with IHOK‐KGM, and involucrin expression was not detected in IHOK‐EF. The expressions of vimentin and snail, an E‐cadherin repressor, were 7.9‐fold and 2.65‐fold higher, respectively, in IHOK‐EF than in IHOK‐KGM (P < 0.05) (Fig. 1B). In immunofluorescence analysis, E‐cadherin and cytokeratin were clearly localized in IHOK‐KGM, but were hardly detected in IHOK‐EF (Fig. 1C and Supplementary Fig. S1). However, snail and vimentin were clearly localized in IHOK‐EF (Fig. 1D and Supplementary Fig. S1). Interestingly, IHOK‐EFKGM expressed E‐cadherin and cytokeratin with reduced snail and vimentin expression (Fig. 1B, C, D, and Supplementary Fig. S1). Taken together, these results indicate that culture conditions can modulate reversible EMT/MET in IHOK.
Proliferation of IHOKs was determined directly by counting cells. Proliferation of IHOK‐EF reduced to 80% and to 40% compared with IHOK‐KGM after 3 days and 7 days, respectively (*P < 0.05 by t‐test). Both expressions of p21 and p27 increased and cyclin D1 expression reduced in IHOK‐EF compared with IHOK‐KGM (Supplementary Fig. S2A, B).
IHOK‐EF CELLS HAVE HIGH MIGRTORY AND INVASIVE ACTIVITIES
To determine whether IHOK‐EF had high migratory and invasive activities, we carried out transwell migration and invasion assays. To exclude proliferation‐dependent effects of IHOK cells, the data were normalized by counting the number of IHOK cells that were separately incubated in the same condition used for migration and invasion assays. Briefly, we used 2 × 104 cells as was the case for migration and invasion assays. After 2 days, the 0.37% of IHOK‐KGM (total number 3.5 × 104), 4.72% of IHOK‐EF (total number 2.75 × 104), and 1.2% of IHOK‐EFKGM (total number 3.0 × 104) migrated, respectively (P < 0.05) (Fig. 2A). To confirm that the in vitro migratory activity of IHOKs is correlated with in vivo dissemination of xenografted IHOKs, we injected three groups of cells including IHOK‐KGM, IHOK‐EF, and IHOK‐EFKGM into zebrafish embryos. IHOK‐EF showed 1.3‐fold higher cell dissemination than IHOK‐KGM (P < 0.05). Cell dissemination was reduced by 13% in IHOK‐EFKGM compared with IHOK‐EF (P < 0.05) (Fig. 2B). For invasive activity, the same number of cells with migratory activity was seeded. After 2 days, the 2.37% of IHOK‐KGM, 8.0% of IHOK‐EF, 2.43% of IHOK‐EFKGM invaded, respectively (P < 0.05) (Fig. 2C). Gelatinolytic activity of MMP‐2 in IHOK‐EF was up‐regulated and 12‐fold higher than that in IHOK‐KGM, but the activity in IHOK‐EFKGM was reduced to the level of IHOK‐KGM (Fig. 2D). Taken together, these results indicate that IHOK‐EF acquired remarkable migratory and invasive activities, which were reversible, due to the changes in culture conditions.
Figure 2.
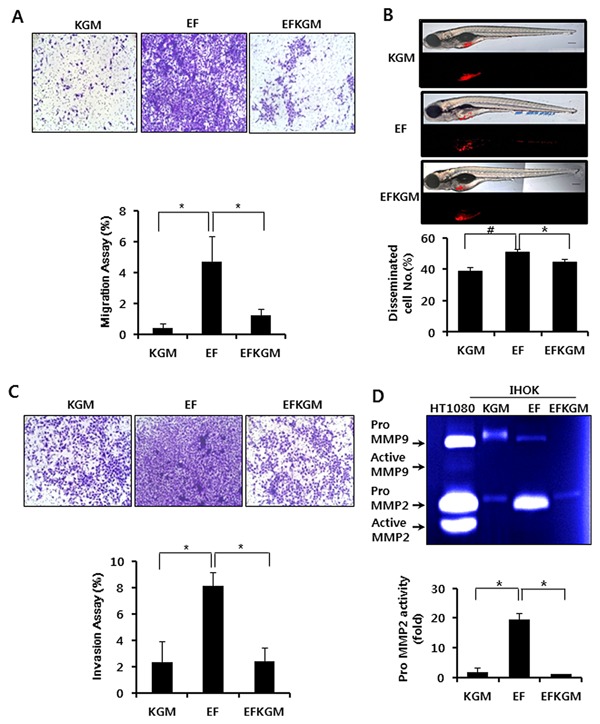
Migratory and invasion activities of IHOKs grown in different culture conditions. (A) Each cell line was seeded on a transwell chamber, and the number of cells that migrated to the other side of the chamber was counted after 48 hr. In the lower chamber, each culture medium was applied as a chemoattractant. Each assay was performed in triplicate (*P < 0.05). (B) Transplanted embryos were incubated in E3 media without methylene blue at 31°C. Four days later, the number of embryos exhibiting cell dissemination was counted (24 larvae/group). Representative fluorescent images (bottom) and merged DIC: fluorescent images (top). Blue arrow indicates cell mass disseminated from the injection site (yolk sac). Bar=200 μm. Data are presented as average ± SD of six independent assays. Student‘s t‐test was used for comparison between experimental groups (# = P < 0.01, * = P < 0.05). (C) For invasion assay, each cell line was seeded on a transwell chamber that was pre‐coated with collagen type I. After 48h, the number of cells that invaded the collagen and moved to the other side of the chamber was counted (*P < 0.05). (D) Each cell line was incubated in its culture medium for 48P < hr. The conditioned media were collected and subjected to gelatin zymography. Gelatin zymography was performed using electrophoresis gels that contained 0.2% gelatin (*P < 0.05).
GENE EXPRESSIONS IN IHOKs WERE ALTERED BY DIFFERENT CULTURE CONDITIONS
To identify whether gene expressions in IHOKs were altered by different culture conditions, we analyzed gene expression profiles by microarray. In the three types of IHOKs, mRNA levels were examined using Agilent Whole Human Genome Array which included 22602 human genes. Among the genes that were up‐regulated or down‐regulated in IHOK‐EF compared with IHOK‐KGM, those with >twofold changes were selected. Among the arrayed human genes, 13.6% (3080 genes) were altered in IHOK‐EF, including 1518 up‐regulated genes and 1562 down‐regulated genes. Figure 3 displays a reversible change in gene expression upon changing the culture medium in which IHOKs were grown: up or down‐regulated genes in IHOK‐EF returned to their original expression levels in IHOK‐EFKGM. Next, we selected E‐cadherin repressors, which were considered as principal mediators of property changes in IHOK, from the microarray data, and compared their expressions in three IHOK cell lines. As can be seen in Supplementary Table 3, five known E‐cadherin repressors including SNAI1, IL‐6, LOXL2, ZEB2, and TWIST1 showed higher expressions in IHOK‐EF than in IHOK‐KGM, and these expressions were reduced in IHOK‐EFKGM compared with IHOK‐EF.
Figure 3.
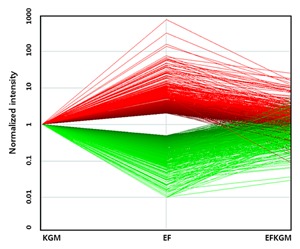
Microarray analysis of gene regulation pattern in IHOKs. Gene regulation levels in the three IHOK cell lines were assessed by microarray analysis. The x‐axis represents the three IHOK cell lines used in the experiment. The y‐axis represents the signal intensities of genes in the three cell lines. Signal intensities of multiple genes in IHOK‐EF and IHOK‐EFKGM were normalized to the signal intensities of genes in IHOK‐KGM. Then, the alterations in the signal intensity of each gene upon changing the culture medium were depicted by connecting the three corresponding dots for IHOK‐KGM, IHOK‐EF, and IHOK‐EFKGM. The red plots indicate upregulated genes, while the green plots indicate downregulated genes in IHOK‐EF compared with IHOK‐KGM.
IL‐6 MODULATES REVERSIBLE EMT IN IHOK CELLS
To validate the microarray expression data for E‐cadherin repressors, we performed real‐time RT‐PCR. Real‐time RT‐PCR data showed that the mRNA expression of IL‐6 was changed by the largest amount upon changing the culture medium, unlike the microarray data (Fig. 4A). Among the five E‐cadherin repressors, IL‐6 showed 55‐fold higher mRNA expression in IHOK‐EF than in IHOK‐KGM, but the expression was markedly reduced in IHOK‐EFKGM. Accordingly, we assumed IL‐6 to be a major contributor to the induction of reversible EMT in IHOK. Next, we examined the secretion level of IL‐6 in the conditioned media of the three IHOKs. Secretion level of IL‐6 was the highest in IHOK‐EF, being 39.3‐fold higher than that in IHOK‐KGM (Fig. 4B). To investigate whether IL‐6 induces reversible EMT in IHOK, we treated IHOK‐KGM with IL‐6. In 11 days, E‐cadherin expression was markedly decreased, while expressions of both snail and vimentin were increased. The down‐regulated expression of E‐cadherin in IL‐6‐treated IHOK‐KGM was reversed when IL‐6 treatment ceased, accompanied by down‐regulation of snail and vimentin (Fig. 4C and D). To evaluate whether IL‐6 is capable of modulating reversible EMT in cancer cell lines, we treated MCF7 breast cancer cells, LOVO colon cancer cells, and YD‐38 oral squamous cell carcinoma cells with IL‐6. As shown in supplementary Figure S3, IL‐6 induced reversible changes in the E‐cadherin expression of LoVo and YD‐38 cancer cells. MCF7 breast cancer cells exhibited a decrease in E‐cadherin expression 2 hr after adding IL‐6, but the expression was restored to the constitutional level after 24 hr
Figure 4.
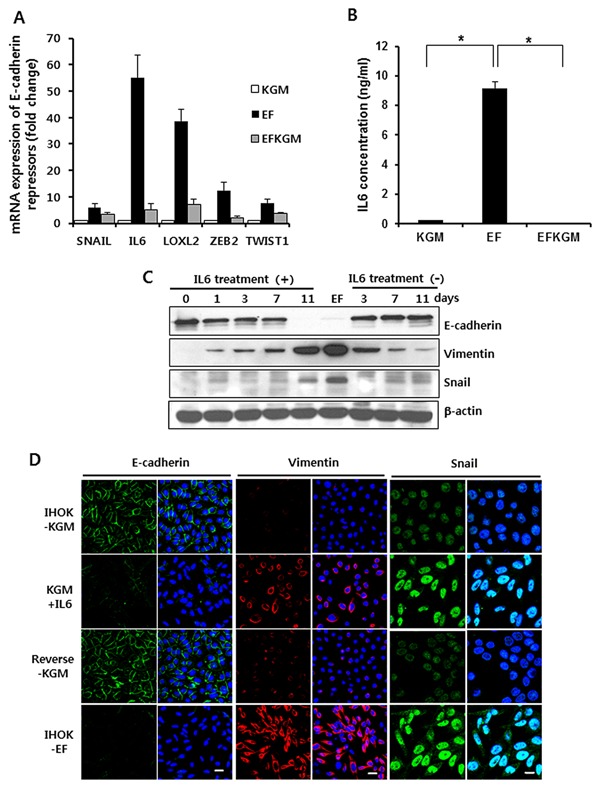
IL‐6‐mediated induction of reversible EMT in IHOKs. (A) mRNA expressions of SNAI1, IL‐6, LOXL2, ZEB2, and TWIST1 were analyzed by real‐time PCR in IHOK‐KGM, EF, and EFKGM cells. Real‐time PCR was carried out using SYBER green I Master, and the results were normalized to the housekeeping gene GAPDH. (B) IHOKs were incubated in each culture medium for 48h for detection of IL‐6. IL‐6 concentration was measured in each conditioned medium by using IL‐6 ELISA Kit (*P < 0.05). (C) IHOK‐KGM was cultured in KGM medium treated with 10 ng/ml IL‐6. Fresh medium containing IL‐6 was added every other day. Then, the cells were cultured again in original KGM medium that lacked IL‐6 for 11 days. (D) Immunofluorescence analysis confirmed the changes in the expression of E‐cadherin, vimentin, and snail in IHOK‐KGM that was treated with IL‐6. Bars indicate 20 μm in the panels of E‐cadherin and vimentin, and bar indicates 10 μm in the panels of snail.
CHOLERA TOXIN INDUCES EMT BY UPREGULATING THE PRODUCTION OF IL‐6
In addition to investigating IL‐6 for its involvement in reversible EMT, we also compared the supplements between KGM and EF‐medium, for changing the culture medium resulted in reversible EMT of IHOKs. Among the supplements to these two media, cholera toxin, and Triiodothyronine (T3) were included only in EF‐medium. For this reason, we cultured IHOK‐KGM with F‐medium, which had the same composition as EF‐medium except the five supplements including cholera toxin and T3, plus either cholera toxin or T3. As each group of cells was cultured for 120 days, the cells treated with cholera toxin lost cell to cell adhesion and showed spindle‐like cell morphology compared with the non‐treated control cells cultured only in F‐medium. However, the cells treated with T3 showed no morphological changes compared with the control cells (Fig. 5A). To examine whether the alteration of cholera toxin‐treated IHOK was correlated with the changes that occurred in IHOK‐EF, we measured the expression of EMT‐associated proteins and cell cycle‐regulatory proteins, as the altered expression of these proteins was characteristic of IHOK‐EF. In the cells treated with cholera toxin, expression of E‐cadherin was down‐regulated and expressions of vimentin and snail were up‐regulated compared with the control cells, suggesting that cholera toxin contributed to the induction of EMT in IHOK. In addition, expression of cyclin D1 was down‐regulated, while expression of p21, a CDK inhibitor, was up‐regulated compared with the control cells (Fig. 5B). These results were compatible with the results of IHOK‐EF shown in the supplementary figure S2A and B. Then, we looked into the relationship between IL‐6 and the EMT of IHOK induced by cholera toxin. First, we measured the secretion level of IL‐6 from the cells treated with cholera toxin or T3. The secretion level of IL‐6 from the cells treated with cholera toxin was 3.6‐fold higher than that from the control cells (P < 0.05) (Fig. 5C). Next, we analyzed whether a neutralizing antibody against IL‐6 can block EMT induced by cholera toxin. As IL‐6 concentration was markedly reduced (Fig. 5D), Cholera toxin‐induced EMT was gradually reversed as could be seen by increased E‐cadherin and reduced snail expressions (Fig. 5E). Therefore, these results suggest that cholera toxin‐mediated increase in the secretion of IL‐6 contributes to the induction of EMT in IHOK.
Figure 5.
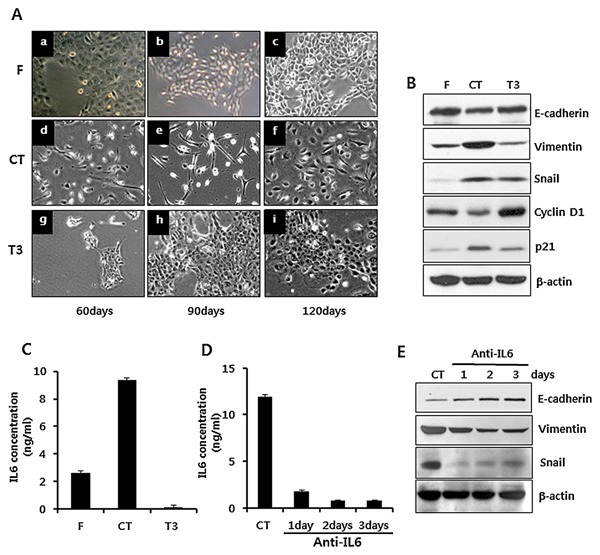
Effect of cholera toxin on the induction of EMT and secretion of IL‐6 in IHOK. (A) IHOK‐KGM cells were cultured in F‐medium (a, b, c), in F‐medium supplemented with 0.01 μg/ml of cholera toxin (d, e, f), and in F‐medium supplemented with 0.2 μg/ml of T3 (g, h, i) for 120 days, respectively. (B) Each group of cells was harvested, lysed, and immunoblotted for detection of EMT‐associated proteins (E‐cadherin, Vimentin, and snail) and proteins related to cell proliferation (Cyclin D1, p21). (C) Cells were incubated for 48 h in each culture medium, and each conditioned medium was analyzed for detection of secreted IL‐6. Data were presented as mean ± SD of three independent wells (*P < 0.05). (D) CT‐ treated IHOKs at the same passage were splitted into 4 dishes, and IL‐6 neutralizing antibody was added to each of the 3 dishes for one, two, or three days, respectively, at a concentration of 100 ng/ml. (E) Through western blot, the cells in the 4 dishes were analyzed for detection of EMT‐associated proteins.
DISCUSSION
This study showed that the changes of culture conditions induce reversible EMT in IHOKs, not only in morphological changes and proliferation but also in migratory and invasive activities (Fig. 1 and Fig. 2). Reversibility of EMT/MET has been documented with respect to a variety of culture conditions. For instance, hypoxia can induce EMT, while reoxygenation triggers MET to reversibly modulate EMT/MET in cancer cells [Jo et al., 2009]. In addition, estrogen promotes reversible EMT‐like transition and collective motility in MCF‐7 breast cancer cells [Planas‐Silva and Waltz, 2007], and high concentrations of glucose induce reversible EMT in human peritoneal mesothelial cells [Yu et al., 2009].
Our data showed that cholera toxin, the major difference between the supplements to KGM and EF‐medium (Supplementary Table 1), induced EMT in IHOK cells (Fig. 5). With regard to morphological changes, cholera toxin induced morphological alteration in a cell type specific manner: In ovarian cancer cells, it induces to polygonal shape through MET process [Han et al., 1999]; In malignant glial cells, it induces neuroglial or neuronal differentiation [Kimura et al., 2001]; In other types of cells including Chinese hamster ovary cells, it induces EMT‐like morphological changes [Rieber et al., 1975; Morinaga et al., 2001]. According to our results, cholera toxin is likely to trigger EMT in IHOK cells.
Cholera toxin has been used as a culture supplement, based on the knowledge that it stimulates the growth of mammary cells [Stampfer, 1982]. However, the conflicting data were reported with regard to cell growth. Cholera toxin enhanced the growth of human normal keratinocytes [Green, 1978] and type II pneumocytes [Uhal et al., 1998], whereas cholera toxin inhibited the proliferation of ovarian [Shaw et al., 2002] and lung cancer cells [Viallet et al., 1990]. On the other hand, accumulating data reported the relationship between cholera toxin and cell cycle regulatory proteins. Cholera toxin induces cell cycle arrest by blocking expression of cell cycle regulatory proteins such as cyclin D1 and CDK2 in malignant glioma cells [Li et al., 2007]. In addition, cAMP, which was elevated by cholera toxin, has been associated with cell cycle regulatory proteins. In cAMP‐induced inhibition of cell cycle progression, cyclin D1 has been major target for cAMP [L'Allemain et al., 1997] and p27, upregulated by cAMP, promotes growth arrest [Kato et al., 1994]. Collectively, the role of cholera toxin with regard to cell cycle may depend on the biological status of the involved cells. In our study, we found that cholera toxin induced EMT and growth inhibition in IHOK cells (Fig. 5).
In our study, we focused on the search for a modulator of EMT. Previous literature has indicated that E‐cadherin repressors play a major role in the induction of EMT by repressing E‐cadherin transcription [Mejlvang et al., 2007]. Thus, we selected five E‐cadherin repressors as potential candidates for EMT modulation among genes whose expressions were significantly altered upon changing culture conditions: SNAI1, IL‐6, LOXL2, ZEB2, and TWIST1. In particular, we focused on IL‐6, for the expression of IL‐6 was the highest among the five E‐cadherin repressors in IHOK‐EF (Fig. 4). We confirmed that IL‐6 significantly contributed to the reversible induction of EMT in IHOK by treating neutralizing antibody of IL‐6 in the media.
We tested whether the reversible EMT by IL‐6 could be induced in other human cancer cell lines such as oral squamous cancer cells, breast, and colon cancer cells. Notably, E‐cadherin expression was responsive to IL‐6 treatment in these cell lines, however, the responsive time point of each cell line was different. Moreover, we could not find further correlation between IL‐6 treatment and the expression of mesenchymal marker proteins and cell cycle‐associated proteins (data not shown). From these results, we considered that IL‐6 is a molecule crucial for the initiation of EMT. However, the responsiveness by IL‐6 treatment may be variable according to the cell types or biological conditions of the involved cells such as proliferation rate, the expression levels of endogenous IL‐6 and its receptors and other molecular factors.
Apart from the EMT‐inducing function of IL‐6 as an E‐cadherin repressor, IL‐6 has also been known to be directly involved in tumorigenesis. With regard to this function of IL‐6, accumulating evidence suggests that IL‐6 promotes cancer progression by inducing invasion [Sansone et al., 2007; Wu et al., 2012]. However, IL‐6 can also act as a cancer‐protective factor in oncogene‐induced cellular senescence, for it promotes the elimination of early neoplastic cells, similar to the transforming growth factor β that performs dual functions in carcinogenesis [Kuilman et al., 2008]. Thus, the cytostatic function of IL‐6 is likely to be context‐dependent as suggested from these data. Likewise, the function of IL‐6 as a molecular modulator of cellular adjustment to an altered environment depends on the context: EMT triggered by IL‐6 might provide better survival opportunity for cells that are in the process of adapting to a new, unfamiliar environment.
In this study, we showed that cholera toxin induced EMT in IHOK by up‐regulating IL‐6 secretion. Neutralization of IL‐6 in cholera toxin‐treated IHOKs made the cells undergo a process of MET at the protein expression level, confirming the role of IL‐6 for inducing EMT in IHOKs. However, we cannot exclude the possibility that other unknown components contained in the basal medium of KGM might be related to this reversible property by enhancing synergistic or antagonistic effects, for the composition of basal KGM medium could not be informed. Additionally, even if cholera toxin did regulate cell proliferation and EMT/MET in IHOKs, the effects of cholera toxin might be concentration‐specific, for the growth of cells was uninhibited and EMT not induced when the cells were treated with 10‐fold higher concentration of cholera toxin (data not shown) compared to the cholera toxin concentration in EF‐medium.
In summary, our study demonstrated that pleiotropic cytokine IL‐6, whose production is regulated by changes in culture conditions, modulates EMT/MET in an autocrine manner to facilitate adjustment of cells to an altered environment. This result will contribute to understand one way of cellular adjustment for surviving in unfamiliar conditions, opening up the possibility of controlling incurable human diseases through modulation of tissue microenvironment.
Supporting information
Supplementary Figure S1.
Supplementary Figure S2.
Supplementary Figure S3.
Supplementary Table S1.
Supplementary Table S2.
Supplementary Table S3.
Supplementary Figures Legend.
ACKNOWLEDGEMENTS
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (2009‐0094027).
All authors are in agreement with submitting and publishing this manuscript in your journal and also declare no competing interests.
REFERENCES
- Abbott DE, Bailey CM, Postovit LM, Seftor EA, Margaryan N, Seftor RE, Hendrix MJ. 2008. The epigenetic influence of tumor and embryonic microenvironments: How different are they?. Cancer Microenviron 1:13–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battifora H 1976. Spindle cell carcinoma: Ultrastructural evidence of squamous origin and collagen production by the tumor cells. Cancer 37:2275–2282. [DOI] [PubMed] [Google Scholar]
- Brabletz T, Jung A, Spaderna S, Hlubek F, Kirchner T. 2005. Opinion: Migrating cancer stem cells—an integrated concept of malignant tumour progression. Nat Rev Cancer 5:744–749. [DOI] [PubMed] [Google Scholar]
- Brabletz T, Spaderna S, Kolb J, Hlubek F, Faller G, Bruns CJ, Jung A, Nentwich J, Duluc I, Domon‐Dell C, Kirchner T, Freund JN. 2004. Down‐regulation of the homeodomain factor Cdx2 in colorectal cancer by collagen type I: An active role for the tumor environment in malignant tumor progression. Cancer Res 64:6973–6977. [DOI] [PubMed] [Google Scholar]
- Chaffer CL, Brennan JP, Slavin JL, Blick T, Thompson EW, Williams ED. 2006. Mesenchymal‐to‐epithelial transition facilitates bladder cancer metastasis: Role of fibroblast growth factor receptor‐2. Cancer Res 66:11271–11278. [DOI] [PubMed] [Google Scholar]
- Cousin B, Ravet E, Poglio S, De Toni F, Bertuzzi M, Lulka H, Touil I, Andre M, Grolleau JL, Peron JM, Chavoin JP, Bourin P, Penicaud L, Casteilla L, Buscail L, Cordelier P. 2009. Adult stromal cells derived from human adipose tissue provoke pancreatic cancer cell death both in vitro and in vivo. PLoS One 4:e 6278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans SW, Beckner SK, Farrar WL. 1987. Stimulation of specific GTP binding and hydrolysis activities in lymphocyte membrane by interleukin‐2. Nature 325:166–168. [DOI] [PubMed] [Google Scholar]
- Ghotra VP, Puigvert JC, Danen EH. 2009. The cancer stem cell microenvironment and anti‐cancer therapy. Int J Radiat Biol 85:955–962. [DOI] [PubMed] [Google Scholar]
- Green H 1978. Cyclic AMP in relation to proliferation of the epidermal cell: A new view. Cell 15:801–811. [DOI] [PubMed] [Google Scholar]
- Han X, Papadopoulos AJ, Jones T, Devaja O, Raju KS. 1999. Cholera toxin‐induced alteration of the phenotype and behaviour of an ovarian carcinoma cell line, SR8. Immunol Cell Biol 77:377–384. [DOI] [PubMed] [Google Scholar]
- Hershko DD, Robb BW, Luo G, Hasselgren PO. 2002. Multiple transcription factors regulating the IL‐6 gene are activated by cAMP in cultured Caco‐2 cells. Am J Physiol Regul Integr Comp Physiol 283:R1140–R1148. [DOI] [PubMed] [Google Scholar]
- Jo M, Lester RD, Montel V, Eastman B, Takimoto S, Gonias SL. 2009. Reversibility of epithelial‐mesenchymal transition (EMT) induced in breast cancer cells by activation of urokinase receptor‐dependent cell signaling. J Biol Chem 284:22825–22833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato JY, Matsuoka M, Polyak K, Massague J, Sherr CJ. 1994. Cyclic AMP‐induced G1 phase arrest mediated by an inhibitor (p27Kip1) of cyclin‐dependent kinase 4 activation. Cell 79:487–496. [DOI] [PubMed] [Google Scholar]
- Kimura M, Hidari KI, Suzuki T, Miyamoto D, Suzuki Y. 2001. Engagement of endogenous ganglioside GM1a induces tyrosine phosphorylation involved in neuron‐like differentiation of PC12 cells. Glycobiology 11:335–343. [DOI] [PubMed] [Google Scholar]
- Kuilman T, Michaloglou C, Vredeveld LC, Douma S, van Doorn R, Desmet CJ, Aarden LA, Mooi WJ, Peeper DS. 2008. Oncogene‐induced senescence relayed by an interleukin‐dependent inflammatory network. Cell 133:1019–1031. [DOI] [PubMed] [Google Scholar]
- L'Allemain G, Lavoie JN, Rivard N, Baldin V, Pouyssegur J. 1997. Cyclin D1 expression is a major target of the cAMP‐induced inhibition of cell cycle entry in fibroblasts. Oncogene 14:1981–1990. [DOI] [PubMed] [Google Scholar]
- Lee HJ, Guo HY, Lee SK, Jeon BH, Jun CD, Park MH, Kim EC. 2005. Effects of nicotine on proliferation, cell cycle, and differentiation in immortalized and malignant oral keratinocytes. J Oral Pathol Med 34:436–443. [DOI] [PubMed] [Google Scholar]
- Lee JM, Dedhar S, Kalluri R, Thompson EW. 2006. The epithelial‐mesenchymal transition: New insights in signaling, development, and disease. J Cell Biol 172:973–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Yin W, Wang X, Zhu W, Huang Y, Yan G. 2007. Cholera toxin induces malignant glioma cell differentiation via the PKA/CREB pathway. Proc Natl Acad Sci U S A 104:13438–13443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mejlvang J, Kriajevska M, Vandewalle C, Chernova T, Sayan AE, Berx G, Mellon JK, Tulchinsky E. 2007. Direct repression of cyclin D1 by SIP1 attenuates cell cycle progression in cells undergoing an epithelial mesenchymal transition. Mol Biol Cell 18:4615–4624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morinaga N, Kaihou Y, Vitale N, Moss J, Noda M. 2001. Involvement of ADP‐ribosylation factor 1 in cholera toxin‐induced morphological changes of Chinese hamster ovary cells. J Biol Chem 276:22838–22843. [DOI] [PubMed] [Google Scholar]
- Nüsslein‐Volhard C, Dahm R. 2002. Zebrafish : A practical approach. Oxford: Oxford University Press. [Google Scholar]
- Niu DF, Kondo T, Nakazawa T, Oishi N, Kawasaki T, Mochizuki K, Yamane T, Katoh R. 2012. Transcription factor Runx2 is a regulator of epithelial‐mesenchymal transition and invasion in thyroid carcinomas. Lab Invest 92:1181–1190. [DOI] [PubMed] [Google Scholar]
- Planas‐Silva MD, Waltz PK. 2007. Estrogen promotes reversible epithelial‐to‐mesenchymal‐like transition and collective motility in MCF‐7 breast cancer cells. J Steroid Biochem Mol Biol 104:11–21. [DOI] [PubMed] [Google Scholar]
- Rieber M, Bacalao J, Alonso G. 1975. Cholera toxin effects on cell growth accompanied by selective alterations in metabolite uptake and modification of cell surface proteins. Cancer Res 35:3009–3013. [PubMed] [Google Scholar]
- Rojas A, Liu G, Coleman I, Nelson PS, Zhang M, Dash R, Fisher PB, Plymate SR, Wu JD. 2011. IL‐6 promotes prostate tumorigenesis and progression through autocrine cross‐activation of IGF‐IR. Oncogene 30:2345–2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sansone P, Storci G, Tavolari S, Guarnieri T, Giovannini C, Taffurelli M, Ceccarelli C, Santini D, Paterini P, Marcu KB, Chieco P, Bonafe M. 2007. IL‐6 triggers malignant features in mammospheres from human ductal breast carcinoma and normal mammary gland. J Clin Invest 117:3988–4002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw TJ, Keszthelyi EJ, Tonary AM, Cada M, Vanderhyden BC. 2002. Cyclic AMP in ovarian cancer cells both inhibits proliferation and increases c‐KIT expression. Exp Cell Res 273:95–106. [DOI] [PubMed] [Google Scholar]
- Shu M, Zhou Y, Zhu W, Wu S, Zheng X, Yan G. 2011. Activation of a pro‐survival pathway IL‐6/JAK2/STAT3 contributes to glial fibrillary acidic protein induction during the cholera toxin‐induced differentiation of C6 malignant glioma cells. Mol Oncol 5:265–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stampfer MR 1982. Cholera toxin stimulation of human mammary epithelial cells in culture. In Vitro 18:531–537. [DOI] [PubMed] [Google Scholar]
- Sullivan NJ, Sasser AK, Axel AE, Vesuna F, Raman V, Ramirez N, Oberyszyn TM, Hall BM. 2009. Interleukin‐6 induces an epithelial‐mesenchymal transition phenotype in human breast cancer cells. Oncogene 28:2940–2947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terasaki T, Shimosato Y, Nakajima T, Tsumuraya M, Morinaga S, Hirohashi S, Yamaguchi K, Kato K, Ichinose H, Nagatsu T. 1986. Changes in cell characteristics due to culture conditions in cell lines from human small cell lung cancer. Jpn J Clin Oncol 16:203–212. [PubMed] [Google Scholar]
- Uhal BD, Papp M, Flynn K, Steck ME. 1998. Cholera toxin stimulates type II pneumocyte proliferation by a cyclic AMP‐independent mechanism. Biochim Biophys Acta 1405:99–109. [DOI] [PubMed] [Google Scholar]
- Vainio S, Lin Y. 2002. Coordinating early kidney development: Lessons from gene targeting. Nat Rev Genet 3:533–543. [DOI] [PubMed] [Google Scholar]
- Viallet J, Sharoni Y, Frucht H, Jensen RT, Minna JD, Sausville EA. 1990. Cholera toxin inhibits signal transduction by several mitogens and the in vitro growth of human small‐cell lung cancer. J Clin Invest 86:1904–1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkenwerder JJ, Palechek PL, Reece JS, Saarinen MA, Arnold MA, Cohen MB, Murhammer DW. 2003. Evaluating prostate cancer cell culturing methods: A comparison of cell morphologies and metabolic activity. Oncol Rep 10:783–789. [PubMed] [Google Scholar]
- Wu DW, Tsai LH, Chen PM, Lee MC, Wang L, Chen CY, Cheng YW, Lee H. 2012. Loss of TIMP‐3 promotes tumor invasion via elevated IL‐6 production and predicts poor survival and relapse in HPV‐infected non‐small cell lung cancer. Am J Pathol 181:1796–1806. [DOI] [PubMed] [Google Scholar]
- Yu MA, Shin KS, Kim JH, Kim YI, Chung SS, Park SH, Kim YL, Kang DH. 2009. HGF and BMP‐7 ameliorate high glucose‐induced epithelial‐to‐mesenchymal transition of peritoneal mesothelium. J Am Soc Nephrol 20:567–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1.
Supplementary Figure S2.
Supplementary Figure S3.
Supplementary Table S1.
Supplementary Table S2.
Supplementary Table S3.
Supplementary Figures Legend.