

ABSTRACT
High bone mass (HBM) can be an incidental clinical finding; however, monogenic HBM disorders (eg, LRP5 or SOST mutations) are rare. We aimed to determine to what extent HBM is explained by mutations in known HBM genes. A total of 258 unrelated HBM cases were identified from a review of 335,115 DXA scans from 13 UK centers. Cases were assessed clinically and underwent sequencing of known anabolic HBM loci: LRP5 (exons 2, 3, 4), LRP4 (exons 25, 26), SOST (exons 1, 2, and the van Buchem's disease [VBD] 52‐kb intronic deletion 3′). Family members were assessed for HBM segregation with identified variants. Three‐dimensional protein models were constructed for identified variants. Two novel missense LRP5 HBM mutations ([c.518C>T; p.Thr173Met], [c.796C>T; p.Arg266Cys]) were identified, plus three previously reported missense LRP5 mutations ([c.593A>G; p.Asn198Ser], [c.724G>A; p.Ala242Thr], [c.266A>G; p.Gln89Arg]), associated with HBM in 11 adults from seven families. Individuals with LRP5 HBM (∼prevalence 5/100,000) displayed a variable phenotype of skeletal dysplasia with increased trabecular BMD and cortical thickness on HRpQCT, and gynoid fat mass accumulation on DXA, compared with both non‐LRP5 HBM and controls. One mostly asymptomatic woman carried a novel heterozygous nonsense SOST mutation (c.530C>A; p.Ser177X) predicted to prematurely truncate sclerostin. Protein modeling suggests the severity of the LRP5‐HBM phenotype corresponds to the degree of protein disruption and the consequent effect on SOST‐LRP5 binding. We predict p.Asn198Ser and p.Ala242Thr directly disrupt SOST binding; both correspond to severe HBM phenotypes (BMD Z‐scores +3.1 to +12.2, inability to float). Less disruptive structural alterations predicted from p.Arg266Cys, p.Thr173Met, and p.Gln89Arg were associated with less severe phenotypes (Z‐scores +2.4 to +6.2, ability to float). In conclusion, although mutations in known HBM loci may be asymptomatic, they only account for a very small proportion (∼3%) of HBM individuals, suggesting the great majority are explained by either unknown monogenic causes or polygenic inheritance. © 2015 The Authors Journal of Bone and Mineral Research published by Wiley Periodicals, Inc. on behalf of American Society for Bone and Mineral Research (ASBMR).
Keywords: LRP5, SOST, ANABOLIC, SEQUENCING, PROTEIN MODELING
Introduction
Worldwide, fewer than 30 families have been reported with low density lipoprotein receptor‐related protein 5 (LRP5) high bone mass (HBM) (MIM# 603506). LRP5, a ubiquitous cell membrane co‐receptor, mediates canonical Wnt signaling and, in bone, facilitates osteoblastic bone formation.1 The 10 documented gain‐of‐function LRP5 mutations reported to date all lie in exons 2, 3, and 4, which collectively code for the first β‐propeller domain, reducing binding affinity with SOST (sclerostin) and Dickkopf 1.2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18 In contrast, loss‐of‐function LRP5 mutations cause osteoporosis pseudoglioma syndrome (OPPG; MIM# 259770), an autosomal recessive condition of congenital blindness and severe childhood osteoporosis with skeletal fragility.19 Heterozygous carriers have been reported to have low bone mineral density (BMD).20 Most OPPG and low BMD–associated mutations have been described in the second and third β‐propeller domains.16 Loss‐of‐function SOST mutations cause sclerosteosis, a rare condition of excessive bone overgrowth (MIM# 269500); a downstream deletion is thought to be responsible for the milder phenotype of van Buchem's disease (VBD) (MIM# 239100).21, 22 Both LRP5‐related and SOST‐related HBM have been associated with bone overgrowth, mandible enlargement, oral tori, cortical thickening, poor buoyancy, and importantly, reduced fracture risk.2 Although sclerosteosis confers a severe phenotype with craniotomy occasionally required to relieve rising intracranial pressure from skull overgrowth, LRP5 HBM has a variable phenotype and may be asymptomatic.2, 3, 4 Recently, LRP4 mutations, thought to impair SOST‐LRP4 interaction, have been reported in a phenotype resembling sclerosteosis.23 Anti‐sclerostin antibodies are now in phase 3 clinical trials,24, 25 and other inhibitors of osteoblastic Wnt antagonists are in development as novel anabolic osteoporosis treatments.26, 27, 28, 29 Such developments exemplify the valuable insights gained from studying rare monogenic conditions. To date, however, no study has employed a systematic approach to establish the frequency or scope of HBM mutations and their associated phenotypes within the general population.
We have previously reported the clinical characteristics of a unique population of adults with unexplained HBM, identified from review of 335,115 historical dual‐energy X‐ray absorptiometry (DXA) scans across 13 UK National Health Service (NHS) centers for BMD Z‐scores or T‐scores ≥+4.30 Within this population, we aimed to determine the genetic causes of HBM by sequencing unrelated HBM cases for mutations in known anabolic HBM loci, namely LRP5 (exons 2, 3, and 4), LRP4 (exons 25 and 26), and SOST (exon 1, the coding region of exon 2, and the VBD 52‐kb intronic deletion occurring 35 kb downstream of SOST). We then aimed to determine the phenotypes associated with such mutations and relate these to predicted three‐dimensional protein models.
Subjects and Methods
Identification of HBM cases
The HBM study is a UK‐based multicentered observational study of adults with unexplained HBM, identified incidentally upon routine clinical DXA scanning. Full details of DXA database screening and participant recruitment have been reported30 (Supporting Information S1). Briefly, DXA databases containing 335,115 DXA scans were searched for individuals with a BMD T‐score or Z‐score ≥+4 at any site within the lumbar spine or hip, at 13 NHS hospitals in England and Wales (nine Hologic, four Lunar). A further two centers with Hologic scanners contributed 23 similar individuals identified prospectively. All 1505 DXA images with BMD T‐score or Z‐score ≥+4 were visually inspected; 962 cases with osteoarthritis and/or other causes of raised BMD were excluded (eg, surgical metalwork, Paget's disease, metastases31). A generalized HBM trait would be expected to affect both spine and hip BMD, though not necessarily to the same extent. Hence we refined our definition of HBM index cases as: (1) L1 Z‐score of ≥+3.2 plus total hip Z‐score of ≥+1.2; or (2) total hip Z‐score ≥+3.2 plus L1 Z‐score of ≥+1.2 (using age and gender‐adjusted BMD Z‐scores). A threshold of +3.2 was consistent with the only published precedent for identifying HBM using DXA;3 and also most appropriately differentiated generalized HBM from artifact.32 A standard deviation of +3.2 would be expected to identify a tail of 0.069% of a normal distribution.33
Of 533 unexplained HBM index cases invited, 258 (48.4%) agreed to participate.30 Index cases were asked to invite their first‐degree relatives and spouse/partner(s) to participate. HBM status was defined in first‐degree relatives as summed L1 plus total hip Z‐score of ≥+3.2.30 Family‐based controls comprised relatives with BMD below this threshold. HBM among spouses was defined as for index cases. Participants were excluded if under 18 years of age, pregnant, or unable to provide written informed consent for any reason. All participants were clinically assessed using a standardized structured history and examination, with phlebotomy for bone biochemistry, bone turnover markers (Supporting Information S2) and DNA collection. DXA scans were performed according to the manufacturer's standard scanning and positioning protocols. Where available, total body (TB) BMD, fat mass (FM) (including android and gynoid FM), and lean mass were measured as reported previously,34 and high‐resolution pQCT (HRpQCT) was performed (Supporting Information S2). Of note, no index cases who reported ever having fractured had radiologic, hematologic, or clinical features consistent with osteopetrosis.30 Written informed consent was obtained for all participants in line with the Declaration of Helsinki35 and this study was approved by the Bath Multi‐centre Research Ethics Committee (REC: 05/Q2001/78) and each NHS Local REC.
Sanger sequencing for HBM mutations
DNA was extracted from peripheral venous blood using standard phenol/chloroform extraction. PCR amplification of LRP5 (exons 2, 3, and 4), LRP4 (exons 25 and 26), and SOST (exon 1, the coding region of exon 2, and the VBD‐associated 52‐kb intronic deletion 35 kb 3′ of SOST) was performed on 50 ng genomic DNA in a reaction mix consisting: 10× Immolase reaction buffer, 10 mM dNTPs, 50 mM MgCl2, 5 μM each primer, 0.5 U Immolase polymerase Taq (Bioline Reagents Ltd, London, UK), and water to final volume of 25 μL. PCR cycling conditions and primer sequences are shown in Supporting Information S3. Samples were Sanger sequenced using standard techniques (BigDye v3.1 chemistry; Life Technologies Corporation, Carlsbad, CA, USA), and capillary sequenced (3130 Genetic Analyzer; Life Technologies Corporation). Electrophoretograms were analyzed using sequence analyses software Genalys v2.0β.36 Variants were examined in online databases: NCBI dbGene/dbSNP (release 135), 1000 Genomes (www.1000genomes.org), UCSC (GRCh37/Hg19), Leiden Open Variation Database (LOVD) (www.lovd.nl), Exome Variant Server (http://evs.gs.washington.edu/EVS), and ExAC (http://exac.broadinstitute.org), using transcripts LRP5: ENSP00000294304, SOST: ENST00000301691, and LRP4: ENSG00000134569. We performed in silico functional prediction using PolyPhen,37 SIFT,38 PMut,39 and Mutation Taster40 bioinformatic prediction algorithms. When the same point mutation was identified in more than one family, haplotypes were compared between index case samples genotyped using an Infinium OmniExpress‐12v1.0 GWAS chip read using an Illumina iScan (Illumina, San Diego, CA, USA), with genotype clustering performed using Illumina BeadStudio software.
Protein modeling
Structural models of the LRP5‐SOST complex were prepared from the homologous structure of the LRP6 complex (PDB code 3SOV).41 Mutations were introduced using the modeling program ICM‐Pro (Molsoft, San Diego, CA, USA) with local minimization to optimize side chain positions within 7 Å of the mutation site.42
Statistical analysis
HBM cases were grouped by mutation type for quantitative analyses in comparison with (1) HBM cases lacking LRP5/SOST/LRP4 mutations and (2) family controls. Descriptive statistics are presented as mean (95% confidence interval [CI]) for continuous and count (percentages) for categorical data. Linear regression was used to analyze continuous variables and generate standardized β coefficients and 95% CIs. Age, gender, menopausal status, and estrogen replacement therapy in women were considered a priori confounders; weight and height were additional potential confounders. Data were managed using Microsoft Access (Microsoft Corp., Redmond, WA, USA) (data entry checks; error rate <0.12%) and analyzed using Stata release 12 statistical software (StataCorp, College Station, TX, USA).
Results
LRP5
We identified two novel missense LRP5 mutations ([c.518C>T; p.Thr173Met], [c.796C>T; p.Arg266Cys]) as well as three previously reported missense LRP5 mutations ([c.593A>G; p.Asn198Ser], [c.724G>A; p.Ala242Thr], [c.266A>G; p.Gln89Arg]), associated with HBM in 11 adults among seven families (Table 1, Supporting Information S4). All LRP5 mutations were heterozygous and segregated with HBM in available pedigrees (Supporting Information S5). Of 11 carrying a heterozygous LRP5 mutation, none had sustained a low‐trauma or moderate‐trauma adult fracture, six reported an inability to float, seven had oral tori, and eight had a noticeably enlarged mandible (Table 1).
Table 1.
Exonic Mutations Identified After Sanger Sequencing of all 258 HBM Index Cases With Clinical Characteristics and In Silico Functional Predictions
| Pedigree | Gene | Mutation | Exon | Amino acid change | Age (years) | Gender | Z‐score total hip | Z‐score L1 | Adult fracture | Enlarged mandible | Tori | Nerve compression | Sinks/ floats (S/F) | Polyphen | SIFT (score) | PMut | Mutation taster | GERP score |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | LRP5 | 593A>G | 3 | Asn198Ser | 30 | M | +12.2 | +8.3 | N | Y a | Y | N | S | Probably damaging | Damaging | Neutral | Disease‐causing | 3.66 |
| 1 | LRP5 | 593A>G | 3 | Asn198Ser | 26 | F | +6.8 | +5.6 | N | Y a | N | N | S | Probably damaging | Damaging | Neutral | Disease‐causing | 3.66 |
| 1 | LRP5 | 593A>G | 3 | Asn198Ser | 66 b | M | +10.2 | +7.8 | N | Y | Y | N | S | Probably damaging | Damaging | Neutral | Disease‐causing | 3.66 |
| 2 | LRP5 | 724G>A | 4 | Ala242Thr | 49 | F | +7.1 | +10.7 | N | Y | Y | N | S | Probably damaging | Damaging | Neutral | Disease‐causing | 3.78 |
| 2 | LRP5 | 724G>A | 4 | Ala242Thr | 21 | M | +6.4 | +8.2 | N | N | n/a | N | F | Probably damaging | Damaging | Neutral | Disease‐causing | 3.78 |
| 3 | LRP5 | 724G>A | 4 | Ala242Thr | 21 | F | +7.1 | +6.0 | N | Y c | Y | Y d | S | Probably damaging | Damaging | Neutral | Disease‐causing | 3.78 |
| 4 | LRP5 | 724G>A | 4 | Ala242Thr | 64 | F | +5.9 | +8.1 | N | Y | Y | Y e | S | Probably damaging | Damaging | Neutral | Disease‐causing | 3.78 |
| 4 | LRP5 | 724G>A | 4 | Ala242Thr | 41 | F | +3.1 | +3.1 | N | Y | Y | N | F | Probably damaging | Damaging | Neutral | Disease‐causing | 3.78 |
| 5 | LRP5 | C796C>T f | 4 | Arg266Cys f | 65 | M | +2.5 | +6.2 | N | Y | Y | N | F | Probably damaging | Tolerated | Pathological | Disease‐causing | ‐0.44 |
| 6 | LRP5 | 266A>G | 2 | Gln89Arg | 69 b | M | +2.4 | +4.6 | N | N | N | Y g | n/a h | Benign | Tolerated | Neutral | Disease‐causing | 4.48 |
| 7 | LRP5 | 518C>T f | 3 | Thr173Met f | 76 | M | +3.6 | +4.2 | Y i | N | N | Y j | F | Probably damaging | Tolerated | Neutral | Disease‐causing | 1.67 |
| 8 | SOST | 530C>A f | 2 | Ser177X f | 70 | F | +1.7 | +3.5 | N | Y | n/a | N | S | n/a | Tolerated | n/a | Disease‐causing | 4.26 |
In silico functional predictions relate to decreases in antagonism of Wnt signaling and hence increased Wnt activity.
HBM = high bone mass; GERP = Genomic Evolutionary Rate Profiling; n/a = not available.
With prognatism.
History of glucocorticoid treatment with oral bisphosphonate use.
Enlarged and asymmetric.
Tightly packed brain gyri on MRI; cranial nerves V and VII mildly impaired.
Conductive deafness.
Novel HBM mutation.
Carpel tunnel syndrome.
Non‐swimmer.
Fibula aged 39, elbow aged 48, both very high impact fractures.
Ulna nerve decompression.
LRP5 HBM quantitative analyses
The 11 HBM cases with LRP5 mutations (“LRP5 HBM cases”) were compared with 347 HBM cases without LRP5 mutations (250 index cases, 94 affected first‐degree relatives, and three spouses who fulfilled HBM index case criteria) (“non‐LRP5 HBM cases”), and 200 family controls. Eight and four HBM cases had TB DXA and HRpQCT performed respectively. LRP5 HBM cases were taller than both non‐LRP5 HBM cases and controls, with larger shoe size and substantially greater BMD at all measured sites, representing greater trabecular density and cortical thickness measured by HRpQCT (Table 2). LRP5 HBM cases were also heavier than controls, with greater fat mass, particularly gynoid fat. After adjustment for age, gender, menopause, and estrogen replacement in women, the LRP5 HBM cases remained substantially taller than both non‐LRP5 HBM cases and controls (Supporting Information S6). Hence, analyses were further adjusted for height; LRP5 HBM cases still had persistently greater BMD at all measured sites, as well as greater gynoid fat mass than controls (Table 3). Further adjustment for total weight highlighted a difference in gynoid fat mass between HBM cases and both controls and non‐LRP5 HBM cases (Supporting Information S7). Although still within the normal reference range, after adjustment mean adjusted calcium, was higher among LRP5 HBM cases; however, bone turnover marker levels were not discernibly different (Table 3).
Table 2.
Clinical Characteristics of LRP5 HBM Cases, Non‐LRP5 HBM Cases, and Family Controls
| LRP5 HBM (n = 11) | Non‐LRP5 HBM (n = 347) | Controls (n = 200) | |
|---|---|---|---|
| Mean (SD) | Mean (SD) | Mean (SD) | |
| Age (years) | 49.5 (21.0) | 61.9 (13.5)* | 54.0 (16.1) |
| Height (cm) | 178.1 (8.6) | 166.3 (8.6)** | 171.1 (10.2)** |
| Weight (kg) | 93.7 (13.5) | 84.8 (17.5) | 82.4 (17.3)* |
| BMI (kg/m2) | 29.5 (2.8) | 30.7 (6.0) | 28.0 (4.9) |
| Shoe size a | 9.8 (2.3) | 7.0 (1.9)** | 7.9 (2.3)* |
| Total hip BMD Z‐score | 5.8 (2.7) | 3.0 (1.1)** | 0.5 (0.9)** |
| L1 BMD Z‐score | 6.4 (2.0) | 3.9 (1.5)** | 0.5 (1.2)** |
| TB BMD (mg/cm2) b | 1.76 (0.32) | 1.33 (0.10)** | 1.21 (0.12)** |
| TB lean mass (kg) b | 55.4 (11.7) | 46.7 (10.1)* | 51.3 (11.4) |
| TB fat mass (kg) b | 37.9 (6.5) | 35.4 (12.8) | 29.0 (11.3)* |
| TB android fat mass (kg) b | 3.72 (0.54) | 3.43 (1.41) | 2.90 (1.26) |
| TB gynoid Fat Mass (kg) b | 6.64 (2.31) | 5.64 (1.85) | 4.85 (1.82)* |
| Glucose c | 6.2 (3.8) | 6.2 (2.2) | 5.6 (1.3) |
| Adjusted calcium | 2.45 (0.07) | 2.41 (0.10) | 2.40 (0.08) |
| Phosphate | 1.12 (0.18) | 1.37 (0.86) | 1.17 (0.44) |
| Alkaline phosphatase | 74.9 (17.7) | 86.7 (33.6) | 85.2 (33.9) |
| P1NP (µg/L) | 44.1 (30.0) | 36.3 (19.9) | 41.1 (23.4) |
| CTX (µg/L) | 0.23 (0.18) | 0.19 (0.13) | 0.24 (0.17) |
| Osteocalcin (total) (µg/L) | 15.7 (8.0) | 15.6 (7.8) | 17.8 (7.8) |
| Tibial trabecular bone density (mg HA/cm3) d | 313.1 (59.8) | 215.1 (40.9)** | 185 (41.5)** |
| Tibial number of trabeculae (1/mm) d | 2.83 (0.24) | 2.31 (0.28)* | 2.25 (0.47)* |
| Tibial trabecular thickness (mm)d | 0.09 (0.02) | 0.08 (0.01) | 0.07 (0.01)* |
| Tibial cortical thickness (mm) d | 2.70 (1.07) | 1.27 (0.43)** | 1.18 (0.33)** |
| n (%) | n (%) | n (%) | |
| Female | 5 (45.5) | 269 (77.5)* | 93 (65.8) |
| Postmenopausal | 2 (40.0) | 216 (80.3)* | 48 (51.6) |
| Estrogen replacement use (ever) | 1 (20.0) | 127 (47.2) | 15 (16.1) |
HBM = high bone mass; TB = total body.
n = 468 for UK shoe size.
Total body DXA measures: n = 8 for LRP5 HBM, 199 for non‐LRP5 HBM, 126 for controls.
n = 247 for finger prick blood glucose.
HRpQCT measures: n = 4 for LRP5 HBM, 59 for non‐LRP5 HBM, 36 for controls. No LRP5 HBM cases reported malignancy; 2 LRP5 HBM cases, 7 non‐LRP5 HBM cases, and 2 controls had ever used oral bisphosphonates.
*p < 0.05, **p < 0.001 when compared with LRP5 HBM case.
Table 3.
Clinical Characteristics of LRP5 HBM Cases, Non‐LRP5 HBM Cases, and Family Controls Adjusted for Age, Gender, Menopausal Status, and Estrogen Replacement Therapy in Women, and Height
| LRP5 HBM (n = 11) mean (95% CI) | Non‐LRP5 HBM (n = 347) mean (95% CI) | Controls (n = 200) mean (95% CI) | |
|---|---|---|---|
| Shoe size a | 8.04 (7.21–8.86) | 7.47 (7.27–7.68) | 7.25 (7.03–7.47) |
| Total hip BMD Z‐score | 6.18 (5.43–6.94) | 2.89 (2.71–3.08)** | 0.54 (0.34–0.75)** |
| L1 BMD Z‐score | 5.97 (5.08–6.87) | 3.62 (3.4–3.84)** | 0.42 (0.17–0.66)** |
| TB BMD (mg/cm2) b | 1.70 (1.64–1.77) | 1.35 (1.33–1.37)** | 1.18 (1.16–1.20)** |
| TB lean mass (kg) b | 48.2 (43.8–52.6) | 49.4 (48.3–50.5) | 47.5 (46.3–48.6) |
| TB fat mass (kg) b | 36.4 (28.3–44.5) | 35.3 (33.3–37.2) | 30.3 (28.1–32.4) |
| TB android fat mass (kg) b | 3.39 (2.44–4.33) | 3.46 (3.22–3.69) | 2.88 (2.62–3.13) |
| TB gynoid fat mass (kg) b | 6.45 (5.29–7.61) | 5.59 (5.31–5.87) | 5.1 (4.79–5.41)* |
| Glucose c | 6.1 (4.3–7.8) | 6.2 (5.7–6.7) | 6.0 (5.4–6.7) |
| Adjusted calcium | 2.47 (2.42–2.53) | 2.41 (2.39–2.42)* | 2.41 (2.40–2.43)* |
| Phosphate | 1.12 (0.72–1.51) | 1.23 (1.14–1.33) | 1.10 (0.99–1.20) |
| Alkaline phosphatase | 74.1 (55.9–92.4) | 81.0 (76.5–85.4) | 84.2 (79.3–89.1) |
| P1NP (µg/L) | 41.2 (28.5–54.0) | 35.7 (32.6–38.8) | 37.6 (34.2–41.1) |
| CTX (µg/L) | 0.22 (0.12–0.31) | 0.19 (0.17–0.22) | 0.23 (0.20–0.25) |
| Osteocalcin (total) (µg/L) | 18.4 (13.4–23.5) | 17.1 (15.9–18.3) | 19.5 (18.1–20.9) |
| Tibial trabecular bone density (mg HA/cm) d | 296.4 (257.0–335.9) | 210.9 (198.5–223.4)** | 175.2 (161.8–188.7)** |
| Tibial number of trabeculae (1/mm) d | 2.67 (2.33–3.00) | 2.28 (2.17–2.38)* | 2.17 (2.06–2.29)* |
| Tibial trabecular thickness (mm) d | 0.09 (0.08–0.11) | 0.08 (0.07–0.08) | 0.07 (0.06–0.07)* |
| Tibial cortical thickness (mm) d | 2.54 (2.18–2.91) | 1.28 (1.16–1.39)** | 1.05 (0.92–1.17)** |
HBM = high bone mass; TB = total body.
n = 468 for UK shoe size.
Total body DXA measures: n = 8 for LRP5 HBM, 199 for non‐LRP5 HBM, 126 for controls.
n = 247 for finger‐prick blood glucose.
HRpQCT measures: n = 4 for LRP5 HBM, 59 for non‐LRP5 HBM, 36 for controls.
*p < 0.05, **p < 0.001 when compared with LRP5 HBM cases.
Individual LRP5 HBM Phenotypes
All clinical cases listed in Table 1 are described in detail in Supporting Information S7. Our most extreme HBM case, with femoral neck T‐score +12.2, had presented at age 19 years when he fainted and hit his head on a toilet seat, and in doing so broke the toilet seat; cranial imaging showed a markedly thickened skull (Fig. 1 A; Supporting Information S5). He has a heterozygous c.593A>G; p.Asn198Ser mutation that is predicted to be functionally deleterious by three of four in silico prediction tools; ie, suggesting decreased antagonism of Wnt signaling with subsequent increased Wnt activity (Table 1).
Figure 1.
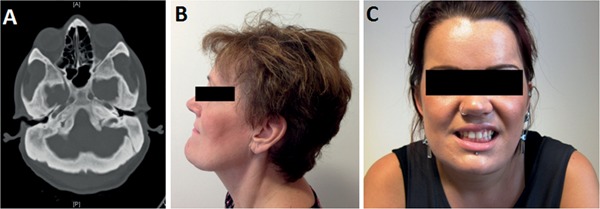
Clinical imaging in LRP5 HBM. (A) Axial computed tomography image showing markedly thickened skull in male aged 30 years with p.Asn198Ser substitution (pedigree 1). (B) Mandible enlargement in female aged 49 years with p.Ala242Thr substitution (pedigree 2). (C) Asymmetric mandible enlargement with partial left cranial nerve V and VII impairment in female aged 21 years with p.Ala242Thr substitution (pedigree 3).
The most frequent LRP5 mutation, c.724G>A; p.Ala242Thr, was identified in three unrelated families (with different LRP5 haplotypes), explaining five HBM cases in total, and is predicted to be functionally deleterious by three of four in silico prediction tools (Table 1, Fig. 1 B, C). Interestingly, the novel mutation c.796C>T; p.Arg266Cys, was identified in a 65 year old man with a 25 year history of ulcerative colitis, for which he had been treated with glucocorticoids almost continuously for 21 years, despite which his HBM persisted; his bone turnover markers were normal (Supporting Information S5).
A heterozygous c.266A>G; p.Gln89Arg mutation was identified in an active man aged 69 years, with mild left hip osteoarthritis; he has never fractured. A novel c.518C>T; p.Thr173Met mutation was identified in a man aged 76 years, with osteoarthritis of knees, hands, and hips (unilateral hip replacement age 66 years), who had sustained two very‐high‐impact fractures age 39 years (fibula) and 48 years (elbow); the latter required ulna nerve decompression 18 years later with ongoing restrictions in the range of movement.
LRP5 protein modeling
The p.Asn198Ser mutation directly affects the SOST interaction site and is predicted to disrupt SOST binding and inhibition, resulting in a severe HBM phenotype (Fig. 2 A). Modeling suggests that the shorter serine side chain is too distant to establish the two hydrogen bonds to SOST N40 (Asn40) that are formed by the wild‐type N198 (Asn198) side chain (Fig. 2 B). The similarly severe p.Ala242Thr mutation is predicted instead to disrupt the core packing of the LRP5 structure, thereby destabilizing the SOST binding site. The larger threonine side chain is likely to introduce steric clashes with the proximal F241 (Phe241) and M282 (Met282) (Fig. 2 C). The alanine at the LRP5 p.Ala242 position is conserved in LRP6 (p.Ala229), suggesting that tight packing in this region is favored. The site of the p.Ala242Thr mutation is only 10 Å from the SOST peptide binding site allowing even minor structural rearrangements to exert a negative effect on this interaction.
Figure 2.
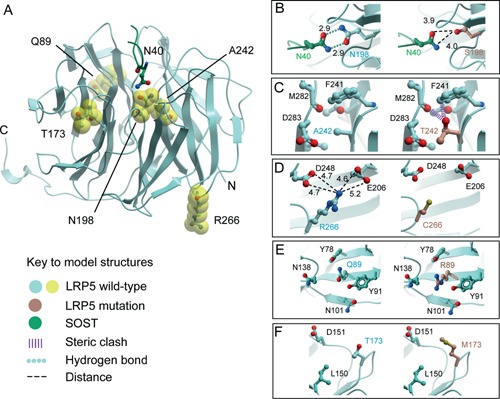
Structural models of the LRP5‐SOST complex (A) with mutations; p.Asn198Ser (B), p.Ala242Thr (C), p.Arg266Cys (D), p.Gln89Arg (E), and p.Thr173Met (F).
The mutations, p.Arg266Cys, p.Gln89Arg, and p.Thr173Met, were associated with less severe HBM phenotypes. p.Arg266 is located on the opposite face of the β‐propeller from the SOST binding site (Fig. 2 A) and is unlikely to directly disrupt its structure or to interfere directly with its binding (Fig. 2 D). Potentially, the introduction of an exposed cysteine residue could induce inappropriate disulphide bond formation resulting in misfolding and aberrant trafficking of the mutant protein.
The p.Gln89Arg mutation introduces an arginine side chain, which is found naturally at the equivalent position in wild‐type LRP6. However, the local packing around this site differs between the two proteins with the absence of a neighboring acidic residue in LRP5. The p.Gln89 position in LRP5 also appears more tightly packed due to the presence of both p.Y78 (Tyr78) and p.Y91 (Tyr91) (Fig. 2 E). Nonetheless, modeling suggests that the mutant arginine side chain can be tolerated. p.Gln89Arg is a reported SNP (rs41494349) with minor allele frequency (MAF) 0.02% in 1000 Genomes (0.1% MAF in East Asians, 0.005% in Europeans (ExAC)).
The p.Thr173Met mutation site lies in one of the β‐propeller loops that line the SOST binding site giving potential for disruption. However, p.Thr173 is distinct from the known peptide site and the side chain is likely to be oriented away from the peptide interface making no direct contact with it (Fig. 2 F). Moreover, the methionine substitution is predicted to be well tolerated. Thus, the less severe phenotypes associated with individuals carrying the p.Arg266Cys, p.Gln89Arg, and p.Thr173Met mutations may be due to less disruptive structural alterations. Models with DKK1 rather than SOST drew similar conclusions.
SOST
We identified a novel heterozygous nonsense variant in exon 2 (c.530C>A; p.Ser177X) in a woman aged 70 years who reported difficulty floating (pedigree 8; Table 1). She had no symptoms of cranial nerve impingement, no syndactyly, and was 166 cm in height. This variant (cDNA.C577A, at chr17:41832822) is listed as rs143571358 in dbSNP135 and ExAC, with MAF of 0.04% and 0.0009% respectively. This base is highly conserved, with a genome evolutionary rate profiling score (GERP) of 4.26 (Table 1). rs143571358 has not been associated with any specific phenotype to date and remains unvalidated in dbSNP. This C>A variant lies within the coding sequence of SOST, and introduces a stop codon at p.177. This is predicted by Mutation Taster to be disease‐causing because the remaining 37 wild‐type amino acids are not incorporated in the mutated sclerostin protein, potentially causing nonsense‐mediated mRNA decay.
We also identified one previously reported SNP in exon 1 in eight HBM cases (rs17882143, c.28G>A; p.Val10Ile, MAF T = 0.047%), predicted to be benign by PolyPhen, tolerated by SIFT, a polymorphism by Mutation Taster, and neutral by PMut; as well as two intergenic SNPs in a further nine HBM cases (8 cases with rs28548107 MAF G = 0.046%; 1 case with rs181372199, MAF T = 0.043%). We found no variants in the 3′ regulatory region of SOST to suggest VBD. No SOST variants were identified in individuals with LRP5 variants (and vice versa).
LRP4
We found no mutations in LRP4 exons 25 and 26. In one HBM case we identified a novel intronic heterozygous variant 24 bases before the start of exon 25 but not in the splice junction (c.3364‐24G>T). The common SNP rs2306033 (MAF 25% in 1000 Genomes) was observed in 56 (21%) HBM cases.
Discussion
This study represents the largest systematic approach to date to identify the genetic cause of HBM, by screening all known HBM loci in a well‐defined population drawn from the general population, and has identified both novel and previously reported variants underlying HBM. We have increased to 13 the number of LRP5 mutations associated with HBM, having identified two novel and three previously reported missense LRP5 mutations, associated with HBM in seven families. The frequency of LRP5 variants in HBM individuals, therefore, is 7 in 258, less than 3%; if our DXA population is representative of the general population, this extrapolates to an overall prevalence of LRP5 HBM mutations in the UK of approximately 5 per 100,000. We identified one person with moderate HBM and a novel heterozygous nonsense SOST mutation predicted to prematurely truncate sclerostin, suggesting her to be a sclerosteosis carrier. However, no cases fulfilled a clear clinical diagnosis of autosomal recessive sclerosteosis with homozygous or compound heterozygous mutations. We did not observe any LRP4 HBM variants in the limited number of exons sequenced. Our findings highlight the rarity of mutations in established HBM loci within the general population, and that the majority of HBM cases remain genetically unexplained.
The clinical variability we observed in LRP5 HBM cases may arise from genotype/phenotype correlation, as suggested by the variable in silico functional consequences presented here. The LRP5 p.Asn198Ser mutation, seen in our most extreme HBM case with hip BMD Z‐scores >+10, has been reported in a family with HBM and deafness, sensorimotor neuropathy, and spinal stenosis,6 features that we did not observe. Our protein modeling shows the direct implications of this mutation on SOST binding, explaining the extreme bone phenotype. A disrupted LRP5‐SOST binding site would be expected to lead to a relative resistance to sclerostin, with secondary increased sclerostin levels, an observation we have previously made in LRP5 HBM.43 Our most frequent LRP5 mutation (p.Ala242Thr), associated with BMD Z‐scores +3.1 to +10.7, has been reported in five previous families (two Portland US, one Sardinia, one France, one China),7, 44 to which we can now add a further three UK families.
We identified an isolated HBM case with an LRP5 p.Arg266Cys variant; although having an allocated SNP ID, this SNP is not validated, has no described MAF in dbSNP, is not listed in ExAC/LOVD, and has not been described previously in association with HBM (or any phenotype). Three of four in silico prediction tools considered this variant to be functionally deleterious, although protein modeling suggested an indirect effect and/or altered protein folding. These less deleterious effects are consistent with a milder, mostly asymptomatic HBM phenotype (BMD Z‐scores +2.5 to +6.5).
LRP5 mutation p.Gln89Arg was identified in one HBM case (no DNA was available from relatives). Interestingly p.Gln89Arg was also identified in one of the first reported LRP5 HBM case series.7 However, this variant has been reported with MAF 8% in the Japanese population.45 Subsequently, p.Gln89Arg was associated with lower (not higher) femoral neck BMD in young Korean men (with MAF 19%)46 and postmenopausal Han Chinese women (with MAF 17%).47 However, this association with low BMD was not seen at the lumbar spine,46, 47 nor in 321 postmenopausal Japanese women, in whom it was instead associated with spinal osteophytes.48 Importantly functional analyses suggest that p.Gln89Arg does not play a functional role in canonical Wnt signaling.49 Whether p.Gln89Arg shares a rare haplotype with a functional BMD allele specifically in white populations is unknown.
LRP5 mutation p.Thr173Met, identified in one isolated HBM case, was previously reported in association with abnormal retinal vasculature/folds in an older British woman diagnosed with familial exudative vitreoretinopathy (FEVR); however, her BMD was not reported.50 Although SIFT and PMut consider this mutation to be tolerated/neutral, respectively, PolyPhen and Mutation Taster predict it to be “probably damaging” and “disease causing,” with a moderate conservation score (GERP 1.67, Table 1). LRP5 HBM mutations are considered fully penetrant; however, phenotypes may vary even within an individual family, as is seen in many genetic conditions; eg, osteogenesis imperfecta.
Originally LRP5 expression studies identified reduced affinity of DKK1 for LRP5 as the mechanism underlying LRP5 HBM.51 The only previous LRP5 protein modeling to date (using p.Asn198Ser and p.Ala242Thr mutations, although without modeling the mutant side chains) predicted that both Wnt inhibitors DKK1 and SOST act through a common site in the first β‐propeller. This supports our models describing both LRP5‐SOST and LRP5‐DKK1 interactions.41 Furthermore, functional studies support p.Asn198Ser diminishing LRP5‐SOST binding; although LRP5‐DKK1 may be differentially affected.52 However, more recently SOST has emerged as the key LRP5 regulator.53 Genetic variance within both SOST and LRP5 are associated with BMD in the general population, hence mechanistic insights can have application to the wider population.54, 55, 56 The extent to which common genetic variation in all BMD‐associated genes, explains the HBM phenotype in non‐LRP5 HBM remains a question of interest.
Although absolute numbers are small, this remains one of the largest mutationally heterogeneous collections of LRP5 HBM cases to date. Though the severity of the clinical phenotype varied according to the precise mutation, as a group, individuals with LRP5 mutations appear to have a more extreme HBM phenotype when compared to other HBM individuals. We have previously reported that HBM individuals have greater trabecular BMD and cortical thickness as assessed by pQCT.57 In the present study we performed HRpQCT in a small subgroup; trabecular BMD and cortical thickness appeared to be higher in LRP5 HBM compared to non‐LRP5 HBM cases. The increased sclerostin levels reported in LRP5 HBM relative to other HBM cases, likely represents a compensatory response.43 Bone turnover appeared similar among LRP5 HBM, non‐LRP5 HBM, and controls (with low coefficients of variation).34 This null finding might reflect the small sample size. However, it is also likely that single time point sampling may not reflect accumulated life‐course exposure to anabolic stimuli. We lacked bone turnover assessment during peak bone mass accrual, during which time the effect of anabolic mutations might be expected to be maximal. Interestingly, two LRP5 HBM cases (p.Arg266Cys; p.Asn198Ser) still had high BMD despite long‐term glucocorticoid use; whether anabolic LRP5 mutations confer a relative resistance to glucocorticoid‐induced osteoporosis remains to be determined, but if so, it would be of great pharmacotherapeutic interest.
Fat mass was increased in individuals with LRP5 HBM mutations, as we previously reported for HBM individuals overall.34 However, we observed a particular preponderance toward a gynoid fat mass distribution, persisting even after adjustment for body weight, in LRP5 HBM compared with both non‐LRP5 HBM and controls. This contrasts with the android fat phenotype we have previously indentified in HBM women (98% of whom have non‐LRP5 HBM).34 Recent mechanistic analyses have shown LRP5 HBM mutations, in some of the individuals reported here, lead to gluteofemoral fat accumulation due to altered LRP5‐dependent transcription in site‐specific depots, which raises interesting metabolic implications for medicines that modulate Wnt activity.58
Our study has limitations. The rarity of LRP5 HBM challenged our sample size, restricted our ability to stratify (eg, by gender), and limited the confidence with which we can make prevalence estimates, it further prevented a clustered analysis, although evidence of intrafamilial clustering has previously been small.30 Furthermore, not all pedigrees had access to total body DXA and HRpQCT assessment, nor were full pedigrees able to be recruited in all cases. Interestingly, three spouses fulfilled HBM index case criteria, suggestive of assortative mating, as reported.30 Sequencing was restricted to exons in which anabolic mutations have been reported; and we have reported all identified variants. It is possible that variants in other β‐propeller regions may contribute to HBM; however, this seems less likely because mutations in the remaining β‐propellers have only been associated with FEVR and OPPG to date. Our approach was a practical one given the size of LRP5 and exon 2 of SOST. We did not sequence for DKK1 mutations; a novel missense mutation in the LRP5 inhibitor DKK1 (c.74Y>F) has recently been reported to segregate with HBM in one Spanish family.59 Nor did we sequence CLCN7 (associated with autosomal dominant osteopetrosis type 2) or genes associated with the more severe autosomal recessive forms of osteopetrosis (eg, TNFSF11s, TCIRG1, and PLEKHM1) because clinical and radiological phenotyping excluded diagnoses of osteopetrosis. The increasing availability and affordability of whole‐exome sequencing will allow comprehensive screening of all known anabolic and osteopetrotic loci simultaneously in similar future studies.
Conclusions
We identified five missense LRP5 mutations and one novel nonsense SOST mutation, in the largest population study of HBM to date. Protein modeling suggests the severity of high BMD corresponds to the degree of predicted LRP5 protein disruption. However, these LRP5 and SOST HBM cases account for only a small proportion (∼3%) of HBM, raising the possibility that either mutations in novel HBM genes or polygenic inheritance is largely responsible for most cases of HBM in the population.
Disclosures
All authors state that they have no conflicts of interest.
Supporting information
Supporting Information.
Acknowledgments
We thank all study participants and staff at our collaborating centers. This study was supported by The Wellcome Trust and NIHR CRN (portfolio number 5163). CLG was funded by a Wellcome Trust Clinical Research Training Fellowship (080280/Z/06/Z), the EU 7th Framework Programme under grant agreement number 247642 (GEoCoDE), a British Geriatric Society travel grant, and is now funded by Arthritis Research UK (grant ref 20000). SH acknowledges Arthritis Research UK support (grant ref 19580). KESP acknowledges the support of Cambridge NIHR Biomedical Research Centre. KAW is supported by the core program of the MRC Nutrition and Bone Health group at MRC Human Nutrition Research, funded by the UK Medical Research Council (Grant code U10590371). EM acknowledges support of the Sheffield Teaching Hospitals Foundation Trust Clinical Research Facility. The SGC is a registered charity (no. 1097737) that receives funds from AbbVie, Bayer, Boehringer Ingelheim, Genome Canada (Ontario Genomics Institute OGI‐055), GlaxoSmithKline, Janssen, Lilly Canada, Novartis Research Foundation, Ontario Ministry of Economic Development and Innovation, Pfizer, Takeda, and Wellcome Trust (092809/Z/10/Z).
Authors' roles: Study design: CG,GDS, MAB, JHT, and ED. Study conduct: CG, SH, KW, MP, JaT, MS, KP, EM, and JHT. Data collection: CG, SH, JP, KW, MP, JaT, MS, JoT, KP, EM, and JHT. Data analysis: CG, LW, KA, MB, GC, KW, MP, EW, AB, WF, MAB, JHT, and ED. Data interpretation: CG, LW, RA, KP, EW, EM, AB, WF, MAB, JHT, and ED. Drafting manuscript: CG, JHT, and ED. Revising manuscript: CG, LW, SH, EM, AB, MB, JHT, and ED. Approving final version: CG, JHT, and ED. CG and AB take responsibility for the integrity of data analysis.
References
- 1. Koay A, Brown MA. Genetic disorders of the LRP5‐Wnt signalling pathway affecting the skeleton. Trends Mol Med. 2005; 11:129–37. [DOI] [PubMed] [Google Scholar]
- 2. Johnson ML, Gong G, Kimberling W, Recker SM, Kimmel DB, Recker RB. Linkage of a gene causing high bone mass to human chromosome 11 (11q12‐13). Am J Hum Genet. 1997; 60(6):1326–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Little RD, Carulli JP, Del Mastro RG, et al. A mutation in the LDL receptor‐related protein 5 gene results in the autosomal dominant high‐bone‐mass trait. Am J Hum Genet. 2002; 70(1):11–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Boyden LM, Mao J, Belsky J, et al. High bone density due to a mutation in LDL‐receptor‐related protein 5. N Engl J Med. 2002; 346(20):1513–21. [DOI] [PubMed] [Google Scholar]
- 5. Whyte MP, Reinus WH, Mumm S. High‐bone mass disease and LRP5. N Engl J Med. 2004; 350:2096–9. [DOI] [PubMed] [Google Scholar]
- 6. Boyden LM, Insogna K, Lifton RP. High bone mass disease and LRP5. N Engl J Med. 2004; 350(20):2098–9. [DOI] [PubMed] [Google Scholar]
- 7. Van Wesenbeeck L, Cleiren E, Gram J, et al. Six novel missense mutations in the LDL receptor‐related protein 5 (LRP5) gene in different conditions with an increased bone density. Am J Hum Genet. 2003; 72(3):763–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Beals RK, McLoughlin SW, Teed RL, McDonald C. Dominant endosteal hyperostosis. Skeletal characteristics and review of the literature. J Bone Joint Surg Am. 2001;83‐A(11): 1643–9. [PubMed] [Google Scholar]
- 9. Kwee ML, Balemans W, Cleiren E, et al. An autosomal dominant high bone mass phenotype in association with craniosynostosis in an extended family is caused by an LRP5 missense mutation. J Bone Miner Res. 2005; 20(7):1254–60. [DOI] [PubMed] [Google Scholar]
- 10. Renton T, Odell E, Drage NA. Differential diagnosis and treatment of autosomal dominant osteosclerosis of the mandible. Br J Oral Maxillofac Surg. 2002; 40(1):55–9. [DOI] [PubMed] [Google Scholar]
- 11. Van Hul E, Gram J, Bollerslev J, et al. Localization of the gene causing autosomal dominant osteopetrosis type I to chromosome 11q12–13. J Bone Miner Res. 2002; 17(6):1111–7. [DOI] [PubMed] [Google Scholar]
- 12. Bollerslev J, Andersen PE Jr. Radiological, biochemical and hereditary evidence of two types of autosomal dominant osteopetrosis. Bone. 1988; 9(1):7–13. [DOI] [PubMed] [Google Scholar]
- 13. Bollerslev J, Nielsen HK, Larsen HF, Mosekilde L. Biochemical evidence of disturbed bone metabolism and calcium homeostasis in two types of autosomal dominant osteopetrosis. Acta Med Scand. 1988; 224(5):479–83. [DOI] [PubMed] [Google Scholar]
- 14. van Wesenbeeck L, Odgren PR, Mackay CA, Van Hul W. Localization of the gene causing the osteopetrotic phenotype in the incisors absent (ia) rat on chromosome 10q32.1. J Bone Miner Res. 2004; 19(2):183–9. [DOI] [PubMed] [Google Scholar]
- 15. Balemans W, Devogelaer JP, Cleiren E, Piters E, Caussin E, Van Hul W. Novel LRP5 missense mutation in a patient with a high bone mass phenotype results in decreased DKK1‐mediated inhibition of Wnt signaling. J Bone Miner Res. 2007; 22(5):708–16. [DOI] [PubMed] [Google Scholar]
- 16. Rickels MR, Zhang X, Mumm S, Whyte MP. Oropharyngeal skeletal disease accompanying high bone mass and novel LRP5 mutation. J Bone Miner Res. 2005; 20(5):878–85. [DOI] [PubMed] [Google Scholar]
- 17. Scopelliti D, Orsini R, Ventucci E, Carratelli D. [Van Buchem disease. Maxillofacial changes, diagnostic classification and general principles of treatment]. Minerva Stomatol. 1999; 48(5):227–34. [PubMed] [Google Scholar]
- 18. Beals RK. Endosteal hyperostosis. J Bone Joint Surg Am. 1976; 58(8):1172–3. [PubMed] [Google Scholar]
- 19. Gong Y, Slee RB, Fukai N, et al. LDL receptor‐related protein 5 (LRP5) affects bone accrual and eye development. Cell. 2001; 107:513–23. [DOI] [PubMed] [Google Scholar]
- 20. Alonso N, Soares DC, McCloskey EV, Summers GD, Ralston SH, Gregson CL. Atypical femoral fracture in osteoporosis pseudoglioma syndrome associated with two novel compound heterozygous mutations in LRP5. J Bone Miner Res. 2015; 30(4):615–20. [DOI] [PubMed] [Google Scholar]
- 21. Staehling‐Hampton K, Proll S, Paeper BW, et al. A 52‐kb deletion in the SOST‐MEOX1 intergenic region on 17q12‐q21 is associated with van Buchem disease in the Dutch population. Am J Med Genet. 2002; 110(2):144–52. [DOI] [PubMed] [Google Scholar]
- 22. Balemans W, Patel N, Ebeling M, et al. Identification of a 52 kb deletion downstream of the SOST gene in patients with van Buchem disease. J Med Genet. 2002; 39(2):91–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Leupin O, Piters E, Halleux C, et al. Bone overgrowth‐associated mutations in the LRP4 gene impair sclerostin facilitator function. J Biol Chem. 2011; 286(22):19489–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Recker RR, Benson CT, Matsumoto T, et al. A randomized, double‐blind phase 2 clinical trial of blosozumab, a sclerostin antibody, in postmenopausal women with low bone mineral density. J Bone Miner Res. 2015; 30(2):216–24. [DOI] [PubMed] [Google Scholar]
- 25. McClung MR, Grauer A, Boonen S, et al. Romosozumab in postmenopausal women with low bone mineral density. N Engl J Med. 2014; 370(5):412–20. [DOI] [PubMed] [Google Scholar]
- 26. Kulkarni NH, Onyia JE, Zeng Q, et al. Orally bioavailable GSK‐3a/b dual inhibitor increases markers of cellular differentiation in vitro and bone mass in vivo. J Bone Miner Res. 2006; 21(6):910–20. [DOI] [PubMed] [Google Scholar]
- 27. Glantschnig H, Hampton R, Wei N, et al. Fully human anti‐DKK1 antibodies increase bone formation and resolve osteopenia in mouse models of estrogen‐deficiency induced bone loss. J Bone Miner Res. 2008; 23:S60–1. [Google Scholar]
- 28. Heiland GR, Zwerina K, Baum W, et al. Neutralisation of Dkk‐1 protects from systemic bone loss during inflammation and reduces sclerostin expression. Ann Rheum Dis. 2010; 69(12):2152–9. [DOI] [PubMed] [Google Scholar]
- 29. Moore WJ, Kern JC, Bhat R, et al. Modulation of Wnt signaling through inhibition of secreted frizzled‐related protein I (sFRP‐1) with N‐substituted piperidinyl diphenylsulfonyl sulfonamides. J Med Chem. 2008; 52(1):105–16. [DOI] [PubMed] [Google Scholar]
- 30. Gregson CL, Steel SA, O'Rourke KP, et al. “ Sink or swim”: an evaluation of the clinical characteristics of individuals with high bone mass. Osteoporos Int. 2012; 23(2):643–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gregson CL, Hardcastle SA, Cooper C, Tobias JH. Friend or foe: high bone mineral density on routine bone density scanning, a review of causes and management. Rheumatology. 2013; 52(6):968–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gregson CL, Steel S, Yoshida K, Reid DM, Tobias JH. An investigation into the impact of osteoarthritic changes on bone mineral density measurements in patients with High Bone Mass. Poster session presented at: Annual Meeting American Society for Bone and Mineral Research (ASBMR); 2008. Sep 13; Montréal, Québec, Canada; Presentation Number: SA257.
- 33. White J, Yeats A, Skipworth G. Tables for statisticians. Cheltenham, UK: Stanley Thornes; 1979.
- 34. Gregson CL, Paggiosi MA, Crabtree N, et al. Analysis of body composition in individuals with high bone mass reveals a marked increase in fat mass in women but not men. J Clin Endocrinol Metab. 2013; 98(2):818–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.World Medical Association (WMA). WMA Declaration of Helsinki. Ethical Principles for Medical Research Involving Human Subjects. 59th WMA General Assembly, Seoul, Korea. 2008 [Internet]. Ferney‐Voltaire, France: WMA; 2008. [cited 2015 Sep 21]. Available from: http://www.wma.net/en/30publications/10policies/b3/17c.pdf.
- 36. Takahashi M, Matsuda F, Margetic N, Lathrop M. Automated identification of single nucleotide polymorphisms from sequencing data. J Bioinform Comput Biol. 2003; 1(2):253–65. [DOI] [PubMed] [Google Scholar]
- 37. Sunyaev S, Ramensky V, Koch I, Lathe W 3rd, Kondrashov AS, Bork P. Prediction of deleterious human alleles. Hum Mol Genet. 2001; 10(6):591–7. [DOI] [PubMed] [Google Scholar]
- 38. Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non‐synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009; 4(7):1073–81. [DOI] [PubMed] [Google Scholar]
- 39. Ferrer‐Costa C, Gelpí JL, Zamakola L, Parraga I, de la Cruz X, Orozco M. PMUT: a web‐based tool for the annotation of pathological mutations on proteins. Bioinformatics. 2005; 21(14):3176–8. [DOI] [PubMed] [Google Scholar]
- 40. Schwarz JM, Rodelsperger C, Schuelke M, Seelow D. MutationTaster evaluates disease‐causing potential of sequence alterations. Nat Methods. 2010; 7(8):575–6. [DOI] [PubMed] [Google Scholar]
- 41. Bourhis E, Wang W, Tam C, et al. Wnt antagonists bind through a short peptide to the first beta‐propeller domain of LRP5/6. Structure. 2011; 19(10):1433–42. [DOI] [PubMed] [Google Scholar]
- 42. Cardozo T, Totrov M, Abagyan R. Homology modeling by the ICM method. Proteins. 1995; 23(3):403–14. [DOI] [PubMed] [Google Scholar]
- 43. Gregson CL, Poole KES, McCloskey EV, et al. Elevated circulating sclerostin concentrations in individuals with high bone mass, with and without LRP5 mutations. J Clin Endocrinol Metab. 2014; 99(8):2897–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wang C, Zhang B‐H., Zhang H, et al. The A242T mutation in the low‐density lipoprotein receptor‐related protein 5 gene in one Chinese family with osteosclerosis. Intern Med. 2013; 52(2):187–92. [DOI] [PubMed] [Google Scholar]
- 45. Okubo M, Horinishi A, Kim D‐H., Yamamoto TT, Murase T. Seven novel sequence variants in the human low density lipoprotein receptor related protein 5 (LRP5) gene. Hum Mutat. 2002; 19(2):186. [DOI] [PubMed] [Google Scholar]
- 46. Koh JM, Jung MH, Hong JS, et al. Association between bone mineral density and LDL receptor‐related protein 5 gene polymorphisms in young Korean men. J Korean Med Sci. 2004; 19(3):407–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zhang Z‐L., Qin Y‐J., He J‐W., et al. Association of polymorphisms in low‐density lipoprotein receptor‐related protein 5 gene with bone mineral density in postmenopausal Chinese women. Acta Pharmacol Sin. 2005; 26(9):1111–6. [DOI] [PubMed] [Google Scholar]
- 48. Urano T, Shiraki M, Narusawa K, et al. Q89R polymorphism in the LDL receptor‐related protein 5 gene is associated with spinal osteoarthritis in postmenopausal Japanese women. Spine. 2007; 32(1):25–9. [DOI] [PubMed] [Google Scholar]
- 49. Mao W, Wordinger RJ, Clark AF. Functional analyses of disease‐associated polymorphism L RP5. Q89R. Mol Vision. 2011; 17:894–902. [PMC free article] [PubMed] [Google Scholar]
- 50. Toomes C, Bottomley HM, Jackson RM, et al. Mutations in LRP5 or FZD4 underlie the common familial exudative vitreoretinopathy locus on chromosome 11q. Am J Hum Genet. 2004; 74(4):721–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ai M, Holmen SL, Van Hul W, Williams BO, Warman ML. Reduced affinity to and inhibition by DKK1 form a common mechanism by which high bone mass‐associated missense mutations in LRP5 affect canonical Wnt signaling. Mol Cell Biol. 2005; 25(12):4946–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Semenov MV, He X. LRP5 mutations linked to high bone mass diseases cause reduced LRP5 binding and inhibition by SOST. J Biol Chem. 2006; 281(50):38276–84. [DOI] [PubMed] [Google Scholar]
- 53. Yorgan TA, Peters S, Jeschke A, et al. The anti‐osteoanabolic function of sclerostin is blunted in mice carrying a high bone mass mutation of Lrp5. J Bone Miner Res. 2015; 30:1175–83. [DOI] [PubMed] [Google Scholar]
- 54. Uitterlinden AG, Arp PP, Paeper BW, et al. Polymorphisms in the sclerosteosis/van Buchem disease gene (SOST) region are associated with bone‐mineral density in elderly whites. Am J Hum Genet. 2004; 75(6):1032–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ferrari SL, Deutsch S, Choudhury U, et al. Polymorphisms in the low‐density lipoprotein receptor‐related protein 5 (LRP5) gene are associated with variation in vertebral bone mass, vertebral bone size, and stature in whites. Am J Hum Genet. 2004; 74(5):866–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Koay MA, Woon PY, Zhang Y, et al. Influence of LRP5 polymorphisms on normal variation in BMD. J Bone Miner Res. 2004; 19(10):1619–27. [DOI] [PubMed] [Google Scholar]
- 57. Gregson CL, Sayers A, Lazar V, et al. The high bone mass phenotype is characterised by a combined cortical and trabecular bone phenotype: findings from a pQCT case‐control study. Bone. 2013; 52(1):380–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Loh NY, Neville MJ, Marinou K, et al. LRP5 regulates human body fat distribution by modulating adipose progenitor biology in a dose‐ and depot‐specific fashion. Cell Metab. 2015; 21(2):262–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Sarrión P, Mellibovsky L, Urreizti R, et al. Genetic analyses of high bone mass cases from the BARCOS cohort of Spanish postmenopausal women. PLoS One. 2014; 9(4):e94607. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information.