

Abstract
Chronic lymphocytic leukemia (CLL) is a common disease with highly variable clinical course. Several recurrent chromosomal alterations are associated with prognosis and may guide risk‐adapted therapy. We have developed a targeted genome‐wide array to provide a robust tool for ascertaining abnormalities in CLL and to overcome limitations of the 4‐marker fluorescence in situ hybridization (FISH). DNA from 180 CLL patients were hybridized to the qChip®Hemo array with a high density of probes covering commonly altered loci in CLL (11q22‐q23, 13q14, and 17p13), nine focal regions (2p15‐p16.1, 2p24.3, 2q13, 2q36.3‐q37.1, 3p21.31, 8q24.21, 9p21.3, 10q24.32, and 18q21.32‐q21.33) and two larger regions (6q14.1‐q22.31 and 7q31.33‐q33). Overall, 86% of the cases presented copy number alterations (CNA) by array. There was a high concordance of array findings with FISH (84% sensitivity, 100% specificity); all discrepancies corresponded to subclonal alterations detected only by FISH. A chromothripsis‐like pattern was detected in eight cases. Three showed concomitant shattered 5p with gain of TERT along with isochromosome 17q. Presence of 11q loss was associated with shorter time to first treatment (P = 0.003), whereas 17p loss, increased genomic complexity, and chromothripsis were associated with shorter overall survival (P < 0.001, P = 0.001, and P = 0.02, respectively). In conclusion, we have validated a targeted array for the diagnosis of CLL that accurately detects, in a single experiment, all relevant CNAs, genomic complexity, chromothripsis, copy number neutral loss of heterozygosity, and CNAs not covered by the FISH panel. This test may be used as a practical tool to stratify CLL patients for routine diagnostics or clinical trials. © 2015 The Authors. Genes, Chromosomes & Cancer Published by Wiley Periodicals, Inc.
INTRODUCTION
Chronic lymphocytic leukemia (CLL) is molecularly and clinically very heterogeneous: some patients have stable disease for many years whereas others show a rapidly progressive course requiring early treatment (Swerdlow et al., 2008; Zenz et al., 2010). Although new targeted therapeutic approaches (i.e., small molecule kinase and BCL2 inhibitors) are improving the outcome, there remains no curative option besides allogeneic stem cell transplantation.
Approximately 80% of CLL cases show genetic alterations at diagnosis, mainly copy number alterations (CNA) rather than chromosomal translocations. The main recurrent CNA are losses of 13q14, 11q22‐q23, and 17p13, and trisomy 12. Currently, as part of routine diagnosis, these four CNAs are tested using fluorescence in situ hybridization (FISH) and based on them, five prognostic groups can be defined (Dohner et al., 2000). Deletion in 13q14 is the most frequent CNA in CLL (∼50% of cases) and is associated with good prognosis when it is found as a sole abnormality (Dohner et al., 2000). Trisomy 12 is present in 10–20% and is associated with NOTCH1 mutations (Balatti et al., 2012; Lopez et al., 2013; Villamor et al., 2013). The 11q22‐q23 losses are rarely found in early‐stage disease but are present in ∼25% of patients with advanced disease; the minimal deleted region harbors ATM, which is mutated in one‐third of cases (Austen et al., 2005), and BIRC3, mutated in a small subset of cases (Rossi et al., 2012). The presence of 11q losses has been associated with more rapid disease progression and extensive lymphadenopathy as well as with shorter time to first treatment (TTT), duration of remission, and overall survival (Dohner et al., 1997). Nevertheless, these associations in the rituximab era are less marked (Hallek et al., 2010; Skowronska et al., 2012; Rosenquist et al., 2013). Finally, 17p13 deletions are found in 4–9% of CLL requiring first‐line therapy; they are found in up to 30–40% patients with relapsed/refractory disease (Zenz et al., 2008, 2011), mainly due to selection of pre‐existing subclones with mutated TP53 (Rossi et al., 2014). Roughly 80% of cases with 17p deletions show TP53 mutations (Zenz et al., 2008). 17p/TP53 alterations are associated with worse prognosis and have important clinical implications at the time of starting the treatment given that these cases are highly chemorefractory to standard cytotoxic regimens (Dohner et al., 1995; Catovsky et al., 2007; Forconi et al., 2008; Dicker et al., 2009; Zenz et al., 2009). Because chemotherapy depends on TP53 it is mandatory to screen for TP53 alterations before initiation of treatment and to consider different therapeutic approaches like alemtuzumab, idelalisib, navetoclax, or ibrutinib (Pospisilova et al., 2012; Farooqui et al., 2015).
Besides 11q and 17p losses, other CNAs reported to confer prognostic value are gains at 2p and 8q and losses at 6q and 8p (Chapiro et al., 2010; Gunnarsson et al., 2010; Brown et al., 2012). Unfortunately, routine FISH testing does not include these regions. Moreover, it has been recently reported that in a series of CLL cases with a normal FISH pattern the presence of CNAs detected by single nucleotide polymorphism (SNP) array (in 22% of cases) was prognostically relevant (Mian et al., 2013). The detection of all these genetic biomarkers may be of paramount importance in clinical practice, since they may identify high risk patients and eventually guide the treatment. Microarray studies of CLL patients have also defined a strong association of increased genomic complexity with poor prognosis (Edelmann et al., 2012; Malek, 2013), represented mainly by cases with a mutated TP53 gene. Clonal evolution (Gunnarsson et al., 2011; Braggio et al., 2012; Knight et al., 2012; Malek, 2013) and chromothripsis (Edelmann et al., 2012) have also been associated with shorter survival in CLL. Overall, these data emphasize the advantages of assessing global genomic complexity by arrays. The only types of genetic alteration that arrays are not able to detect are balanced translocations, which are rare in CLL and have not been associated to poor clinical behavior, except if they are accompanied by a 17p loss (Baliakas et al., 2014).
Array‐based platforms are now widely used in routine diagnosis, especially for constitutional diseases, and are gaining popularity in assessing cancer samples. Many studies have demonstrated that FISH and arrays have more than 90% concordance and that arrays better define gains and losses (Schwaenen et al., 2004; Pfeifer et al., 2007; Gunn et al., 2008; Sargent et al., 2009; Kay et al., 2010; O'Malley et al., 2011; Ouillette et al., 2011; Rinaldi et al., 2011; Edelmann et al., 2012; Knight et al., 2012). The few discordances observed in these studies are usually due to insufficient probe density and subclonality. Another advantage of arrays versus FISH is that only arrays are able to detect copy‐number neutral loss of heterozygosity (CNN‐LOH) of mutated TP53, for instance (Ouillette et al., 2011), and also 11q losses not targeting ATM, gains of chromosome 12 not involving the centromere, and 13q losses not targeting DLEU2, which are the standard FISH probes used in CLL samples (Gunn et al., 2009; Hagenkord et al., 2010).
In the present study, we describe the development of a custom array for CLL that interrogates the most significant genetic alterations, is particularly enriched in CLL‐associated regions, and that captures the complex patterns of aberrations, including chromothripsis. This tool may guide the risk‐stratification and therapeutic decisions according to the genomic alterations or genomic complexity of each patient.
MATERIALS AND METHODS
Patients
The study included 133 patients from the Hospital Clinic (Barcelona) with the diagnosis of CLL according to the WHO criteria (Swerdlow et al., 2008) (Table 1). All patients gave informed consent in agreement with the Institutional Review Board and according to the International Cancer Genome Consortium guidelines (Hudson et al., 2010). In addition, a second series of 47 CLL patients from another institution (University of Leicester, UK, with genetic analyses performed at the University of Kiel) was studied. Fifty‐seven cases (32%) were sampled at the time of diagnosis and the remaining were obtained 7–696 months after diagnosis, with a median of 177 months. Overall, patients were predominantly males (64%), with a median age of 62 years, and most of them presented with Binet stage A (72%). Adverse prognostic factors were detected in about half of the patients: IGHV unmutated status in 62% and positive expression of CD38 and ZAP‐70 in 38 and 44% of cases, respectively. At 10 years of follow‐up, 83% patients (95% confidence interval [CI]: 71–95%) had received CLL‐specific treatment, and the overall survival (OS) of the entire cohort was 39% (95% CI: 27–51%). Number of cases and techniques applied to each of the series are detailed in Supporting Information Figure S1.
Table 1.
Main Clinical Characteristics and Outcome of 180 CLL Patients
| Patient characteristics | N | % |
|---|---|---|
| Age, years | ||
| Median | 62 | |
| Range | 33–98 | |
| Gender | ||
| Male | 115 | 64 |
| Female | 65 | 36 |
| Binet stage | ||
| A | 130 | 72 |
| B | 37 | 21 |
| C | 12 | 7 |
| ZAP 70 expression | ||
| Negative (≤20%) | 60 | 56 |
| Positive (>20%) | 48 | 44 |
| CD38 expression | ||
| Negative (≤30%) | 93 | 62 |
| Positive (>30%) | 58 | 38 |
| IGHV mutational status | ||
| Mutated | 59 | 38 |
| Unmutated | 96 | 62 |
| Mutations | ||
| NOTCH1 | 26 | 22 |
| SF3B1 | 13 | 12 |
| TP53 | 18 | 14 |
| Treated patients | 133 | 74 |
| Deceased patients | 83 | 47 |
| Patients treated at 10 yr (95% CI) | – | 83% (71–95%) |
| Overall survival at 10 yr (95% CI) | – | 39% (27–51%) |
CI, confidence interval; yr, years.
Molecular and Cytogenetic Analyses
DNA was extracted from mononuclear blood cells from CLL patients by using a Qiagen kit (Qiagen, Valencia, CA). All cases had more than 50% tumor cells. IGHV, TP53, NOTCH1, and SF3B1 mutation analyses were performed using direct Sanger sequencing as previously described (Quesada et al., 2012; Villamor et al., 2013; Delgado et al., 2014). Conventional cytogenetics (CC) was performed on G and/or R banded chromosomes obtained after short term culture (usually up to 72 h) without stimulation or with stimulation with e.g. Phorbol 12‐Myristate 13‐Acetate or DSP30 (Shi et al., 2013). Results were described according to the International System for Human Cytogenetic Nomenclature (Shaffer et al., 2013). Only cytogenetic data from samples obtained within 6 months from the DNA sampling date were considered. The FISH panel used interrogated the commonly deleted regions in 11q22, 13q14.3, and 17p13 as well as (partial) trisomy 12 (Abbott Molecular, Des Plaines, IL). For the detection of chromosomal aberrations affecting the TERT locus, FISH was applied using a three color break‐apart assay (RP11‐678B2 spectrum green, RP11‐117B23 diethylaminocoumarin, and RP11‐356C5 spectrum orange) (Nagel et al., 2010). At least one hundred nuclei were examined for each probe and were evaluated in accordance with the diagnostic cut‐offs of the respective laboratories.
Copy Number Arrays
A custom oligonucleotide‐based comparative genomic hybridization (CGH) array was designed based on SurePrint G3 Human CGH 8x60K microarray using eArray software (Agilent Technologies, Palo Alto, CA) (Table 2 and Supporting Information Table S1). In the version v1.0 of the array the selected oligonucleotide probes targeted some focal genomic regions at high coverage (7.7–8.4 probes per Kb), some commonly altered regions at medium/high coverage (0.4–1.8 probes per Kb), and the whole genome at a lower coverage (backbone). With this design, the backbone was composed of an average of one probe every 115 kb outside the regions of interest. The probe density was higher in 13q14.3, 11q22‐q23, and 17p13 (Fig. 1A) and even more enriched in specific genes related to lymphoid neoplasms (MTAP/CDKN2A/CDKN2B, BCL2L11, BCL11A, REL, BCL2, MALT1, MYC, NMYC, and SP110/SP140/SP100); medium coverage was applied to relatively large regions spanning 6q14.1‐q22.31, 7q31.33‐q33, and 11q22.3‐q23.3. The array was also enriched in probes covering two immunoglobulin loci (IGH at 14q32 and IGK at 2p11) for B‐cell clonality control. All catalogued germline copy number variants were removed (Database of Genomic Variants: http://dgv.tcag.ca/dgv/app/home). The series of 133 patients (and one sequential sample from case 102) from Barcelona (Spain) were hybridized with the version 1.0 outsourced to qGenomics (www.qgenomics.com). Then we designed version v3.0, which included three new focal CNA (2q13, 3p21.31, and 10q24.32) and 10340 SNP probes from the Agilent catalog for LOH detection. Regions of potential CNN‐LOH 6q, 8p, 11q, 13q, and 17p were interrogated with 23% (2384) of this set of SNP probes, whereas a backbone of the genome was covered with the remaining probes (7962) (Table 3). The series of 47 CLL patients from Leicester (UK) was hybridized with the v3.0 design outsourced to qGenomics.
Table 2.
Chromosomal Regions Enriched in the qChip®Hemo Array (GRCh37/hg19) for Detection of Copy Number Alterations
| Chr | Start | End | Size | Region (genes) | Densitya |
|---|---|---|---|---|---|
| chr2 | 60676446 | 60782012 | 105566 | 2p16.1 (BLC11A) | Very high |
| chr2 | 61104447 | 61151158 | 46711 | 2p16.1 (REL) | Very high |
| chr2 | 111878490 | 111926022 | 47533 | 2q13 (BCL2L11) | Very high |
| chr2 | 231090445 | 231223847 | 133402 | 2q37.1 (SP140) | Very high |
| chr8 | 128747629 | 128753930 | 6301 | 8q24.21 (MYC) | Very high |
| chr11 | 108082602 | 108252729 | 170127 | 11q22.3 (ATM) | Very high |
| chr13 | 50556688 | 50699677 | 142989 | 13q14.2 (DLEU2, DLEU1, MIR16‐1, MIR15A) | Very high |
| chr17 | 7563917 | 7591659 | 27742 | 17p13.1 (TP53) | Very high |
| chr2 | 16050000 | 16150000 | 100000 | 2p24.3 (MYCN) | High |
| chr2 | 60500000 | 61500000 | 1000000 | 2p16.1‐p15 (BCL11A, PAPOLG, REL PUS10, PEX13) | High |
| chr2b | 111376353 | 111977326 | 600973 | 2q13 (BUB1, ACOXL, BCL2L11) | High |
| chr2 | 230055752 | 231666905 | 1611153 | 2q36.3‐q37.1 (DNER, TRIP12, FBXO36, SLC16A14, SP110, SP140, SP100) | High |
| chr2 | 89118885 | 89438885 | 320000 | 2p11.2 (IGK) | High |
| chr3b | 46996537 | 48502973 | 1506436 | 3p21.31 (SETD2,MAP4, CDC25A, FBXW12) | High |
| chr9 | 21798721 | 22125806 | 327085 | 9p21.3 (MTAP, CDKN2A, CDKN2B) | High |
| chr10b | 103818600 | 104498019 | 679419 | 10q24.32 (GBF1, NFKB2, PSD, FBXL5, SUFU) | High |
| chr13 | 48007849 | 54010887 | 6003038 | 13q14.2‐q14.3 | High |
| chr14 | 106328955 | 106808955 | 480000 | 14q32.33 (IGH) | High |
| chr17 | 6500001 | 10700000 | 4199999 | 17p13.1 | High |
| chr18 | 56250406 | 61070906 | 4820500 | 18q21.32‐q21.33 (MALT1, ZNF532, SEC11C, PIGN, TNFRSF11A, ZCCHC2, PHLPP1, BCL2) | High |
| chr6 | 78000000 | 120000000 | 42000000 | 6q14.1‐q22.31 | Intermediate |
| chr7 | 124449746 | 136150979 | 11701233 | 7q31.33‐q33 | Intermediate |
| chr11 | 106000000 | 118000000 | 12000000 | 11q22.3‐q23.3 | Intermediate |
Very high coverage: 7.7–8.4 probes x Kb; high: 0.7–1.8 probes x Kb; intermediate: 0.1–0.2 probes x Kb.
Regions added to v3.0 and not enriched in v1.0.
Figure 1.
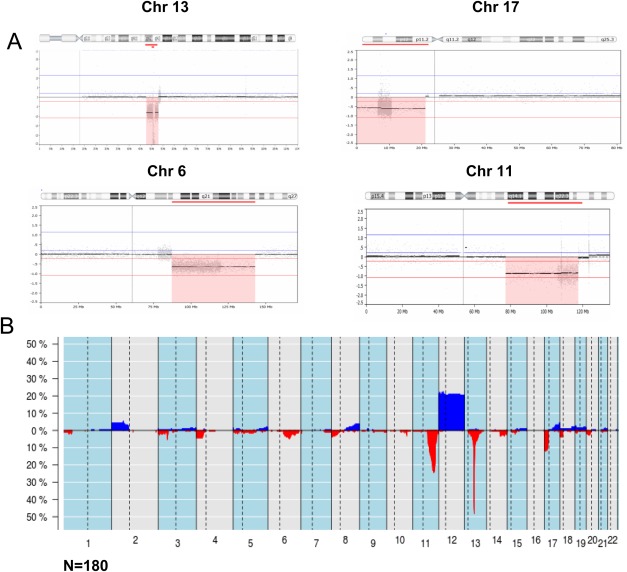
Whole‐genome overview of copy number alterations detected by qChip®Hemo array. (A) Chromosomal view of four altered regions highlighted with a red box. Probes for losses of 13q, 17p, 6q, and 11q14‐q23 are enriched in the array. Gray dots represent individual probes and the higher density of probes in these regions as compared with the backbone coverage can be observed. (B) Frequency plot of copy number alterations in 180 CLL patients. On the X‐axis the chromosomes are represented horizontally, on the Y‐axis the percentage of cases showing the copy number alterations (gains in blue, losses in red).
Table 3.
Chromosomal Regions Enriched in the qChip®Hemo Array v3.0 (GRCh37/hg19) for Detection of Copy Number Neutral Loss of Heterozigosity
| Chr | Start | End | Size | Region | No. SNP probes | Density (probe/Mb) |
|---|---|---|---|---|---|---|
| chr6 | 76070412 | 149814761 | 73744 | 6q14‐q25.1 | 1046 | 14 |
| chr8 | 285123 | 28752104 | 28467 | 8p23.3‐p21.1 | 662 | 23 |
| chr11 | 103864934 | 119876696 | 16012 | 11q22.3‐q23.3 | 252 | 16 |
| chr13 | 44049120 | 59049915 | 15001 | 13q14.11‐q21.1 | 185 | 12 |
| chr17 | 72082 | 16177386 | 16105 | 17p13.3‐p11.2 | 238 | 15 |
| Backbone | – | – | – | whole‐genome | 7962 | 3 |
For each hybridization 300–600 ng of DNA were used. Array hybridization and scanning procedures were performed as previously described (Salaverria et al., 2013). As quality control metric we used DLR spread (probe‐to‐probe log ratio noise of an array) and all samples had low values (mean 0.17; range 0.106–0.35) indicating high quality of hybridization. The qChip®Hemo array data have been deposited to GEO under the accession GSE66923.
A subset of 34 samples was additionally analyzed using Affymetrix Genome‐Wide Human SNP6.0 microarrays (SNP6.0) (Affymetrix, Santa Clara, CA), outsourced to Centro Nacional de Genotipado CEGEN (www.cegen.org ) following the manufacturer's procedures as previously described (Royo et al., 2012). Nexus 7.5 Discovery Edition (Biodiscovery, El Segundo, CA) was used for global analysis and visualization of results from both platforms. Array data were visually inspected for possible subclonal alterations. Coordinates are given according to the GRCh37/hg19 assembly.
Statistical Methods
To correlate data from qChip®Hemo, FISH, and karyotyping crosstab analysis (Fisher's exact test) and Cohen's kappa coefficient were used to measure the agreement between the techniques for four categorical variables (11q, 13q, and 17p losses and trisomy 12). TTT was calculated from the date of sampling to the date of frontline treatment or last follow‐up, and OS was calculated from the date of sampling to the date of death or last follow‐up. TTT and OS were plotted using the Kaplan–Meier method and the log‐rank test was applied. Multivariate analysis was performed with the stepwise proportional hazards model (Cox model) assessing that the covariates used in the model did not violate the proportional hazard assumption. The 37 cases with post‐treatment samples were excluded from the survival analyses. PASW Statistics 18.0 (SPSS, Hong Kong S.A.R) was used. All statistical tests were two‐sided and the level of statistical significance was set at 0.05.
RESULTS
Landscape of Alterations and Complexity of CLL Genomes Detected by a qChip®Hemo Array
Of the 180 cases hybridized with qChip®Hemo, 154 (86%) had CNA. In total, 511 alterations were identified (mean 2.8 CNAs per sample; range 0–28), 377 being deletions and 134 gains (Fig. 1B and Supporting Information Table S2). Recurrent alterations usually tested in CLL were identified in 143 cases (79%): loss in 13q14.3 (48%), loss in 11q22‐q23, including the ATM gene (24%), loss in 17p13, including the TP53 gene (12%), and trisomy 12 (21%). Interestingly, 35 of the 43 (81%) cases with loss of 11q included also the BIRC3 gene, and an additional case showed only deletion of BIRC3 but no loss of ATM (case 184). The qChip®Hemo detected homozygous deletions of 13q14.3 in 22 (25%) of the samples with this alteration (Supporting Information Figs. S2A and S2B). Besides 13q14.3, a single homozygous deletion was found (D961, 11q23.2‐q23.3, size 1.6 Mb and telomeric to the ATM gene) and only one case (D1016) showed amplifications (three discontinuous amplifications in 12p13.3, 12p12.3, and 12p12.1). Other recurrent CNAs were losses of 6q (7%), gains of 2p (6%), focal loss of 3p21 (5%), and 4p losses, 8p losses, and 8q gains (4% each). Among the 47 cases hybridized qChip®Hemo v3.0 (including SNP probes) only two cases showed copy CNN‐LOH of 11q, no other CNN‐LOH was detected in 6q, 8p, 13q, or 17p.
From patient 102, two sequential samples were studied: at diagnosis and at clinical progression 4 years later. The case showed no CNAs at diagnosis but a complex genetic profile with 17 alterations at progression, including 17p loss (Supporting Information Fig. S3).
Of note, using qChip®Hemo array analysis we identified eight cases (8/180; 4%) with multiple lesions in single chromosomes, a pattern suggestive of chromothripsis (at least seven switches between two or more copy number states detected on an individual chromosome) (Edelmann et al., 2012; Rausch et al., 2012). Six of the cases had a deletion in 17p, with concomitant mutation of TP53 in three of these. Interestingly, in three of the eight cases with the chromothripsis‐like pattern we observed a shattered 5p arm characterized by alternating losses and gains, including gain of the TERT (Figs. 2A and 2B). To confirm the alterations at the TERT locus we performed FISH with a three color break‐apart assay (Nagel et al., 2010). Consistent with the array results, one case had a gain of the TERT locus in 30% of the nuclei analyzed (D1016). The other two cases (D1089 and 094) showed gain of the TERT locus in 86% and 62% of the nuclei, respectively, and loss of the centromeric region with breakpoints closely centromeric of TERT. Interestingly, gain of TERT in case 094 showed a tandem duplication pattern (Fig. 2C). Two cases without 5p alterations showed a normal FISH pattern, i.e. two normal copies of the gene (D961 and D990).
Figure 2.

Chromothripsis of chromosome 5 detected by qChip®Hemo array and TERT analysis. (A) An overview of whole‐genome alterations of 3 CLL patients with chromothripsis is shown in the upper panel. Chromosomes 1 to 22 are represented horizontally (gains in blue, losses in red). (B) Chromosome 5 of the three affected patients; 5p was involved by chromothripsis in all cases and extended to 5q in one patient. The TERT gene, included in the minimaly gained region, is highlighted by a square. All cases showed concomitantly gain of 17q and loss of 17p (with mutation of TP53 only in case D1016). (C) Interphase FISH analysis confirmed gain of TERT in the three cases with gain. FISH results of case D1016 showed three signals for RP11‐678B2 (green), RP11‐117B23 (blue), and RP11‐356C5 (orange); FISH results of case D1089 and 094 showed three signals for RP11‐678B2 (green) and RP11‐117B23 (blue) and one signal for RP11‐365C5 (orange), and case 094 showed an amplification or tandem duplication pattern. Arrows indicate TERT signals.
Comparison of the Custom Array with FISH, Metaphase Cytogenetics, and SNP6.0 Array
We next compared alterations detected by qChip®Hemo array and FISH in 106 cases. The median percentage and range of altered cells by FISH was 81% (range 20–99) for 11q loss, 62% (range 20–90) for trisomy 12, 62% (range 10–100) for 13q loss, and 30% (range 10–100) for 17p (Supporting Information Tables S3 and S4). Concordance measured by kappa coefficient showed perfect agreement for loss of 11q and trisomy 12 (κ = 1.00), strong for 13q loss (κ = 0.806), and moderate for 17p loss (κ = 0.598). Overall, there was a high specificity (100% for all regions tested) and sensitivity (mean: 84%; 100% for 11q loss and trisomy 12, 84% for 13q loss, and 50% for 17p loss). All discrepancies corresponded to subclonal alterations detected only by FISH in 10–22% of the cells in 10 cases with 13q14 FISH deletion and 10–17% of the cells in 13 cases with 17p FISH deletion. Among the 26 cases without CNAs by array, four cases had only deletion of 13q14 by FISH in 12, 12, 14, and 22% of the cells, respectively.
Comparison between the qChip®Hemo array and CC results was performed using only data from the 64 cases with abnormal karyotypes (Supporting Information Tables S3 and S5 and Fig. S4). The concordance was strong for 11q loss and trisomy 12 (κ = 0.806 and 0.779) and good for 17p loss (κ = 0.706). Thirteen of the 15 (87%) discrepancies were CNAs detected by array but not by CC (five deletions of 11q, 5 trisomies 12, and 3 deletions of 17p). In two cases, the 17p deletions were not detected by array. Deletion in 13q was observed in 18 cases by CC, 14 of them were also detected by array but four subclonal deletions were only detected by CC (in 2, 3, 4, and 6 metaphases, respectively).
The combination of CC and the qChip®Hemo array information identified that common losses in 13q and 17p were associated with recurrent translocations involving these regions: nine translocations involving 13q with chromosomes 3, 7, 9, 11, 14, 16, and 17 as partners had loss of the minimal region in 13q14.3 in all cases except two. Similarly, nine translocations involving chromosome 17 with chromosomes 4, 5, 13, 15, and 18 as partners resulted in TP53 loss in all cases except two. Interestingly, 3 cases had translocations involving 3p, and one of them (case D1024) had a focal loss of 2.7 Mb, including the SETD2 and MAP4 genes, due to an unbalanced translocation with 13q14, t(3;13)(p22;q14). Interestingly, a MALT1‐MAP4 fusion has been reported in diffuse large B‐cell lymphoma (Murga Penas et al., 2006). No translocations of 11q were found, although it was the second most frequently lost region. Three cases harbored the t(14;18)(q32;q21) but no losses were observed in the 18q region.
The results of the qChip®Hemo array were next compared with the SNP6.0 array results in 34 cases. Both platforms reliably detected the same CNAs (Supporting Information Fig. S5), and only two minor discrepancies were observed: a biallelic loss in 13q14.3 that was detected as a monoallelic loss by SNP6.0 (Supporting Information Fig. S2C) and a small and subclonal loss in 13q14.2 (35 Kb) in case 038 detected only by qChip®Hemo (due to a high coverage of the region in the targeted array: 326 probes in qChip®Hemo and 12 probes in SNP6.0).
Gene Mutations
High risk mutations in NOTCH1, SF3B1, and TP53 were detected in 22, 12, and 14% of the patients, respectively. In relation of common CNAs, trisomy 12 was more common in cases with NOTCH1 mutations than in cases with NOTCH1 wild‐type (P = 0.01); 13q14 losses were more frequent in cases with wild‐type NOTCH1 compared with those with NOTCH1 mutations (P = 0.015); and 17p loss was associated with TP53 mutations (P < 0.001) (Supporting Information Fig. S6). Of the 26 cases without CNAs by array eight had mutations in NOTCH1 and four had SF3B1 mutations; none had TP53 mutations.
Clinical Impact
We next investigated whether the presence of CNA detected by qChip®Hemo array had a significant impact on patient outcome. CLL patients with 11q losses had significantly shorter TTT (P = 0.003; hazard ratio [HR] 2.4; 95% confidence interval [CI] 1.3–4.3); patients with two or more CNAs also had shorter TTT (P = 0.001; HR 2.6; 95% CI 1.5–4.4) (Supporting Information Fig. S7). Moreover, outcome analysis showed that patients with 17p deletions detected by array analysis had a significantly shorter survival than patients without 17p loss (5‐year OS, 19% versus 60%, P < 0.001). However, patients with 17p loss detected only by FISH in low percentage of cells (<17%) behaved similarly to patients without 17p loss (Figs. 3A and 3B). Additionally, patients with chromothripsis‐like patterns had poor prognosis compared with patients without such a pattern (5‐year OS 0% versus 56%; P = 0.02) (Fig. 3C). Finally, stratification of patients according to number of CNAs showed that chromosomal complexity correlated with prognosis (>6 CNAs vs. 4–6 CNAs vs. 0–3 CNAs; 5‐year OS 19, 45, and 61%, respectively; P = 0.001) (Fig. 3D). In a multivariate analyses assessing OS (including 17p13 deletions, presence of chromothripsis, and genomic complexity [>6 CNAs]), only deletions of 17p13 (HR 3.5; 95% CI: 1.7–7.2; P = 0.001) showed independent prognostic impact. CNA in CLL patients analyzed by qChip®Hemo array could recapitulate the genetic model of CLL originally developed by Dohner et al., 2000 based on FISH results (P < 0.001) with increasing survival times according to five genetic categories (17p loss, 11q loss, trisomy 12, no alterations, and 13q loss) (Supporting Information Fig. S8).
Figure 3.
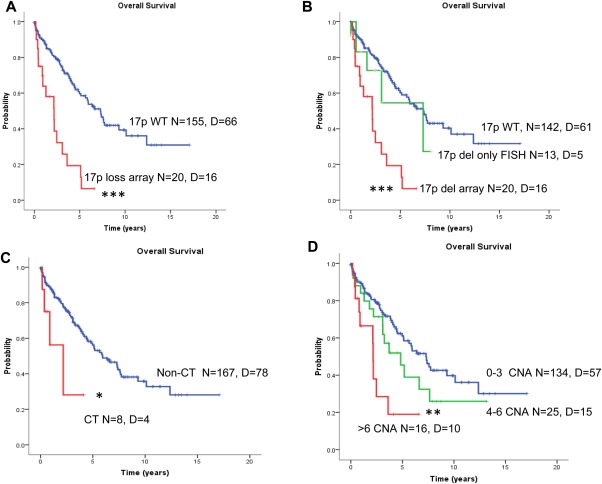
Overall survival (OS) analysis of CLL patients using the qChip®Hemo array. (A) Patients with 17p loss had a significantly shorter OS compared with patients without the 17p deletion. (B) Patients with 17p deletion detected by array had shorter OS compared with patients with 17p deletion detected only by FISH (10–17% cells in all cases), and patients without 17p deletion. (C) Patients with chromothripsis show significantly shorter OS compared with cases without. (D) Kaplan–Meier plot of OS of CLL patients according to increasing number of CNAs (0–3, vs. 4–6 vs. >6 alterations). (*P < 0.05; **P < 0.01; ***P < 0.001). CT, chromothripsis; D, deceased; N, number; WT, wild‐type (not deleted). [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
DISCUSSION
During the last decade, FISH has represented a powerful tool to assess the main four genomic alterations in CLL and has also been a very useful technique to predict the patient's outcome (Dohner et al., 2000). More recently, it has been used to guide therapy in a subset of patients (Hallek et al., 2008). Karyotype analysis can identify translocations and high genomic complexity in addition to common chromosomal losses, but has the limitation of the low mitotic rate of the CLL cells in culture. Several recent reports have shown that low and high resolution arrays are able to detect characteristic CLL alterations and to provide additional prognostic information compared with standard FISH using four markers (deletion of 11q, 13q, and 17p, and trisomy 12) (Chapiro et al., 2010; Gunnarsson et al., 2010, 2011; Edelmann et al., 2012; Mian et al., 2013). Moreover, Mian et al., 2013 found that large alterations could also further predict the outcome in the substantial subgroup (20%) of CLL patients with a normal FISH for the 11q22‐q23, 13q14, chromosome 12, and 17p13 regions. In addition, several studies show that the presence of genomic complexity in CLL is important for identifying high‐risk patients, which could have been missed or misclassified by using the standard FISH CLL panel alone. In that sense, the array that we have developed has been able to detect the more frequent regions altered in CLL genomes, with expected frequencies as well as other more focal alterations. Moreover, 11q losses and the presence of two or more CNAs detected by the array were related to shorter TTT, whereas 17p loss, increased genomic complexity, and chromothripsis were associated with shorter overall survival. Furthermore, as has been previously reported (Dicker et al., 2009; Delgado et al., 2014), CLL cases with 17p13 alterations more frequently have complex karyotypes and poor overall survival. Interestingly, in the present study, we demonstrate that patients with 17p loss detected only by FISH in minor subpopulations (<17%), but not detected by array, behave similarly to patients without 17p loss. This suggests that the lower sensitivity of the array compared to FISH in detecting these small subclones with alteration may not have a direct impact on worse survival.
We here show that qChip®Hemo array data are concordant with FISH and high density array data, and moreover, that this array can detect other alterations (not covered by the FISH panel) with prognostic significance and also complex patterns of alterations in single chromosomes (suggestive of chromothripsis) that cannot be detected by FISH. This phenomenon was detected in eight cases of the present study (4.4% incidence) and was related to poor overall survival. Chromothripsis was initially reported in two CLL cases by whole‐genome sequencing (Campbell et al., 2010; Bassaganyas et al., 2013) and has been recently reported to identify a subset of CLL patients with worse prognosis (Edelmann et al., 2012). Of note, three of the cases with a chromothripsis‐like pattern showed a highly altered 5p arm including gains of TERT. This gene at 5p13.33 encodes the telomerase reverse‐transcriptase TERT that is rarely targeted by somatic chromosomal translocations and amplifications in B‐cell neoplasms (Nagel et al., 2010). These data and our findings suggest that deregulation of the TERT gene by chromosomal abnormalities leading to increased telomerase activity might contribute to B‐cell lymphomagenesis or even to catastrophic chromosomal reorganizations. However, further functional experiments will be needed to address this hypothesis. Noteworthy, alterations in the composition of telomeric proteins (including TERT) have been reported to be involved in the pathogenesis of CLL (Poncet et al., 2008). Moreover, susceptibility SNPs in the TERT and POT1 genes, both involved in telomere function, have been described in a large series of CLL patients (Speedy et al., 2014).
Previous reports of targeted CN arrays for the analysis of CLL have not been applied in a clinical setting mainly because they did not fully achieve the goal of targeting the main CLL alterations. In 2004 and 2007, the first BAC/PAC array‐CGH CLL platforms for clinical purposes were developed (Schwaenen et al., 2004; Patel et al., 2008). Despite their low resolution, both platforms showed high sensitivity and specificity compared with FISH. In 2009, a customized Agilent 4*44K array targeting 15 CLL commonly altered regions was reported (Sargent et al., 2009) with an average spacing of 5‐11 kb in CLL regions. Another BAC array was reported in 2011 (Kolquist et al., 2011; Schultz et al., 2011). However, the major drawback for the clinical utility of this array was the lack of sufficient resolution to identify 13q14.3 and ATM losses. Recently, a new CLL customized Agilent 4*44K has been developed (Houldsworth et al., 2014), including 20 regions recurrently showing imbalances in CLL. The application of this platform to a large series of CLL cases revealed that gains of 2p, 3q, and 8q and losses of 8p have prognostic significance. However, this array has not been compared with FISH data.
The qChip®Hemo array that we have designed and tested allows an exhaustive study of CNAs in a single experiment requiring only 300–600 ng of tumor DNA, without the need of cell culture, and can analyze, in a single experiment, genome‐wide alterations. The qChip®Hemo array is also a less time‐consuming and subjective technique compared with FISH. Additionally, with the introduction of a set of SNP probes in the latest version of the qChip®Hemo array also CNN‐LOH could be assessed. Finally, the data reported herein support the qChip®Hemo as a cost‐effective tool that could replace FISH in a clinical setting of patients with CLL or other B‐cell neoplasms.
In conclusion, the present platform constitutes a robust, sensitive, and standardized tool based on a microarray that is able to detect recurrent CNAs in CLL and to properly capture their potential clinical impact. The similar overall performance, easier workflow, dramatic reduction of specialized hands‐on time (crucial in cost calculations), and the whole‐genome screening capacity make qChip®Hemo highly applicable for molecular testing of CLL, with the possibility to detect 17p and 11q losses, and also additional alterations with clinical impact (i.e, loss of 6q or 8p, gain of 2p of 8q, presence of high genomic complexity, and chromothripsis). The information provided by the array can be useful in the clinical setting by providing the clinicians objective and rapid information of genomic alterations. Risk‐adapted therapy of CLL cases stratified based on their genomic alterations may be applied, with the option to vary the therapeutic strategy. However, the prognostic impact of new, focal and less frequent alterations, as well as chromothripsis, would require further studies in prospective clinical trials as well as the implementation of information about mutations of driver genes (i.e. NOTCH1, SF3B1, and TP53) with potential prognostic relevance.
Supporting information
Supporting Information
Supporting Information Table S2
ACKNOWLEDGMENTS
The authors are indebted to the Genomics core facility of the IDIBAPS and to HCB‐IDIBAPS Biobank‐Tumor Bank and Hematopathology Collection for sample procurement. They are grateful to Miriam Prieto, Cándida Gómez, Amparo Arias, Silvia Martín, and Cristina Capdevila for excellent technical assistance and Nathalie Villahoz and Carmen Muro for excellent work in the coordination of the CLL Spanish Consortium. The authors thank the staff of the cytogenetic and molecular cytogenetic laboratories from the Institute of Human Genetics, Kiel, in particular Dr. Stefan Gesk, Dr. Lana Harder, Reina Zühlke‐Jenisch, Claudia Becher, and Margret Ratjen for their excellent support. They are also very grateful to all patients with CLL who have participated in this study. This work was partially developed at the Centro Esther Koplowitz (CEK), Barcelona, Spain.
Supported by: Fondo de Investigaciones Sanitarias, Instituto de Salud Carlos III (ISCIII), Grant numbers: PI11/01177, PI14/00571; Worldwide Cancer Research; Grant number: 12‐0142; Marató de TV3; Grant number: TV3‐Cancer/2013410; Generalitat de Catalunya Suport Grups de Recerca; Grant number: 2013‐SGR‐378; Red Temática de Investigación Cooperativa en Cáncer (RTICC), Grant numbers: RD12/0036/0036, RD12/0036/0023, RD12/0036/0004, RD12/0036/0069; Subprograma Juan de la Cierva, Grant number: JCI‐2011‐10232; Miguel Servet Contract, Grant number: CP13/00159; the Spanish Ministry of Science and Innovation (MICINN) through the ISCIII —International Cancer Genome Consortium for Chronic Lymphocytic Leukemia (ICGC‐CLL Genome Project); Institució Catalana de Recerca i Estudis Avançats" (ICREA) of the Generalitat de Catalunya; European Regional Development Fund “Una manera de fer Europa” Alexander von Humboldt Post‐doctoral Fellowship.
REFERENCES
- Austen B, Powell JE, Alvi A, Edwards I, Hooper L, Starczynski J, Taylor AM, Fegan C, Moss P, Stankovic T. 2005. Mutations in the ATM gene lead to impaired overall and treatment‐free survival that is independent of IGVH mutation status in patients with B‐CLL. Blood 106:3175–3182. [DOI] [PubMed] [Google Scholar]
- Balatti V, Bottoni A, Palamarchuk A, Alder H, Rassenti LZ, Kipps TJ, Pekarsky Y, Croce CM. 2012. NOTCH1 mutations in CLL associated with trisomy 12. Blood 119:329–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baliakas P, Iskas M, Gardiner A, Davis Z, Plevova K, Nguyen‐Khac F, Malcikova J, Anagnostopoulos A, Glide S, Mould S, Stepanovska K, Brejcha M, Belessi C, Davi F, Pospisilova S, Athanasiadou A, Stamatopoulos K, Oscier D. 2014. Chromosomal translocations and karyotype complexity in chronic lymphocytic leukemia: A systematic reappraisal of classic cytogenetic data. Am J Hematol 89:249–255. [DOI] [PubMed] [Google Scholar]
- Bassaganyas L, Bea S, Escaramis G, Tornador C, Salaverria I, Zapata L, Drechsel O, Ferreira PG, Rodriguez‐Santiago B, Tubio JM, Navarro A, Martin‐Garcia D, Lopez C, Martinez‐Trillos A, Lopez‐Guillermo A, Gut M, Ossowski S, Lopez‐Otin C, Campo E, Estivill X. 2013. Sporadic and reversible chromothripsis in chronic lymphocytic leukemia revealed by longitudinal genomic analysis. Leukemia 27:2376–2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braggio E, Kay NE Vanwier S, Tschumper RC, Smoley S, Eckel‐Passow JE Sassoon T, Barrett M, Van Dyke DL, Byrd JC, Jelinek DF, Shanafelt TD Fonseca R. 2012. Longitudinal genome‐wide analysis of patients with chronic lymphocytic leukemia reveals complex evolution of clonal architecture at disease progression and at the time of relapse. Leukemia 26:1698–1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JR, Hanna M, Tesar B, Werner L, Pochet N, Asara JM, Wang YE, Dal CP, Fernandes SM, Thompson C, Macconaill L, Wu CJ, Van de PY, Correll M, Regev A, Neuberg D, Freedman AS. 2012. Integrative genomic analysis implicates gain of PIK3CA at 3q26 and MYC at 8q24 in chronic lymphocytic leukemia. Clin Cancer Res 18:3791–3802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell PJ, Yachida S, Mudie LJ, Stephens PJ, Pleasance ED, Stebbings LA, Morsberger LA, Latimer C, McLaren S, Lin ML, McBride DJ, Varela I, Nik‐Zainal SA Leroy C, Jia M, Menzies A, Butler AP, Teague JW, Griffin CA, Burton J, Swerdlow H, Quail MA, Stratton MR, Iacobuzio‐Donahue C Futreal PA. 2010. The patterns and dynamics of genomic instability in metastatic pancreatic cancer. Nature 467:1109–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catovsky D, Richards S, Matutes E, Oscier D, Dyer MJ, Bezares RF, Pettitt AR Hamblin T, Milligan DW, Child JA, Hamilton MS, Dearden CE, Smith AG, Bosanquet AG, Davis Z, Brito‐Babapulle V, Else M, Wade R, Hillmen P. 2007. Assessment of fludarabine plus cyclophosphamide for patients with chronic lymphocytic leukaemia (the LRF CLL4 Trial): A randomised controlled trial. Lancet 370:230–239. [DOI] [PubMed] [Google Scholar]
- Chapiro E, Leporrier N, Radford‐Weiss I, Bastard C, Mossafa H, Leroux D, Tigaud I, De BM, Terre C, Brizard F, Callet‐Bauchu E, Struski S, Veronese L, Fert‐Ferrer S, Taviaux S, Lesty C, Davi F, Merle‐Beral H, Bernard OA, Sutton L, Raynaud SD, Nguyen‐Khac F. 2010. Gain of the short arm of chromosome 2 (2p) is a frequent recurring chromosome aberration in untreated chronic lymphocytic leukemia (CLL) at advanced stages. Leuk Res 34:63–68. [DOI] [PubMed] [Google Scholar]
- Delgado J, Salaverria I, Baumann T, Martinez‐Trillos A, Lee E, Jimenez L, Navarro A, Royo C, Santacruz R, Lopez C, Payer AR, Colado E, Gonzalez M, Armengol L, Colomer D, Pinyol M, Villamor N, Aymerich M, Carrio A, Costa D, Clot G, Gine E, Lopez‐Guillermo A, Campo E, Bea S. 2014. Genomic complexity and IGHV mutational status are key predictors of outcome of chronic lymphocytic leukemia patients with TP53 disruption. Haematologica 99:e231–e234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dicker F, Herholz H, Schnittger S, Nakao A, Patten N, Wu L, Kern W, Haferlach T, Haferlach C. 2009. The detection of TP53 mutations in chronic lymphocytic leukemia independently predicts rapid disease progression and is highly correlated with a complex aberrant karyotype. Leukemia 23:117–124. [DOI] [PubMed] [Google Scholar]
- Dohner H, Fischer K, Bentz M, Hansen K, Benner A, Cabot G, Diehl D, Schlenk R, Coy J, Stilgenbauer S. 1995. p53 gene deletion predicts for poor survival and non‐response to therapy with purine analogs in chronic B‐cell leukemias. Blood 85:1580–1589. [PubMed] [Google Scholar]
- Dohner H, Stilgenbauer S, James MR, Benner A, Weilguni T, Bentz M, Fischer K, Hunstein W, Lichter P. 1997. 11q deletions identify a new subset of B‐cell chronic lymphocytic leukemia characterized by extensive nodal involvement and inferior prognosis. Blood 89:2516–2522. [PubMed] [Google Scholar]
- Dohner H, Stilgenbauer S, Benner A, Leupolt E, Krober A, Bullinger L, Dohner K, Bentz M, Lichter P. 2000. Genomic aberrations and survival in chronic lymphocytic leukemia. N Engl J Med 343:1910–1916. [DOI] [PubMed] [Google Scholar]
- Edelmann J, Holzmann K, Miller F, Winkler D, Buhler A, Zenz T, Bullinger L, Kuhn MW, Gerhardinger A, Bloehdorn J, Radtke I, Su X, Ma J, Pounds S, Hallek M, Lichter P, Korbel J, Busch R, Mertens D, Downing JR, Stilgenbauer S, Dohner H. 2012. High‐resolution genomic profiling of chronic lymphocytic leukemia reveals new recurrent genomic alterations. Blood 120:4783–4784. [DOI] [PubMed] [Google Scholar]
- Farooqui MZ, Valdez J, Martyr S, Aue G, Saba N, Niemann CU, Herman SE, Tian X, Marti G, Soto S, Hughes TE, Jones J, Lipsky A, Pittaluga S, Stetler‐Stevenson M, Yuan C, Lee YS, Pedersen LB, Geisler CH, Calvo KR, Arthur DC, Maric I, Childs R, Young NS, Wiestner A. 2015. Ibrutinib for previously untreated and relapsed or refractory chronic lymphocytic leukaemia with TP53 aberrations: A phase 2, single‐arm trial. Lancet Oncol 16:169–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forconi F, Rinaldi A, Kwee I, Sozzi E, Raspadori D, Rancoita PM, Scandurra M, Rossi D, Deambrogi C, Capello D, Zucca E, Marconi D, Bomben R, Gattei V, Lauria F, Gaidano G, Bertoni F. 2008. Genome‐wide DNA analysis identifies recurrent imbalances predicting outcome in chronic lymphocytic leukaemia with 17p deletion. Br J Haematol 143:532–536. [DOI] [PubMed] [Google Scholar]
- Gunn SR, Mohammed MS, Gorre ME, Cotter PD, Kim J, Bahler DW, Preobrazhensky SN, Higgins RA, Bolla AR, Ismail SH, de JD, Eldering E, van Oers MH, Mellink CH, Keating MJ, Schlette EJ, Abruzzo LV, Robetorye RS. 2008. Whole‐genome scanning by array comparative genomic hybridization as a clinical tool for risk assessment in chronic lymphocytic leukemia. J Mol Diagn 10:442–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunn SR, Hibbard MK, Ismail SH, Lowery‐Nordberg M, Mellink CH, Bahler DW, Abruzzo LV, Enriquez EL, Gorre ME, Mohammed MS, Robetorye RS. 2009. Atypical 11q deletions identified by array CGH may be missed by FISH panels for prognostic markers in chronic lymphocytic leukemia. Leukemia 23:1011–1017. [DOI] [PubMed] [Google Scholar]
- Gunnarsson R, Isaksson A, Mansouri M, Goransson H, Jansson M, Cahill N, Rasmussen M, Staaf J, Lundin J, Norin S, Buhl AM, Smedby KE, Hjalgrim H, Karlsson K, Jurlander J, Juliusson G, Rosenquist R. 2010. Large but not small copy‐number alterations correlate to high‐risk genomic aberrations and survival in chronic lymphocytic leukemia: A high‐resolution genomic screening of newly diagnosed patients. Leukemia 24:211–215. [DOI] [PubMed] [Google Scholar]
- Gunnarsson R, Mansouri L, Isaksson A, Goransson H, Cahill N, Jansson M, Rasmussen M, Lundin J, Norin S, Buhl AM, Smedby KE, Hjalgrim H, Karlsson K, Jurlander J, Geisler C, Juliusson G, Rosenquist R. 2011. Array‐based genomic screening at diagnosis and during follow‐up in chronic lymphocytic leukemia. Haematologica 96:1161–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagenkord JM, Monzon FA, Kash SF, Lilleberg S, Xie Q, Kant JA. 2010. Array‐based karyotyping for prognostic assessment in chronic lymphocytic leukemia: Performance comparison of Affymetrix 10K2.0, 250K Nsp, and SNP6.0 arrays. J Mol Diagn 12:184–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallek M, Cheson BD, Catovsky D, Caligaris‐Cappio F, Dighiero G, Dohner H, Hillmen P, Keating MJ, Montserrat E, Rai KR, Kipps TJ. 2008. Guidelines for the diagnosis and treatment of chronic lymphocytic leukemia: A report from the International Workshop on Chronic Lymphocytic Leukemia updating the National Cancer Institute‐Working Group 1996 guidelines. Blood 111:5446–5456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallek M, Fischer K, Fingerle‐Rowson G, Fink AM, Busch R, Mayer J, Hensel M, Hopfinger G, Hess G, von GU, Bergmann M, Catalano J, Zinzani PL, Caligaris‐Cappio F, Seymour JF, Berrebi A, Jager U, Cazin B, Trneny M, Westermann A, Wendtner CM, Eichhorst BF, Staib P, Buhler A, Winkler D, Zenz T, Bottcher S, Ritgen M, Mendila M, Kneba M, Dohner H, Stilgenbauer S. 2010. Addition of rituximab to fludarabine and cyclophosphamide in patients with chronic lymphocytic leukaemia: A randomised, open‐label, phase 3 trial. Lancet 376:1164–1174. [DOI] [PubMed] [Google Scholar]
- Houldsworth J, Guttapalli A, Thodima V, Yan XJ, Mendiratta G, Zielonka T, Nanjangud G, Chen W, Patil S, Mato A, Brown JR, Rai K, Chiorazzi N, Chaganti RS. 2014. Genomic imbalance defines three prognostic groups for risk stratification of patients with chronic lymphocytic leukemia. Leuk Lymphoma 55:920–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson TJ, Anderson W, Artez A, Barker AD, Bell C, Bernabe RR, Bhan MK, Calvo F, Eerola I, Gerhard DS, Guttmacher A, Guyer M, Hemsley FM, Jennings JL, Kerr D, Klatt P, Kolar P, Kusada J, Lane DP, Laplace F, Youyong L, Nettekoven G, Ozenberger B, Peterson J, Rao TS, Remacle J, Schafer AJ, Shibata T, Stratton MR, Vockley JG, Watanabe K, Yang H, Yuen MM, Knoppers BM, Bobrow M, Cambon‐Thomsen A, Dressler LG, Dyke SO, Joly Y, Kato K, Kennedy KL, Nicolas P, Parker MJ, Rial‐Sebbag E, Romeo‐Casabona CM, Shaw KM, Wallace S, Wiesner GL, Zeps N, Lichter P, Biankin AV, Chabannon C, Chin L, Clement B, de AE, Degos F, Ferguson ML, Geary P, Hayes DN, Hudson TJ, Johns AL, Kasprzyk A, Nakagawa H, Penny R, Piris MA, Sarin R, Scarpa A, Shibata T, van d V, Futreal PA, Aburatani H, Bayes M, Botwell DD, Campbell PJ, Estivill X, Gerhard DS, Grimmond SM, Gut I, Hirst M, Lopez‐Otin C, Majumder P, Marra M, McPherson JD, Nakagawa H, Ning Z, Puente XS, Ruan Y, Shibata T, Stratton MR, Stunnenberg HG, Swerdlow H, Velculescu VE, Wilson RK, Xue HH, Yang L, Spellman PT, Bader GD, Boutros PC, Campbell PJ, Flicek P, Getz G, Guigo R, Guo G, Haussler D, Heath S, Hubbard TJ, Jiang T, Jones SM, Li Q, Lopez‐Bigas N, Luo R, Muthuswamy L, Ouellette BF, Pearson JV, Puente XS, Quesada V, Raphael BJ, Sander C, Shibata T, Speed TP, Stein LD, Stuart JM, Teague JW, Totoki Y, Tsunoda T, Valencia A, Wheeler DA, Wu H, Zhao S, Zhou G, Stein LD, Guigo R, Hubbard TJ, Joly Y, Jones SM, Kasprzyk A, Lathrop M, Lopez‐Bigas N, Ouellette BF, Spellman PT, Teague JW, Thomas G, Valencia A, Yoshida T, Kennedy KL, Axton M, Dyke SO, Futreal PA, Gerhard DS, Gunter C, Guyer M, Hudson TJ, McPherson JD, Miller LJ, Ozenberger B, Shaw KM, Kasprzyk A, Stein LD, Zhang J, Haider SA, Wang J, Yung CK, Cross A, Liang Y, Gnaneshan S, Guberman J, Hsu J, Bobrow M, Chalmers DR, Hasel KW, Joly Y, Kaan TS, Kennedy KL, Knoppers BM, Lowrance WW, Masui T, Nicolas P, Rial‐Sebbag E, Rodriguez LL, Vergely C, Yoshida T, Grimmond SM, Biankin AV, Bowtell DD, Cloonan N, deFazio A, Eshleman JR, Etemadmoghadam D, Gardiner BA, Kench JG, Scarpa A, Sutherland RL, Tempero MA, Waddell NJ, Wilson PJ, McPherson JD, Gallinger S, Tsao MS, Shaw PA, Petersen GM, Mukhopadhyay D, Chin L, DePinho RA, Thayer S, Muthuswamy L, Shazand K, Beck T, Sam M, Timms L, Ballin V, Lu Y, Ji J, Zhang X, Chen F, Hu X, Zhou G, Yang Q, Tian G, Zhang L, Xing X, Li X, Zhu Z, Yu Y, Yu J, Yang H, Lathrop M, Tost J, Brennan P, Holcatova I, Zaridze D, Brazma A, Egevard L, Prokhortchouk E, Banks RE, Uhlen M, Cambon‐Thomsen A, Viksna J, Ponten F, Skryabin K, Stratton MR, Futreal PA, Birney E, Borg A, Borresen‐Dale AL, Caldas C, Foekens JA, Martin S, Reis‐Filho JS, Richardson AL, Sotiriou C, Stunnenberg HG, Thoms G, van d V, van't VL, Calvo F. 2010. International network of cancer genome projects. Nature 464:993–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kay NE, Eckel‐Passow JE, Braggio E, Vanwier S, Shanafelt TD, Van Dyke DL, Jelinek DF, Tschumper RC, Kipps T, Byrd JC, Fonseca R. 2010. Progressive but previously untreated CLL patients with greater array CGH complexity exhibit a less durable response to chemoimmunotherapy. Cancer Genet Cytogenet 203:161–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight SJ, Yau C, Clifford R, Timbs AT, Sadighi AE, Dreau HM, Burns A, Ciria C, Oscier DG, Pettitt AR, Dutton S, Holmes CC, Taylor J, Cazier JB, Schuh A. 2012. Quantification of subclonal distributions of recurrent genomic aberrations in paired pre‐treatment and relapse samples from patients with B‐cell chronic lymphocytic leukemia. Leukemia 26:1564–1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolquist KA, Schultz RA, Slovak ML, McDaniel LD, Brown TC, Tubbs RR, Cook JR, Theil KS, Cawich V, Valentin C, Minier S, Neill NJ, Byerly S, Morton SA, Sahoo T, Ballif BC, Shaffer LG. 2011. Evaluation of chronic lymphocytic leukemia by oligonucleotide‐based microarray analysis uncovers novel aberrations not detected by FISH or cytogenetic analysis. Mol Cytogenet 4:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez C, Delgado J, Costa D, Villamor N, Navarro A, Cazorla M, Gomez C, Arias A, Munoz C, Cabezas S, Baumann T, Rozman M, Aymerich M, Colomer D, Pereira A, Cobo F, Lopez‐Guillermo A, Campo E, Carrio A. 2013. Clonal evolution in chronic lymphocytic leukemia: Analysis of correlations with IGHV mutational status, NOTCH1 mutations and clinical significance. Genes Chromosomes Cancer 52:920–927. [DOI] [PubMed] [Google Scholar]
- Malek SN. 2013. The biology and clinical significance of acquired genomic copy number aberrations and recurrent gene mutations in chronic lymphocytic leukemia. Oncogene 32:2805–2817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mian M, Rinaldi A, Mensah AA, Rossi D, Ladetto M, Forconi F, Marasca R, Uhr M, Stussi G, Kwee I, Cavalli F, Gaidano G, Zucca E, Bertoni F. 2013. Large genomic aberrations detected by SNP array are independent prognosticators of a shorter time to first treatment in chronic lymphocytic leukemia patients with normal FISH. Ann Oncol 24:1378–1384. [DOI] [PubMed] [Google Scholar]
- Murga Penas EM, Kawadler H, Siebert R, Frank M, Ye H, Hinz K, Becher C, Hummel M, Barth TF, Bokemeyer C, Stein H, Trumper L, Moller P, Marynen P, Du MQ, Yang X, Hansmann ML, Dierlamm J. 2006. A novel fusion of the MALT1 gene and the microtubule‐associated protein 4 (MAP4) gene occurs in diffuse large B‐cell lymphoma. Genes Chromosomes Cancer 45:863–873. [DOI] [PubMed] [Google Scholar]
- Nagel I, Szczepanowski M, Martin‐Subero JI, Harder L, Akasaka T, Ammerpohl O, Callet‐Bauchu E, Gascoyne RD Gesk S, Horsman D, Klapper W, Majid A, Martinez‐Climent JA, Stilgenbauer S, Tonnies H, Dyer MJ, Siebert R. 2010. Deregulation of the telomerase reverse transcriptase (TERT) gene by chromosomal translocations in B‐cell malignancies. Blood 116:1317–1320. [DOI] [PubMed] [Google Scholar]
- O'Malley DP, Giudice C, Chang AS, Chang D, Barry TS, Hibbard MK, Chen R, Chen ST. 2011. Comparison of array comparative genomic hybridization (aCGH) to FISH and cytogenetics in prognostic evaluation of chronic lymphocytic leukemia. Int J Lab Hematol 33:238–244. [DOI] [PubMed] [Google Scholar]
- Ouillette P, Collins R, Shakhan S, Li J, Peres E, Kujawski L, Talpaz M, Kaminski M, Li C, Shedden K, Malek SN. 2011. Acquired genomic copy number aberrations and survival in chronic lymphocytic leukemia. Blood 118:3051–3061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel A, Kang SH, Lennon PA, Li YF, Rao PN, Abruzzo L, Shaw C, Chinault AC, Cheung SW. 2008. Validation of a targeted DNA microarray for the clinical evaluation of recurrent abnormalities in chronic lymphocytic leukemia. Am J Hematol 83:540–546. [DOI] [PubMed] [Google Scholar]
- Pfeifer D, Pantic M, Skatulla I, Rawluk J, Kreutz C, Martens UM, Fisch P, Timmer J, Veelken H. 2007. Genome‐wide analysis of DNA copy number changes and LOH in CLL using high‐density SNP arrays. Blood 109:1202–1210. [DOI] [PubMed] [Google Scholar]
- Poncet D, Belleville A, t'kint de RC, Roborel de CA Ben SE, Merle‐Beral H, Callet‐Bauchu E, Salles G, Sabatier L, Delic J, Gilson E. 2008. Changes in the expression of telomere maintenance genes suggest global telomere dysfunction in B‐chronic lymphocytic leukemia. Blood 111:2388–2391. [DOI] [PubMed] [Google Scholar]
- Pospisilova S, Gonzalez D, Malcikova J, Trbusek M, Rossi D, Kater AP, Cymbalista F, Eichhorst B, Hallek M, Dohner H, Hillmen P, van OM, Gribben J, Ghia P, Montserrat E, Stilgenbauer S, Zenz T. 2012. ERIC recommendations on TP53 mutation analysis in chronic lymphocytic leukemia. Leukemia 26:1458–1461. [DOI] [PubMed] [Google Scholar]
- Quesada V, Conde L, Villamor N, Ordonez GR, Jares P, Bassaganyas L, Ramsay AJ, Bea S, Pinyol M, Martinez‐Trillos A, Lopez‐Guerra M, Colomer D, Navarro A, Baumann T, Aymerich M, Rozman M, Delgado J, Gine E, Hernandez JM, Gonzalez‐Diaz M, Puente DA, Velasco G, Freije JM, Tubio JM, Royo R, Gelpi JL, Orozco M, Pisano DG, Zamora J, Vazquez M, Valencia A, Himmelbauer H, Bayes M, Heath S, Gut M, Gut I, Estivill X, Lopez‐Guillermo A, Puente XS, Campo E, Lopez OC. 2012. Exome sequencing identifies recurrent mutations of the splicing factor SF3B1 gene in chronic lymphocytic leukemia. Nat Genet 44:47–52. [DOI] [PubMed] [Google Scholar]
- Rausch T, Jones DT, Zapatka M, Stutz AM, Zichner T, Weischenfeldt J, Jager N, Remke M, Shih D, Northcott PA, Pfaff E, Tica J, Wang Q, Massimi L, Witt H, Bender S, Pleier S, Cin H, Hawkins C, Beck C, von DA, Hans V, Brors B, Eils R, Scheurlen W, Blake J, Benes V, Kulozik AE, Witt O, Martin D, Zhang C, Porat R, Merino DM, Wasserman J, Jabado N, Fontebasso A, Bullinger L, Rucker FG, Dohner K, Dohner H, Koster J, Molenaar JJ, Versteeg R, Kool M, Tabori U, Malkin D, Korshunov A, Taylor MD, Lichter P, Pfister SM, Korbel JO. 2012. Genome sequencing of pediatric medulloblastoma links catastrophic DNA rearrangements with TP53 mutations. Cell 148:59–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinaldi A, Mian M, Kwee I, Rossi D, Deambrogi C, Mensah AA, Forconi F, Spina V, Cencini E, Drandi D, Ladetto M, Santachiara R, Marasca R, Gattei V, Cavalli F, Zucca E, Gaidano G, Bertoni F. 2011. Genome‐wide DNA profiling better defines the prognosis of chronic lymphocytic leukaemia. Br J Haematol 154:590–599. [DOI] [PubMed] [Google Scholar]
- Rosenquist R, Cortese D, Bhoi S, Mansouri L, Gunnarsson R. 2013. Prognostic markers and their clinical applicability in chronic lymphocytic leukemia: Where do we stand?. Leuk Lymph 54:2351–2364. [DOI] [PubMed] [Google Scholar]
- Rossi D, Fangazio M, Rasi S, Vaisitti T, Monti S, Cresta S, Chiaretti S, Del G, Fabbri I, Bruscaggin G, Spina A, Deambrogi V, Marinelli C, Fama M, Greco R, Daniele M, Forconi G, Gattei F, Bertoni V, Deaglio F, Pasqualucci S, Guarini L, la‐Favera A, Foa R, Gaidano RG. 2012. Disruption of BIRC3 associates with fludarabine chemorefractoriness in TP53 wild‐type chronic lymphocytic leukemia. Blood 119:2854–2862. [DOI] [PubMed] [Google Scholar]
- Rossi D, Khiabanian H, Spina V, Ciardullo C, Bruscaggin A, Fama R, Rasi S, Monti S, Deambrogi C, De PL, Wang J, Gattei V, Guarini A, Foa R, Rabadan R, Gaidano G. 2014. Clinical impact of small TP53 mutated subclones in chronic lymphocytic leukemia. Blood 123:2139–2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Royo C, Navarro A, Clot G, Salaverria I, Gine E, Jares P, Colomer D, Wiestner A, Wilson WH, Vegliante MC, Fernandez V, Hartmann EM, Trim N, Erber WN, Swerdlow SH, Klapper W, Dyer MJ, Vargas‐Pabon M, Ott G, Rosenwald A, Siebert R, Lopez‐Guillermo A, Campo E, Bea S. 2012. Non‐nodal type of mantle cell lymphoma is a specific biological and clinical subgroup of the disease. Leukemia 26:1895–1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salaverria I, Royo C, Carvajal‐Cuenca A, Clot G, Navarro A, Valera A, Song JY, Woroniecka R, Rymkiewicz G, Klapper W, Hartmann EM, Sujobert P, Wlodarska I, Ferry JA, Gaulard P, Ott G, Rosenwald A, Lopez‐Guillermo A, Quintanilla‐Martinez L, Harris NL, Jaffe ES, Siebert R, Campo E, Bea S. 2013. CCND2 rearrangements are the most frequent genetic events in cyclin D1(‐) mantle cell lymphoma. Blood 121:1394–1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sargent R, Jones D, Abruzzo LV, Yao H, Bonderover J, Cisneros M, Wierda WG, Keating MJ, Luthra R. 2009. Customized oligonucleotide array‐based comparative genomic hybridization as a clinical assay for genomic profiling of chronic lymphocytic leukemia. J Mol Diagn 11:25–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz RA, Delioukina M, Gaal K, Bedell V, Smith DD, Forman SJ, McDaniel LD, Ballif BC, Shaffer LG, Slovak ML. 2011. Evaluation of chronic lymphocytic leukemia by BAC‐based microarray analysis. Mol Cytogenet 4:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwaenen C, Nessling M, Wessendorf S, Salvi T, Wrobel G, Radlwimmer B, Kestler HA, Haslinger C, Stilgenbauer S, Dohner H, Bentz M, Lichter P. 2004. Automated array‐based genomic profiling in chronic lymphocytic leukemia: Development of a clinical tool and discovery of recurrent genomic alterations. Proc Natl Acad Sci USA 101:1039–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaffer LG, McGowan‐Jordan J, Schmid M. 2013. ISCN 2013: An International System for Human Cytogenetic Nomenclature. Basel: Karger. [Google Scholar]
- Shi M, Cipollini MJ, Crowley‐Bish PA, Higgins AW, Yu H, Miron PM. 2013. Improved detection rate of cytogenetic abnormalities in chronic lymphocytic leukemia and other mature B‐cell neoplasms with use of CpG‐oligonucleotide DSP30 and interleukin 2 stimulation. Am J Clin Pathol 139:662–669. [DOI] [PubMed] [Google Scholar]
- Skowronska A, Parker A, Ahmed G, Oldreive C, Davis Z, Richards S, Dyer M, Matutes E, Gonzalez D, Taylor AM, Moss P, Thomas P, Oscier D, Stankovic T. 2012. Biallelic ATM inactivation significantly reduces survival in patients treated on the United Kingdom Leukemia Research Fund Chronic Lymphocytic Leukemia 4 trial. J Clin Oncol 30:4524–4532. [DOI] [PubMed] [Google Scholar]
- Speedy HE, Di Bernardo MC, Sava GP, Dyer MJ, Holroyd A, Wang Y, Sunter NJ, Mansouri L, Juliusson G, Smedby KE, Roos G, Jayne S, Majid A, Dearden C, Hall AG, Mainou‐Fowler T, Jackson GH, Summerfield G, Harris RJ, Pettitt AR, Allsup DJ, Bailey JR, Pratt G, Pepper C, Fegan C, Rosenquist R, Catovsky D, Allan JM, Houlston RS. 2014. A genome‐wide association study identifies multiple susceptibility loci for chronic lymphocytic leukemia. Nat Genet 46:56–60. [DOI] [PubMed] [Google Scholar]
- Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J, Vardiman JW, editors. 2008. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon: IARC. [Google Scholar]
- Villamor N, Conde L, Martinez‐Trillos A, Cazorla M, Navarro A, Bea S, Lopez C, Colomer D, Pinyol M, Aymerich M, Rozman M, Abrisqueta P, Baumann T, Delgado J, Gine E, Gonzalez‐Diaz M, Hernandez JM, Colado E, Payer AR Rayon C, Navarro B, Jose TM, Bosch F, Quesada V, Puente XS, Lopez‐Otin C, Jares P, Pereira A, Campo E, Lopez‐Guillermo A. 2013. NOTCH1 mutations identify a genetic subgroup of chronic lymphocytic leukemia patients with high risk of transformation and poor outcome. Leukemia 27:1100–1106. [DOI] [PubMed] [Google Scholar]
- Zenz T, Krober A, Scherer K, Habe S, Buhler A, Benner A, Denzel T, Winkler D, Edelmann J, Schwanen C, Dohner H, Stilgenbauer S. 2008. Monoallelic TP53 inactivation is associated with poor prognosis in chronic lymphocytic leukemia: Results from a detailed genetic characterization with long‐term follow‐up. Blood 112:3322–3329. [DOI] [PubMed] [Google Scholar]
- Zenz T, Habe S, Denzel T, Mohr J, Winkler D, Buhler A, Sarno A, Groner S, Mertens D, Busch R, Hallek M, Dohner H, Stilgenbauer S. 2009. Detailed analysis of p53 pathway defects in fludarabine‐refractory chronic lymphocytic leukemia (CLL): dissecting the contribution of 17p deletion, TP53 mutation, p53‐p21 dysfunction, and miR34a in a prospective clinical trial. Blood 114:2589–2597. [DOI] [PubMed] [Google Scholar]
- Zenz T, Mertens D, Kuppers R, Dohner H, Stilgenbauer S. 2010. From pathogenesis to treatment of chronic lymphocytic leukaemia. Nat Rev Cancer 10:37–50. [DOI] [PubMed] [Google Scholar]
- Zenz T, Mertens D, Dohner H, Stilgenbauer S. 2011. Importance of genetics in chronic lymphocytic leukemia. Blood Rev 25:131–137. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information Table S2