

Abstract
The expression of the tumor suppressor p53 can influence the bioactivation of, and DNA damage induced by, the environmental carcinogen benzo[a]pyrene, indicating a role for p53 in its cytochrome P450 (CYP)‐mediated biotransformation. The carcinogen 2‐amino‐1‐methyl‐6‐phenylimidazo[4,5‐b]pyridine (PhIP), which is formed during the cooking of food, is also metabolically activated by CYP enzymes, particularly CYP1A2. We investigated the potential role of p53 in PhIP metabolism in vivo by treating Trp53(+/+), Trp53(+/−) and Trp53(−/−) mice with a single oral dose of 50 mg/kg body weight PhIP. N‐(Deoxyguanosin‐8‐yl)‐2‐amino‐1‐methyl‐6‐phenylimidazo[4,5‐b]pyridine (PhIP‐C8‐dG) levels in DNA, measured by liquid chromatography‐tandem mass spectrometry, were significantly lower in liver, colon, forestomach and glandular stomach of Trp53(−/−) mice compared to Trp53(+/+) mice. Lower PhIP‐DNA adduct levels in the livers of Trp53(−/−) mice correlated with lower Cyp1a2 enzyme activity (measured by methoxyresorufin‐O‐demethylase activity) in these animals. Interestingly, PhIP‐DNA adduct levels were significantly higher in kidney and bladder of Trp53(−/−) mice compared to Trp53(+/+) mice, which was accompanied by higher sulfotransferase (Sult) 1a1 protein levels and increased Sult1a1 enzyme activity (measured by 2‐naphthylsulfate formation from 2‐naphthol) in kidneys of these animals. Our study demonstrates a role for p53 in the metabolism of PhIP in vivo, extending previous results on a novel role for p53 in xenobiotic metabolism. Our results also indicate that the impact of p53 on PhIP biotransformation is tissue‐dependent and that in addition to Cyp1a enzymes, Sult1a1 can contribute to PhIP‐DNA adduct formation.
Keywords: tumor suppressor p53, heterocyclic aromatic hydrocarbon, PhIP, carcinogen metabolism, DNA adduct formation, cytochrome P450, sulfotransferases, mouse model, mass spectrometry
Short abstract
What's new?
Many environmental carcinogens must be metabolized to form reactive electrophilic species capable of damaging DNA. The tumour suppressor p53 was shown to influence the cytochrome P450 (CYP)‐mediated biotransformation of the environmental carcinogen benzo[a]pyrene. The suspected carcinogen 2‐amino‐1‐methyl‐6‐phenylimidazo[4,5‐b]pyridine (PhIP), which is formed during the cooking of food, is also metabolically activated by CYP enzymes. This study shows that expression of p53 influences the biotransformation of PhIP. Using a gene knockout mouse model, the authors show that the impact of p53 on PhIP‐DNA adduct formation is tissue‐dependent and that, in addition to cytochrome P450 enzymes, sulfotransferases contribute to the bioactivation of PhIP.
The environment is becoming increasingly polluted with chemicals that play an important role in many human cancers. Genotoxic carcinogens from the environment can damage DNA by binding covalently to it, which can lead to mutations. The tumor suppressor TP53, which encodes p53, plays a crucial role during cellular response to DNA damage by regulating various cellular processes such as cell cycle arrest, apoptosis and DNA repair.1 Under genotoxic stress, cellular levels of p53 protein increase via post‐transcriptional mechanisms and the ability of p53 to bind specific DNA sequences is activated. p53 function disabled by mutations in the TP53 coding sequence might lead to the development of tumors.2
Many environmental carcinogens require metabolism (e.g., catalysed by cytochrome P450 [CYP] enzymes) to form reactive electrophilic species capable of covalently binding to DNA (i.e., DNA adduct formation). We found recently in human cells that p53 expression is linked to CYP1A1‐mediated metabolic activation of the environmental carcinogen benzo[a]pyrene (BaP).3 BaP‐DNA adduct levels in TP53(−/−) cells were significantly lower than in TP53(+/+) cells, which correlated with BaP‐induced CYP1A1 expression in these cells. Bypass of the need for metabolic activation by treating cells with BaP‐7,8‐dihydrodiol‐9,10‐epoxide (BPDE), the activated metabolite of BaP, resulted in similar adduct levels in all cell lines, regardless of p53 status. In contrast, in in‐vitro experiments BaP‐DNA adduct formation was higher in livers and kidneys of Trp53(−/−) mice compared to Trp53(+/+) mice, while treating with BPDE resulted in similar adduct levels in both mouse lines.4 The higher BaP‐DNA adduct levels in the livers of BaP‐treated Trp53(−/−) mice correlated with higher Cyp1a1 enzyme activity in these animals.
Heterocyclic aromatic amines (HAAs) are carcinogenic compounds formed during cooking of meat, fish and poultry.5 Among these HAAs is 2‐amino‐1‐methyl‐6‐phenylimidazo[4,5‐b]pyridine (PhIP), which is mutagenic in several screening assays6 and which induces colon, prostate and mammary gland tumors in rats, which correspond to the principal sites in humans associated with Western diet‐related cancer, implying that it could be a human carcinogen.5, 7 The International Agency for Research on Cancer (IARC) has classified PhIP as possibly carcinogenic to humans (Group 2B).5 HAAs, including PhIP, form DNA adducts after metabolic activation and CYPs (particularly CYP1A2 but also CYP1A1) are the most important enzymes in the initial oxidation of PhIP, leading to the formation of N‐hydroxy‐PhIP (N‐OH‐PhIP) (see Supporting Information Fig. 1).6 Further metabolism by N‐acetyltransferases (NATs) or sulfotransferases (SULTs) converts N‐OH‐PhIP into esters capable of undergoing heterolytic cleavage to produce a nitrenium ion, which is the ultimate reactive species that reacts with DNA.8 The major covalent DNA adduct detected in vivo resulting from exposure of experimental animals and humans to PhIP is N‐(deoxyguanosin‐8‐yl)‐2‐amino‐1‐methyl‐6‐phenylimidazo[4,5‐b]pyridine (PhIP‐C8‐dG).9
In this study, we investigated the impact of Trp53 status on the bioactivation of PhIP in Trp53(+/+), Trp53( +/− ) and Trp53(−/−) mice. These gene knock‐out mouse models have been shown to be particularly useful for studying Trp53 in carcinogenesis because p53 function is highly cell‐type specific.2 PhIP‐C8‐dG was detected in DNA and quantified using liquid chromatography‐tandem mass spectrometry (LC‐MS/MS) selected reaction monitoring (SRM) mode. Tissue‐specific expression and activity of xenobiotic‐metabolising enzymes (XMEs) involved in PhIP metabolism were compared with DNA adduct formation in the same tissue.
Material and Methods
Carcinogen and adduct standard
Synthesis of PhIP was performed at the Biochemical Institute for Environmental Carcinogens, Prof. Dr. Gernot Grimmer‐Foundation (Grosshansdorf, Germany) according to a method described previously.10 The synthesis of PhIP‐C8‐dG and [13C10]PhIP‐C8‐dG was performed as reported.11
Animal treatment
All animal experiments were conducted in accordance with the law at the Leiden University Medical Center, Leiden, The Netherlands, after approval by the institutional ethics committee. Trp53(+/+), Trp53(+/−) and Trp53(−/−) male C57BL/6 mice were generated, housed, and genotyped as reported.4, 12 Groups of male Trp53(+/+), Trp53(+/−) and Trp53(−/−) mice (3 months old; 25–30 g; n = 4/group) were treated with a single oral dose of 50 mg/kg body weight (bw) of PhIP dissolved in corn oil following a treatment protocol used previously to study PhIP metabolism.10 Control mice (n = 4) received solvent (corn oil) only. Animals were killed 24 hrs after treatment and their tissues (liver, lung, kidney, colon, small intestine, bladder, forestomach and glandular stomach) were collected, snap‐frozen in liquid nitrogen, and stored at −80°C until further analysis.
DNA adduct analysis by LC‐ESI‐MS/MS
DNA from whole tissue was isolated by a standard phenol‐chloroform extraction method. An LC‐ESI‐MS/MS method to detect and quantify PhIP‐C8‐dG adducts in DNA was adapted as described previously (see Supporting Information File 1).11
Preparation of microsomal and cytosolic samples
Hepatic and renal microsomal and cytosolic fractions (n = 4) were isolated as described4 in Supporting Information File 1.
Measurement of Cyp1a enzyme activity in hepatic and renal microsomes
Hepatic and renal microsomal samples were characterised for Cyp1a1/2 activity by determining 7‐ethoxyresorufin O‐deethylation (EROD) activity and for Cyp1a2 by determining 7‐methoxyresorufin O‐demethylation (MROD) activity.13 Cyp1a enzyme activity was also measured with 3‐cyano‐7‐ethoxycoumarin (CEC) as substrate.14 Detailed descriptions of methods are given in Supporting Information File 1.
Expression of Sult1a1 and Nat1/2 protein by western blotting
Microsomal and cytosolic proteins were separated using NuPage 4–12% Bis‐Tris sodium‐dodecyl sulfate (SDS)‐polyacrylamide gels (Life Technologies), and Western blotting was carried out as reported previously.4, 15 Sult1a1 and Nat1/2 forms were detected with antisera raised in rabbit against bacterial inclusion bodies of human SULT1A or NAT2 (Supporting Information File 1).14
Measurement of Sult1a enzyme activity in renal cytosols
Renal cytosolic samples were characterised for Sult1a activity by monitoring the formation of p‐nitrophenol from a 5′‐phosphoadenosine 3′‐phosphosulfate (PAPS)‐regenerating system (Supporting Information File 1).16
Results
PhIP‐induced DNA adduct formation in Trp53(+/+), Trp53(+/−) and Trp53(−/−) mice
The formation of PhIP‐C8‐dG adducts was determined in various organs by LC‐MS/MS (Fig. 1). Figure 1a shows the LC‐ESI‐MS/MS collision induced dissociation (CID) product ion spectrum of PhIP‐C8‐dG standard whereas Figures 1b and 1c show typical LC‐MS/MS SRM ion chromatograms of the internal [13C10]PhIP‐C8‐dG alone or after spiking liver DNA isolated from Trp53(+/+) mice treated with 50 mg/kg bw PhIP, respectively. No PhIP‐C8‐dG was detectable in control (untreated) mice (data not shown). PhIP‐C8‐dG adduct levels were significantly lower in liver, forestomach, glandular stomach, and colon of Trp53(−/−) mice compared to Trp53(+/+) mice while adduct levels in kidney and bladder of Trp53(−/−) mice were significantly higher than in Trp53(+/+) mice (Fig. 1 d). For liver adduct levels were ∼2.1‐fold lower in Trp53(−/−) mice than in Trp53(+/+) mice whereas for kidney they were ∼4.8‐fold higher. Of note, no influence of p53 on PhIP‐DNA adduct formation was observed in Trp53(+/−) mice, except for glandular stomach in which similar adduct levels were found in Trp53(+/−) and Trp53(−/−) mice (Fig. 1 d).
Figure 1.
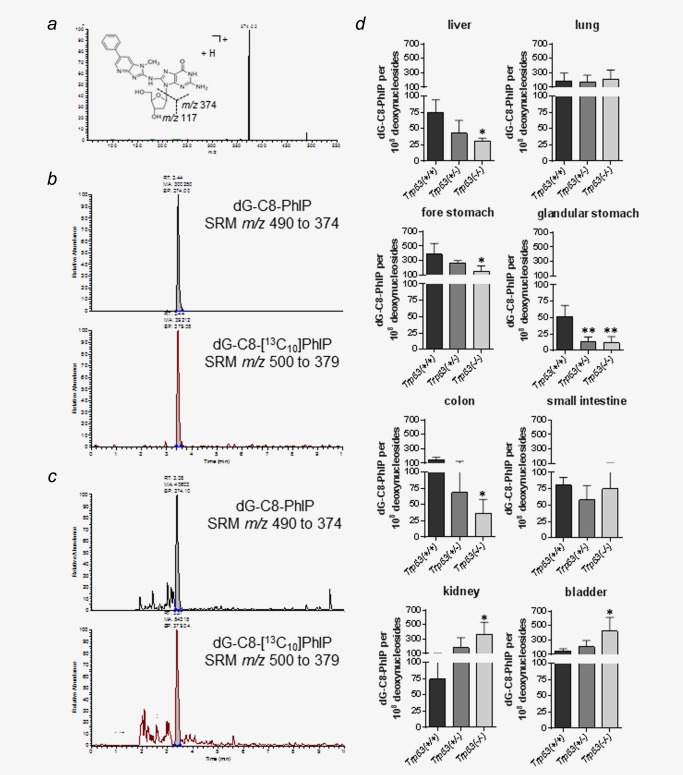
(a) Positive LC‐ESI‐MS/MS collision induced dissociation (CID) product ion spectrum of PhIP‐C8‐dG standard (1 ng/ml). The spectrum was acquired after continuous infusion at a flow rate of 10 μL/min with a syringe pump. Typical LC‐MS/MS SRM ion chromatograms for the determination of PhIP‐C8‐dG in (b) a mixture of PhIP‐C8‐dG standard (1 ng/ml) and 0.49 ng (1000 fmol) of [13C10]PhIP‐C8‐dG internal standard on column and in (c) 50 μg liver DNA isolated from Trp53(+/+) mice treated with 50 mg/kg bw PhIP and 0.49 ng (1000 fmol) of [13C10]PhIP‐C8‐dG internal standard on column. The SRM transitions monitored were m/z 490 to 374 for PhIP‐C8‐dG and m/z 500 to 379 for [13C10]PhIP‐C8‐dG. The analytical column was eluted isocratically at a flow rate of 200 μL/min with water/acetonitrile (0.1% formic acid) (85:15, v/v). (d) Quantitative LC‐ESI‐MS/MS analysis of PhIP‐C8‐dG in various tissues of Trp53(+/+), Trp53(+/−) and Trp53(−/−) mice after exposure to PhIP. Values are the mean ± SD (n = 4). Statistical analysis was performed by one‐way ANOVA followed by Tukey post‐hoc test (*p < 0.05, **p < 0.01; different from Trp53(+/+) mice). [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Expression of PhIP metabolising enzymes in Trp53(+/+), Trp53(+/−) and Trp53(−/−) mice
In rodents, CYP‐mediated N‐oxidation of PhIP occurs primarily in the liver which leads via N‐OH‐PhIP to the formation of PhIP‐C8‐dG in DNA. Metabolic activation of PhIP is catalysed mainly by CYP1A2, but also CYP1A1.6 Thus we studied Cyp1a1 and Cyp1a2 enzyme activity in hepatic microsomal fractions isolated from Trp53(+/+), Trp53(+/−) and Trp53(−/−) mice. MROD activity, which is considered a measure for Cyp1a2 enzyme activity, was significantly lower (∼2.7‐fold) in the liver of PhIP‐treated Trp53(−/−) mice relative to PhIP‐treated Trp53(+/+) mice (Fig. 2a) which correlates with the levels of PhIP‐DNA adducts in the livers of these animals. EROD and CEC activities are considered measures of Cyp1a activity, measuring both Cyp1a1 and Cyp1a2 enzyme activity. First, we found that PhIP treatment led to a significant induction of Cyp1a enzyme activity in livers of Trp53(+/+) and Trp53(+/−) mice, but not in Trp53(−/−) mice (Fig. 2b). Second, we observed that Cyp1a enzyme activity in PhIP‐treated Trp53(−/−) mice was lower than in Trp53(+/−) and Trp53(+/+) mice, although statistical significance was observed only in the samples measured with the CEC assay (Fig. 2c). Collectively these results show that lower hepatic Cyp1a enzyme activity in Trp53(−/−) mice correlates well with lower PhIP‐DNA adduct formation in the liver of these animals.
Figure 2.
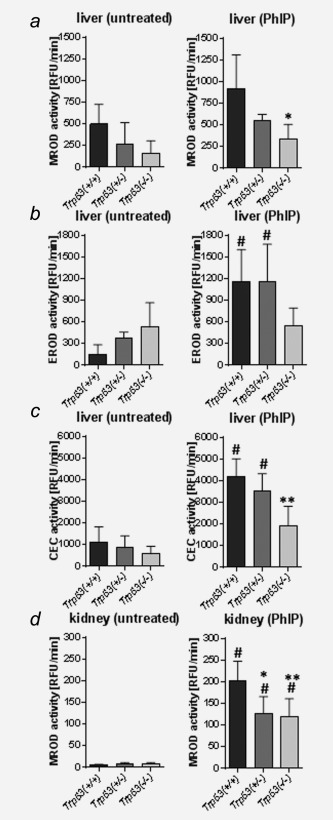
Measurement of microsomal enzymes metabolising PhIP in the livers and kidneys isolated from Trp53(+/+), Trp53(+/−) and Trp53(−/−) mice. Cyp1a enzyme activity was measured as (a) MROD, (b) EROD as well as (c) CEC activity in hepatic microsomes and (d) MROD activity in renal microsomes isolated from control (untreated) mice (left panel) or mice treated with PhIP (right panel). Values are the mean ± SD of three independent determinations; 4 animals per genotype were analysed in three separate experiments. RFU, relative fluorescence unit. Statistical analysis was performed by one‐way ANOVA followed by Tukey post‐hoc test (# p < 0.05, versus control [untreated] mice; *p < 0.05, **p < 0.01, different from PhIP‐treated Trp53(+/+) mice).
As Trp53 status also impacted on PhIP‐DNA adduct formation in the kidney (compare Fig. 1 d) we next studied the expression and activity of PhIP metabolising enzymes in renal microsomal and cytosolic fractions. Due to the role of Cyp1a2 as major phase I enzyme activating PhIP we measured MROD activity in renal microsomes. We found that Cyp1a2 enzyme activity was strongly induced after PhIP treatment in all mouse lines (Fig. 2d). Interestingly, MROD activity was ∼two‐fold lower in Trp53(+/−) and Trp53(−/−) mice relative to Trp53(+/+) mice (Fig. 2d) which did not correlate with PhIP‐DNA adduct formation in this organ (compare Fig. 1 d).
Previous studies have shown that the Cyp1a‐mediated metabolite N‐OH‐PhIP can be further activated by phase II enzymes such as SULTs and NATs (see Supporting Information Fig. 1).8 SULTs and NATs can convert N‐OH‐PhIP into esters capable of undergoing heterolytic cleavage to produce a PhIP‐nitrenium ion which can bind covalently to DNA forming PhIP‐C8‐dG. Using Western blotting we found higher Sult1a1 protein levels in the kidney of Trp53(−/−) mice compared to Trp53(+/+) mice (Fig. 3 a). No such changes in Sult1a1 protein levels were observed in hepatic cytosolic samples, where there was no detectable expression. This finding was unexpected as liver has been reported to be a primary site of Sult1a1 expression.17 As the antibody was raised against bacterial inclusion bodies of human SULT1A, it is possible that the affinity to detect mouse Sult1a1 protein may be lower. However, differences in the organ‐specific distribution of Sults in mice have been described.17
Figure 3.
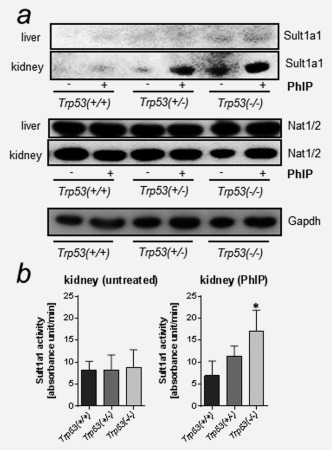
Measurement of cytosolic enzymes metabolising PhIP in the livers and kidneys isolated from Trp53(+/+), Trp53(+/−) and Trp53(−/−) mice. (a) Western blot analysis of Sult1a1 and Nat1/2 in hepatic and renal cytosols. Representative images of the Western blotting are shown and at least duplicate analysis was performed from independent experiments. Gapdh protein expression was used as loading control for the cytosolic fractions and a representative blot is shown. (b) Sult1a enzyme activity was measured using a colorimetric assay with 2‐ naphthol. Values are the mean ± SD of three independent determinations; four animals per genotype were analysed in three separate experiments. Statistical analysis was performed by one‐way ANOVA followed by Tukey post‐hoc test (*p < 0.05, different from PhIP‐treated Trp53(+/+) mice).
No change in Nat1/2 protein levels were observed between mouse lines in hepatic and renal cytosolic samples (Fig. 3 a). This suggests that Trp53 status does not modulate the expression of Nats in either liver or kidney of these mice. As N‐OH‐PhIP is activated by SULT1A enzymes (i.e., SULT1A1 and SULT1A2 in humans)8 we determined Sult1a1 enzyme activity in renal cytosols isolated from Trp53(+/+), Trp53(+/−) and Trp53(−/−) mice. We found that Sult1a enzyme activity was significantly increased in PhIP‐treated Trp53(−/−) mice compared to Trp53(+/−) and Trp53(+/+) mice exposed to PhIP (Fig. 3 b). This increase in Sult1a enzyme activity in Trp53(−/−) mice was in line with higher Sult1a1 protein levels (compare Fig. 3 a) and correlated with enhanced PhIP‐DNA adduct formation in the kidney of these animals (see Fig. 1 d).
Discussion
In this study, we have used Trp53(+/+), Trp53(+/−) and Trp53(−/−) mice to investigate the effect of p53 on PhIP‐DNA adduct formation in vivo and to examine the expression and activity of key enzymes involved in PhIP metabolism. We have conducted an in vivo study in order to take into account the roles of route of administration, absorption, renal clearance and tissue‐specific expression and activity of XMEs.18 PhIP‐DNA adduct formation was observed in all tissues investigated confirming a previous study involving multiple PhIP doses.10 However, the degree of DNA binding in various tissues of Trp53(+/+) mice was different between the two studies which is likely to be attributed to the different protocols (i.e., a single dose of 50 mg/kg bw PhIP versus five daily doses). Interestingly, PhIP‐induced DNA adduct formation was altered differently in multiple organs of Trp53(−/−) mice relative to Trp53(+/+) mice suggesting that the influence of p53 is highly tissue‐specific. Formation of PhIP‐C8‐dG adducts were significantly lower in liver, forestomach, glandular stomach and colon of Trp53(−/−) mice compared to Trp53(+/+) mice while adduct levels in kidney and bladder of Trp53(−/−) mice were significantly higher than in Trp53(+/+) mice. In lung and small intestine no difference in PhIP‐DNA adduct formation was observed between mouse lines.
Mainly, Cyp1a2, but also Cyp1a1, are considered to be the key enzymes responsible for N‐oxidation of PhIP to N‐OH‐PhIP,6 while N‐OH‐PhIP can be further activated by Nats and Sults.8 Our data showed that lower PhIP‐DNA adduct levels in the livers of Trp53(−/−) mice relative to Trp53(+/+) mice correlated with lower hepatic Cyp1a2 enzyme activity in the former animals. In contrast, higher PhIP‐C8‐dG levels in the kidney of Trp53(−/−) mice relative to Trp53(+/+) mice correlated with higher Sult1a1 protein levels and higher Sult1a enzyme activity in the former animals. Thus, our results not only confirm a previous study4 demonstrating an emerging role for p53 in Cyp‐mediated carcinogen metabolism in vivo but they also demonstrate the potential impact of p53 function on other XMEs (i.e., Sult1a1) in carcinogen metabolism in vivo.
Studies investigating carcinogen‐DNA adduct formation in transgenic mice with altered Trp53 status are sparse.4, 19 Carmichael et al.19 observed a ∼twofold increase in diethylstilbestrol (DES)‐induced DNA adduct formation in Trp53 (+/−) mice compared to Trp53(+/+), while Trp53(−/−) mice were not examined. The authors concluded that small differences in the protein expression of several Cyp enzymes could have contributed to the alterations in DES‐induced adduct levels.19 We recently found that DNA adduct formation induced by BaP was significantly higher in liver and kidney of Trp53(−/−) mice than Trp53(+/+) mice while 3‐nitrobenzanthrone (3‐NBA)‐induced DNA adduct formation was not altered in these mouse lines.4 These results could be explained by different metabolic pathways leading to DNA adduct formation. CYP1A1 is considered the key enzyme responsible for the metabolic activation of BaP18 while the most efficient enzyme to activate 3‐NBA is NAD(P)H:quinone oxidoreductase (NQO1).20 Higher BaP‐DNA adduct levels in the livers of Trp53(−/−) mice correlated with increased Cyp1a enzyme activity in these animals.4 In addition, significantly higher amounts of BaP metabolites were formed ex vivo in hepatic microsomes from BaP‐pretreated Trp53(−/−) mice compared to wild‐type animals.4 No differences were observed for 3‐NBA‐DNA adduct formation between Trp53(+/+), Trp53(+/−) or Trp53(−/−) mice, which was in line with unchanged Nqo1 enzyme activity.4
In human cells, p53 has been shown to impact on CYP1A1‐mediated bioactivation of BaP, but not on the metabolic activation of 3‐NBA by NQO1.3, 21 In human cells, in vitro loss of p53 function (i.e., TP53(−/−) cells) resulted in considerably lower BaP‐DNA adduct levels compared to TP53(+/+) cells after BaP exposure.3, 21 We found that BaP‐induced CYP1A1 expression in human cells was regulated through p53 binding to a p53 response element (p53RE) in the regulatory region of CYP1A1,3 thereby enhancing its transcription. Others have found that in human cells p53 can induce the activity of CYP3A4 or UGT2B7 via its binding to p53REs and subsequent enhancement of gene transcription.22, 23 However, the role of p53 in CYP1A1‐mediated metabolism of BaP is different in vitro and in vivo.3, 4 While the induction of Cyp1a1 via p53 binding to a p53RE in the regulatory region of Cyp1a1 fails to explain the impact of p53 on BaP metabolism in the liver in vivo,4 this mechanism could possibly be relevant for p53 function on Cyp1a2‐mediated PhIP metabolism in the liver (present study).
Our results indicate that Cyp1a2 does not have the same role in DNA adduct formation by PhIP in the kidney. Here, higher adduct levels in Trp53(−/−) mice than in Trp53(+/+) can be explained by enhanced expression and activity of Sults (e.g., Sult1a1). There is evidence of the expression of multiple forms of Sult enzymes in mouse kidney, mainly Sult1d1, but also Sult1a1, which have been linked to carcinogen‐DNA adduct formation in the kidney.8, 24 To our knowledge this is the first study that suggests that Sult‐mediated carcinogen activation can be linked to p53 function. The exact mechanism remains to be determined, but these results confirm an impact of p53 on both Phase I and Phase II enzymes.
In summary, we found that p53 functions influence the bioactivation of PhIP in vivo, indicating that gene‐environmental interactions need to be taken into account with regard to carcinogen metabolism. Results to date indicate that the cellular impact of p53 on carcinogen metabolism depends on the agent studied and the XMEs involved. Future investigations will need to assess whether other environmental carcinogens activated by Sults depend on p53 function and how p53 regulates the expression of XMEs in vivo.
Supporting information
Supporting Information
Supporting Information
Acknowledgement
David Phillips and Volker Arlt are members of the COMSIG (Causes Of Mutational SIGnatures) consortium. The opinions expressed here do not necessarily represent those of the US Food and Drug Administration.
References
- 1. Hollstein M, Hainaut P. Massively regulated genes: the example of TP53. J Pathol 2010;220:164–73. [DOI] [PubMed] [Google Scholar]
- 2. Donehower LA. Insights into wild‐type and mutant p53 functions provided by genetically engineered mice. Hum Mutat 2014;35:715–27. [DOI] [PubMed] [Google Scholar]
- 3. Wohak LE, Krais AM, Kucab JE, et al. Carcinogenic polycyclic aromatic hydrocarbons induce CYP1A1 in human cells via a p53 dependent mechanism. Arch Toxicol, in press. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Krais AM, Speksnijder EN, Melis JPM, et al. The impact of p53 on DNA damage and metabolic activation of the environmental carcinogen benzo[a]pyrene: effects in Trp53(+/+), Trp53(+/‐) and Trp53(‐/‐) mice. Arch Toxicol 2015, in press. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. IARC. Some naturally occuring substances: food items and constituents, heterocyclic aromatic amines and mycotoxins. IARC Monogr Eval Carcinog Risk Hum 1993;56. [Google Scholar]
- 6. Gooderham NJ, Zhu H, Lauber S, et al. Molecular and genetic toxicology of 2‐amino‐1‐methyl‐6‐phenylimidazo[4,5‐b]pyridine (PhIP). Mutat Res 2002;506507:91–9. [DOI] [PubMed] [Google Scholar]
- 7. Dingley KH, Curtis KD, Nowell S, et al. DNA and protein adduct formation in the colon and blood of humans after exposure to a dietary‐relevant dose of 2‐amino‐1‐methyl‐6‐phenylimidazo[4,5‐b]pyridine. Cancer Epidemiol Biomarkers Prev 1999;8:507–12. [PubMed] [Google Scholar]
- 8. Dobbernack G, Meinl W, Schade N, et al. Altered tissue distribution of 2‐amino‐1‐methyl‐6‐phenylimidazo[4,5‐b]pyridine‐DNA adducts in mice transgenic for human sulfotransferases 1A1 and 1A2. Carcinogenesis 2011;32:1734–40. [DOI] [PubMed] [Google Scholar]
- 9. Snyderwine EG, Yu M, Schut HA, et al. Effect of CYP1A2 deficiency on heterocyclic amine DNA adduct levels in mice. Food Chem Toxicol 2002;40:1529–33. [DOI] [PubMed] [Google Scholar]
- 10. Arlt VM, Singh R, Stiborova M, et al. Effect of hepatic cytochrome P450 (P450) oxidoreductase deficiency on 2‐amino‐1‐methyl‐6‐phenylimidazo[4,5‐b]pyridine‐DNA adduct formation in P450 reductase conditional null mice. Drug Meta Dispos 2011;39:2169–73. [DOI] [PubMed] [Google Scholar]
- 11. Singh R, Arlt VM, Henderson CJ, et al. Detection and quantitation of N‐(deoxyguanosin‐8‐yl)‐2‐amino‐1‐methyl‐6‐phenylimidazo[4,5‐b]pyridine adducts in DNA using online column‐switching liquid chromatography tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci 2010;878:2155–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jacks T, Remington L, Williams BO, et al. Tumor spectrum analysis in p53‐mutant mice. Curr Biol 1994;4:1–7. [DOI] [PubMed] [Google Scholar]
- 13. Mizerovska J, Dracinska H, Frei E, et al. Induction of biotransformation enzymes by the carcinogenic air‐pollutant 3‐nitrobenzanthrone in liver, kidney and lung, after intra‐tracheal instillation in rats. Mutat Res 2011;720:34–41. [DOI] [PubMed] [Google Scholar]
- 14. Martin FL, Patel II, Sozeri O, et al. Constitutive expression of bioactivating enzymes in normal human prostate suggests a capability to activate pro‐carcinogens to DNA‐damaging metabolites. Prostate 2010;70:1586–99. [DOI] [PubMed] [Google Scholar]
- 15. Kucab JE, Phillips DH, Arlt VM. Metabolic activation of diesel exhaust carcinogens in primary and immortalized human TP53 knock‐in (Hupki) mouse embryo fibroblasts. Environ Mol Mutagen 2012;53:207–17. [DOI] [PubMed] [Google Scholar]
- 16. Frame LT, Ozawa S, Nowell SA, et al. A simple colorimetric assay for phenotyping the major human thermostable phenol sulfotransferase (SULT1A1) using platelet cytosols. Drug Meta Dispos 2000;28:1063–8. [PubMed] [Google Scholar]
- 17. Alnouti Y, Klaassen CD. Tissue distribution and ontogeny of sulfotransferase enzymes in mice. Toxicol Sci 2006;93:242–55. [DOI] [PubMed] [Google Scholar]
- 18. Arlt VM, Stiborova M, Henderson CJ, et al. Metabolic activation of benzo[a]pyrene in vitro by hepatic cytochrome P450 contrasts with detoxification in vivo: experiments with hepatic cytochrome P450 reductase null mice. Carcinogenesis 2008;29:656–65. [DOI] [PubMed] [Google Scholar]
- 19. Carmichael PL, Mills JJ, Campbell M, et al. Mechanisms of hormonal carcinogenesis in the p53+/− hemizygous knockout mouse: studies with diethylstilbestrol. Toxicol Pathol 2001;29 Suppl:155–60. [DOI] [PubMed] [Google Scholar]
- 20. Stiborova M, Martinek V, Svobodova M, et al. Mechanisms of the different DNA adduct forming potentials of the urban air pollutants 2‐nitrobenzanthrone and carcinogenic 3‐nitrobenzanthrone. Chem Res Toxicol 2010;23:1192–201. [DOI] [PubMed] [Google Scholar]
- 21. Hockley SL, Arlt VM, Jahnke G, et al. Identification through microarray gene expression analysis of cellular responses to benzo(a)pyrene and its diol‐epoxide that are dependent or independent of p53. Carcinogenesis 2008;29:202–10. [DOI] [PubMed] [Google Scholar]
- 22. Goldstein I, Rivlin N, Shoshana OY, et al. Chemotherapeutic agents induce the expression and activity of their clearing enzyme CYP3A4 by activating p53. Carcinogenesis 2013;34:190–8. [DOI] [PubMed] [Google Scholar]
- 23. Hu DG, Rogers A, Mackenzie PI. Epirubicin upregulates UDP glucuronosyltransferase 2B7 expression in liver cancer cells via the p53 pathway. Mol Pharmacol 2014;85:887–97. [DOI] [PubMed] [Google Scholar]
- 24. Sachse B, Meinl W, Glatt H, et al. The effect of knockout of sulfotransferases 1a1 and 1d1 and of transgenic human sulfotransferases 1A1/1A2 on the formation of DNA adducts from furfuryl alcohol in mouse models. Carcinogenesis 2014;35:2339–45. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information