

Abstract
Mesenchymal stem cells (MSCs) undergo a decline in function following ex vivo expansion and exposure to irradiation. This has been associated with accumulation of DNA damage and has important implications for tissue engineering approaches or in patients receiving radiotherapy. Therefore, interventions, which limit accumulation of DNA damage in MSC, are of clinical significance. We were intrigued by findings showing that zoledronate (ZOL), an anti‐resorptive nitrogen containing bisphosphonate, significantly extended survival in patients affected by osteoporosis. The effect was too large to be simply due to the prevention of fractures. Moreover, in combination with statins, it extended the lifespan in a mouse model of Hutchinson Gilford Progeria Syndrome. Therefore, we asked whether ZOL was able to extend the lifespan of human MSC and whether this was due to reduced accumulation of DNA damage, one of the important mechanisms of aging. Here, we show that this was the case both following expansion and irradiation, preserving their ability to proliferate and differentiate in vitro. In addition, administration of ZOL before irradiation protected the survival of mesenchymal progenitors in mice. Through mechanistic studies, we were able to show that inhibition of mTOR signaling, a pathway involved in longevity and cancer, was responsible for these effects. Our data open up new opportunities to protect MSC from the side effects of radiotherapy in cancer patients and during ex vivo expansion for regenerative medicine approaches. Given that ZOL is already in clinical use with a good safety profile, these opportunities can be readily translated for patient benefit. Stem Cells 2016;34:756–767
Keywords: Stem cells, DNA damage, Aging, Radiation, mTOR, Bisphosphonates
Significance Statement.
We have shown that zoledronate, a drug already used in patients with osteoporosis and cancer metastasis to bone, is able to attenuate accumulation of DNA damage in mesenchymal stem cells following exposure to radiation and following extensive culturing in vitro. By doing so it maintain their function, i.e. their numbers and ability to differentiate. Given the good safety profile of zoledronate, new potential applications for the protection of mesenchymal stem cells from the side effects of radiotherapy in cancer patients and during ex vivo expansion required for tissue engineering applications may be readily exploited for patient benefit.
Introduction
Ex vivo expansion or exposure to irradiation has been shown to be associated with reduced mesenchymal stem cells (MSC) function and accumulation of DNA damage 1, 2, 3, 4, 5. For example, MSC expanded in vitro or exposed to irradiation were reported to have decreased ability to initiate osteoblastic differentiation 1, 5, 6. Similarly, mice exposed to irradiation have been shown to undergo depletion of mesenchymal progenitors in their bone marrow, and increased their differentiation to adipogenesis 2, 6.
These effects can limit clinical efficacy. For example, bone tissue engineering strategies are being developed to overcome some of the limitations of autologous and allogeneic bone grafts for the repair of large bone defects 7. This usually involves the use of MSC which have been expanded in culture combined with other cell types, growth factors, and carriers (scaffolds or hydrogels). However, the decreased ability for osteoblast differentiation occurring with ex vivo expansion of MSC is one of the barriers to successful tissue regeneration 4, 7, 8. Ionizing radiation is used to treat pelvic cancers. Stress fractures or bone fragmentation are a significant problem arising from this type of therapy 9, 10. The risk of hip fracture for a woman undergoing pelvic irradiation for the treatment of cancers such as cervix, anus, or rectum is three times that of the population of women who do not receive irradiation and these fractures are difficult to heal 11. Total body irradiation (TBI) is used as conditioning regime in hematopoietic stem cell transplants and has been shown to increase adipogenesis in the bone marrow microenvironment 2. However, increased adipogenesis suppresses hematopoiesis, potentially affecting hematopoietic recovery following bone marrow transplant 12. Thus, interventions, which attenuate accumulation of DNA damage in MSC during ex vivo expansion or radiation therapy, are of clinical importance and an unmet need.
In this study, we identified zoledronate (ZOL) as a potential intervention to limit DNA damage accumulation. ZOL is one of the most potent of the nitrogen‐containing bisphosphonates (BPs), used clinically for the treatment of osteoporosis, Paget's disease and cancer induced bone loss 13. It acts by inhibiting the enzyme farnesyl diphosphate synthase (FPPS) in the mevalonate pathway, thereby preventing the prenylation of many GTPases, including RAS and RAS homolog enriched in brain (RHEB) 13, 14. More recently, bisphosphonates have been shown to have marked and unexpected effects on survival. ZOL in combination with statins produced a remarkable improvement of progeroid features and increased lifespan in a murine model of Hutchinson‐Gilford Progeria Syndrome (HGPS) 15. In a double blind placebo‐controlled clinical trial of patients with hip fractures, Lyles et al. found a 28% reduction in mortality in the patients receiving ZOL compared with controls 16. This effect was too large to be simply the result of prevention of secondary fractures 17.
This prompted us to ask the question whether ZOL was able to extend survival of MSCs during expansion and irradiation and whether this was related to attenuation of accumulation of DNA damage, one of the important mechanisms of cellular aging and radiation damage. In an effort to identify the mechanism of action we reasoned that inhibition of Mammalian Target of Rapamycin (mTOR) signaling was a potential target. It is implicated in lifespan extension in model organisms 18 and requires prenylated proteins such as RAS and RHEB, whose activity may be modulated by ZOL. In addition, downstream effectors of mTOR, such as Forkhead Box O3 (FOXO3A), have been recognized as important effectors for the recruitment of DNA damage response proteins, such as the protein kinase Ataxia Telangiectasia Mutated (ATM) 19. Therefore, we asked whether mTOR was involved in the enhanced DNA repair activity by ZOL. In this study, we provide evidence that ZOL‐treated MSCs extends their lifespan and retain their function following ex vivo expansion and exposure to irradiation by inhibition of the mTOR signaling.
Materials and Methods
Chemicals
ZOL (kindly provided by FHE), was dissolved in phosphate buffer saline (PBS) at 1.45 mg/ml to obtain a 5‐mM concentration and further diluted to the required concentrations in MSC medium, comprised of Dulbecco's Modified Eagle's Medium (DMEM; GIBCO, Paisley, U.K.) supplemented with 10% fetal bovine serum (FBS Hyclone, Thermo Scientific, Northumberland, U.K.) for in vitro studies. The enantiomeric pair of pyrindine bisphosphonates, PG‐1014491 (1R,6S)‐isomer of cis‐2‐azabicyclo[4.3.0]nonane‐8,8‐diphosphonic and PG‐1014493 (1R,6S)‐isomer of cis‐2‐azabicyclo[4.3.0]nonane‐8,8‐diphosphonic (kindly provided by Prof. Robert Boeckman, University of Rochester, Rochester, NY) 20 were also dissolved in PBS and further diluted to obtain the required concentration of 1 µM in MSC medium for in vitro studies. Trans, trans farnesol (FOH) (Sigma Aldrich, Dorset, U.K.) and geranylgeraniol (GGOH) (Sigma Aldrich, Dorset, U.K.) were dissolved in ethanol (EtOH) at 7.33 mg/ml and 9.58 mg/ml, respectively, to obtain a 33‐mM concentration and further diluted to a final concentration of 33 µM in MSC medium for in vitro studies. The mTOR inhibitors, Rapamycin (Enzo Life Sciences, Exeter, U.K.) and KU‐0063794 (Selleck, Suffolk, U.K.) were dissolved in dimethyl sulfoxide (DMSO) and used at a final concentration of 10 nM for in vitro while the PI3K/Akt inhibitor, LY294002 (Cell Signalling, Denver, Massachusetts) was dissolved in DMSO and further diluted to a final concentration of 10 µM.
Isolation and Culture of MSCs
Human MSCs (hMSC) were derived from bone marrow (BM) harvested from the pelvis of young patients (2–15 years old) undergoing osteotomy for reasons other than metabolic disorders at Sheffield Children's Hospital, U.K. Bone marrow was obtained following informed written parental consent in accordance with local research ethical committee approval and the declaration of Helsinki principles. The bone marrow was collected in MSC medium containing 0.01% of penicillin/streptomycin (Sigma, Dorset, U.K.), and 0.1% heparin. Bone marrow mononuclear cells (MNC) were isolated by density gradient centrifugation at 800g for 20 minutes using Lymphocyte separation medium (1.077g/l, PAA Laboratories, Somerset, U.K.). After two washes with PBS (Gibco, Paisley, U.K.), the cells were plated at 8,000 MNC per square centimeter in MSC medium and incubated at 37°C in 5% carbon dioxide in air. After 48 hours, the nonadherent cells were removed and the medium was changed weekly until cells were confluent. Cells were then harvested using 0.05% Trypsin‐1 mM EDTA (Gibco, Paisley, U.K.) and replated at 1,000 per square centimeter. Cultures were fed twice weekly and the number of population doublings (PD) was calculated as log N/Log2, where N is a ratio of number of cells at confluence and the number of cells at the start of culture. The assay for the number of colony forming units fibroblast (CFU‐F) generated from established cultures was obtained by plating hMSC at 10 cells per square centimeter in duplicate and incubating for 14 days at 37°C in 5% CO2 in air. Cultures were stained using Wright's Giemsa and purple stained colonies consisting of a minimum of 50 cells were counted as individual CFU‐fibroblast (CFU‐F). The assay for the number of CFU osteoblasts (CFU‐O) was obtained by plating 20 cells per square centimeter from established cultures in MSC medium supplemented with osteogenic supplements composed of 0.05 mM l‐Ascorbic Acid (Sigma Aldrich, St. Louis, Missouri), 10 mM β glycerophosphate (Sigma Aldrich, St. Louis, Missouri), and 100 nM dexamethasone (Sigma Aldrich, St. Louis, Missouri). Cells were maintained for 14 days at 37°C in 5% CO2 in air and fed twice weekly. At day 14, colonies were stained for alkaline phosphatase (ALP) enzymatic activity using 86R ALP kit (Sigma Aldrich St Louis, Missouri) according to the manual instructions. The colonies comprising of at least 40 cells with a definite centre of origin and stained for ALP were considered as one CFU‐O. CFU adipocyte (CFU‐A) were obtained by plating hMSC from established cultures at limiting dilutions ranging from 105 to 6.25 × 103 (eight wells per dilution) at final volume of 100 µl in a 96‐well plate as described previously 4, 21.
Differentiation of hMSC
hMSC seeded at 1.2 × 103 and 2.8 × 103 per cm2 were induced to undergo osteogenic and adipogenic differentiation, respectively, using osteogenic and adipogenic supplements as specified above. Cells were fed twice weekly either with osteogenic medium or with adipogenic medium and after 2 weeks, total RNA was extracted using RNAqueous 4PCR Kits (Ambion, Warrington, U.K.) according to manufacturer's instructions. Two micrograms of total RNA was used for reverse transcription using the first Strand cDNA Synthesis Kit (GE Healthcare, Buckinghamshire, U.K.). Quantitative real‐time polymerase chain reactions (RT‐PCR) were performed using SYBR green PCR Master Mix (Eurogentec, Romsey, U.K.) and 0.1 µM primers (Supporting Information Table S1). PCR amplification was carried out according to the following conditions: 50°C for 2 minutes, 95°C for 10 minutes (1 cycle); 95°C for 15 seconds, 60°C for 1 minute, (40 cycles). The data were analyzed using SDS 2.0 software.
DNA Damage Assessment by γH2AX Immunostaining and Comet Assay
DNA damage was induced by exposing hMSC to a 137Cs Gamma source. DNA double strand breaks were identified by γH2AX staining. Briefly, cells were fixed with 4% paraformaldehyde, permeabilized with 0.5% Triton‐X (Sigma, Dorset, U.K.), and blocked with 5% normal goat serum (DAKO, Glostrup, Denmark) in PBS, followed by incubation overnight at 4°C with primary antibody, anti‐phospho‐histone H2AX (Ser139) (Millipore, Billerica, Massachusetts) used at 1:800 in 5% normal goat serum. Cells were then incubated in anti‐mouse fluorescein isothiocyanate (FITC) conjugated secondary antibody (Insight Biotechnology, Santa Cruz, Middlesex, UK) at 1:200 in PBS for 1 hour at room temperature. Slides were mounted using VectaShield containing 4′,6‐diamidino‐2‐phenylindole (DAPI) to stain the nuclei blue. Cells were viewed using a 63 × objective in an Inverted Zeiss LSM 510 NLO microscope. Nuclei with DNA damage γH2AX foci were scored using ImageJ 1.45 software (http://rsbweb.nih.gov/ij/). Single‐cell gel electrophoresis alkaline comet assay was also used to assess cellular DNA damage. Briefly, cells were washed in cold PBS, suspended at 1:10 ratio in 0.5% low‐melting point agarose (37°C) on precoated slides, lysed, and subjected horizontal electrophoresis under alkaline conditions. After 30 minutes of low voltage electrophoresis, slides were stained with ethidium bromide and visualized under a fluorescent microscope. DNA damage was scored by classification of tail content in five categories (CC1‐CC5) based on increasing tail moment. Overall score was calculated as percentage of cells in each comet category for each treatment CC5 signifying maximum DNA damage.
Cell Cycle Analysis
hMSC cultures treated in presence or absence of ZOL for 3 days were irradiated (3Gy) and fixed for cell cycle analysis 4 hours post irradiation. Briefly 106 cells were fixed in prechilled 100% EtOH added dropwise and incubated on ice for 15 minutes. At the time of analysis, the cells were centrifuged, washed in PBS and stained with a freshly made solution containing 0.1 mg/ml propidium iodide (Sigma, Dorset, UK), 0.1% Triton x‐100 and 0.2 mg/ml ribonuclease A (Sigma, Dorset, UK) in PBS. All samples were incubated for 30 minutes at room temperature in the dark. Cell cycle distribution was determined by flow cytometry (LSR II BD Biosciences, Oxford, UK) for 10000 events at a flow rate not exceeding 500 cells per second and data were analyzed using FlowJo software.
Western Blotting
Cells were lysed with mammalian cell lysis buffer supplemented with 2% protease and 1% phosphatase inhibitor cocktails (Sigma, Dorset, U.K.) to obtain total lysates. For nuclear and cytoplasmic fractions, the homogenate (total lysate) was centrifuged at 1,500g for 5 minutes to sediment the nuclei. The supernatant was then resedimented at 15,000g for 5 minutes, and then the resulting supernatant formed the cytoplasmic fraction. The nucleic sediment was washed three times and resuspended in PBS containing 0.5% NP‐40 to extract nuclear proteins. The extracted material was sedimented at 15,000g for 10 minutes and the resulting supernatant was termed the nuclear fraction. Forty micrograms of total cell or fractionated lysate was separated using a 10% tris glycine gel and transferred to Immobilon‐P membranes (Millipore Corp., Billerica, MA). Membranes were blocked using 5% dry milk for all antibodies with the exception of p‐FOXO3A, FOXO3A, p‐ATM, and ATM, which were blocked in 5% bovine serum albumin (BSA; Sigma, Dorset, U.K.). Membranes were then incubated with primary antibodies anti‐mTOR, AKT, P70S6K, FOXO3A, and their phosphorylated forms p‐mTOR, p‐AKT, p‐P70S6K, p‐FOXO3A, p‐ATM (Cell Signaling Technology, Denver, Massachusetts), Ku70, Ku80 (Thermo Scientific, France), and RAD51(Santa Cruz Biotechnology, Heidelberg, Germany) at 1:1,000. GAPDH (Abcam, Cambridge, U.K.) was used at 1:30,000, LAMIN B1 (Invitrogen, Paisley, U.K.) and RAP1A (SantaCruz, Middlesex, U.K.) at 1:3,000 and βACTIN (Abcam, Cambridge, U.K.) at 1:10,000. Secondary HRP‐conjugated anti‐mouse and anti‐rabbit IgG were use at 1:30,000 and 1:1,000, respectively (DAKO, Glostrup, Denmark). Detection was performed using ECL reaction kits according to manufacturer's instruction (GE Healthcare, Buckinghamshire, U.K.) and quantification of the signal was performed using ImageJ 1.45 software (http://rsbweb.nih.gov/ij/).
Knockdown of FOXO3a Using Small Interfering RNA
hMSC were cultured in 24‐well plates (5 × 104 cells per well) up to 50% confluence and transfected with FOXO3a siRNA (5′GAGCUCUUGGUGGAUCAUCTT3′) or nonsilencing siRNA (5′‐CGUACGCGGAAUACUUCGA‐3′) at 100 nM (Cell Signaling, Denver, Massachusetts) using transfection reagent Lipofectamine 2000 (Invitrogen, Paisley, U.K.) according to manufacturer's instructions. Cells were either treated with ZOL (1 µM) or PBS 6 hours following addition of siRNA and 72 hours later killed to confirm knockdown of FOXO3a by either Western blotting as described above or immunofluorescence.
For analysis by confocal microscopy hMSC were fixed with 4% paraformaldehyde for 10 minutes at room temperature. Cells were permeabilized in 0.5% TritonX‐100, blocked with PBS containing 2% BSA, and incubated with antibody specific to FOXO3a (1:300) or ATM‐pS1981 (1:200) followed by Cy3 (red) conjugated anti‐rabbit (1:200) and Alexa 488 (green) conjugated anti‐mouse (1:200) secondary antibodies. Slides were mounted using Vecta Shield containing DAPI to stain the nuclei blue. Cells were viewed using a 63 × objective in an Inverted Zeiss LSM 510 NLO microscope. The images were analyzed using the NIH ImageJ 1.45 software (http://rsbweb.nih.gov/ij/) Colocalization of the two proteins was shown as the merged images.
In Vivo Studies Using C57Bl/6 Mice
Mice were injected with ZOL (125 µg/kg) or PBS and 3 days later irradiated at 3Gy or 12Gy and assayed for the presence of DNA damage foci or the number of clonogenic progenitors 24 hours later. To assay the number of murine CFU‐F and CFU‐O, freshly isolated murine bone marrow cells were plated in six‐well culture plates at density of 52 × 103 cells per square centimeter, in duplicates in 4 ml of murine MSC complete medium or osteogenic differentiation medium, respectively. Plates were incubated for 14 days at 37°C in 5% carbon dioxide in air with medium change every 3 days for 2 weeks. At day 14, the plates were stained as described earlier.
Data Analysis
Statistical analysis was performed on GraphPad Prism 6 (version 6.0.4). Data were expressed as mean ± SD and statistical comparisons were made by either one‐way analysis of variance (ANOVA) or two‐way ANOVA followed by post hoc analysis using Bonferroni post hoc test or Sidak post‐test for multiple comparisons. If comparisons were made between two groups, Student's t‐test was used. A p < 0.05 was considered statistically significant.
Results
ZOL Extends the Life Span of hMSC and Prevents Accumulation of DNA Damage with Ex Vivo Expansion
To test whether ZOL increased lifespan of human bone marrow‐derived MSCs (hMSC), these were expanded in the presence or absence of ZOL (1 µM) until they stopped proliferating for 3 weeks, after which they were considered senescent. While control cultures stopped proliferating after 27–31 PD, cultures grown in the presence of ZOL proliferated up to 37–42 PD (Fig. 1A, 1B). During expansion, they retained spindle shaped fibroblast morphology for longer times compared with untreated cultures (Fig. 1C) and showed reduced levels of senescent markers p16 and p21 at passage 12 (Fig. 1D; Supporting Information Fig. S1A). More importantly, hMSC cultures expanded in ZOL showed a higher content of clonogenic cells (CFU‐F, CFU‐O and CFU‐A) compared with untreated hMSCs when replated at low density at passage 8 (Fig. 1E, n = 3 p < 0.001) and showed an increased ability to differentiate towards the osteogenic and adipogenic lineages. Indeed a significant increase in expression levels of osteoblast markers, core‐binding factor subunit alpha‐1, ALP, osteopontin, and osteocalcin (Fig. 1F; Supporting Information Fig. S1B–S1C; n = 3), was seen in cultures expanded in the presence of ZOL and induced to differentiate to the osteogenic lineage after having washed off the drug. Similarly, cultures exposed to adipogenic supplements showed higher number of adipogenic vacuoles and a significant increase in adipogenic differentiation markers, peroxisome proliferator‐activated receptor γ (PPAR γ) and lipoprotein lipase, in ZOL expanded cultures compared with PBS (Fig. 1G; Supporting Information Fig. S1D–S1E).
Figure 1.
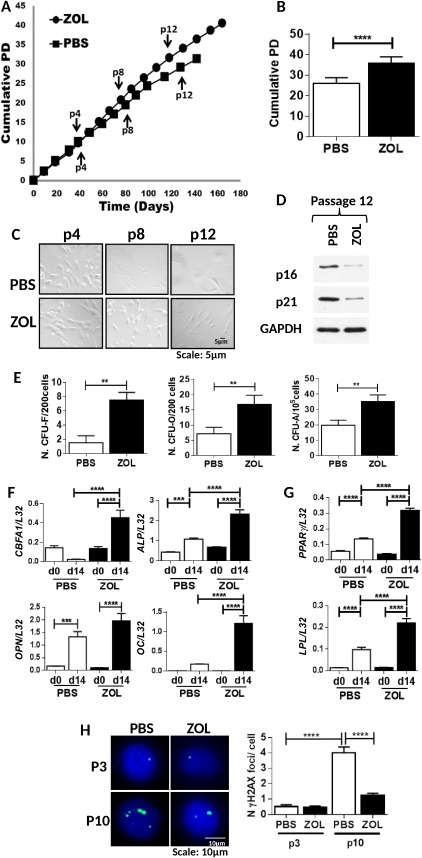
Zoledronate (ZOL) extends the life span of human mesenchymal stem cell (hMSC) and prevents accumulation of DNA damage with ex vivo expansion. (A): A representative example of cumulative population doublings (PD) of hMSC with time in culture grown in the presence (circle) or absence (square) of ZOL at 1 µM (n = 9). (B): Total number of PD; (C) Representative examples of hMSC morphology when cultured in presence or absence of ZOL at passage (p) 4, 8, and 12. (D): A representative example of expression of p16, p21, and glyceraldehyde 3‐phosphate dehydrogenase (GAPDH; bottom panel) by Western blot analysis in hMSC cultured in presence or absence of ZOL at 1 µM at passage 12. (E): Number of colony forming unit‐fibroblast (CFU‐F), CFU‐osteoblast (CFU‐O), and CFU‐adipocytes (CFU‐A) in hMSC cultures at passage 8 in the presence or absence of ZOL (n = 6). (F): hMSC exposed to osteogenic differentiation supplements for 14 days and assessed for expression of osteogenic differentiation markers core‐binding factor subunit alpha, alkaline phosphatase, osteopontin, and osteocalcin. (G) hMSC exposed to adipogenic differentiation supplements for 14 days and assessed for expression of adipogenic differentiation markers peroxisome proliferator‐activated receptor γ, lipoprotein lipase. All markers were normalized to ribosomal protein L32 (n = 3). (H): A representative example of γH2AX foci (green) and 4′, 6‐diamidino‐2‐phenylindole‐stained nuclei (blue) in phosphate buffer saline and ZOL‐treated hMSC at passage 3 and 10 (left panel) 63×; Number of DNA damage foci enumerated at passage 3 and 10 in hMSC cultured in the presence or absence of ZOL (n = 3) (right panel). All data are presented as mean ± SD and analyzed by t‐tests or for multiple comparisons by one way analysis of variance with Bonferroni multiple comparison post hoc test *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001. Abbreviations: CFU, colony forming unit; LPL, lipoprotein lipase; OC, osteocalcin; OPN, osteopontin; PBS, phosphate buffer saline; PD, population doublings; PPAR γ, peroxisome proliferator‐activated receptor γ; ZOL, zoledronate.
To determine whether this was associated with decreased accumulation of DNA damage, γH2AX DNA damage foci were enumerated at passage 3 (8–10 PD) and after 10 passages (24–29 PD) in hMSC cultured in the presence or absence of ZOL (Fig. 1H, n = 3). Both PBS‐ and ZOL‐treated cultures showed low levels of DNA damage foci at early passage, whereas at late passage a significant increase in the number of foci was observed in PBS‐treated cultures, but not in ZOL‐treated hMSC. These results suggest that ZOL extends the lifespan of hMSC following in vitro expansion and delays the loss of clonogenic and differentiation ability observed with cellular aging 4 and this is associated with reduced accumulation of DNA damage.
ZOL Enhances DNA Repair in hMSC Following Irradiation and Rescues Their Function
To determine whether ZOL was able to enhance the repair of DNA damage, hMSC were exposed to ZOL for 3 days and then exposed to 1, 3, 5, 7 Gy irradiation and assayed at 0, 4, 12, 24, 48 hours post‐irradiation for the presence of DNA damage foci. A significant increase in DNA damage foci was seen in all cultures immediately after irradiation at all doses of radiation with the exception of cells exposed to 7 Gy, which were not viable. No significant difference in the number of foci was observed between irradiated cultures exposed to ZOL compared with controls at this time point (Fig. 2A; Supporting Information Fig. S2A, S2B, n = 3). Starting at 4 hours post irradiation, a significant decrease in γH2AX foci was observed in cultures treated with ZOL compared with control cultures and the difference was more pronounced at the later time points and at the lower doses of irradiation (Fig. 2A; Supporting Information Fig. S2A, S2B). Similarly, ZOL‐treated cultures showed a significant percentage of cells in CC1 comet category with no comet tail moment compared with untreated hMSC that had very few cells in the same category. Moreover, cells with Comet tail moment in CC3–CC5 were significantly reduced in ZOL treatment compared with untreated hMSC after irradiation (IR) (Fig. 2B). This was accompanied by protection of the clonogenic ability of hMSC at 1 and 3 Gy (Fig. 2C; Supporting Information Fig. S2C, n = 3) and partial protection at 5 Gy (Supporting Information Fig. S2C, n = 3). Moreover, the lifespan of cells exposed to 3 Gy of irradiation in the presence of ZOL was consistently longer not only to those exposed to irradiation in absence of ZOL but also to those not exposed to irradiation. This is despite ZOL was removed immediately after IR and cells were expanded in MSC medium (Fig. 2D, n = 3). The same cultures showed enhanced differentiation ability to osteogenic and adipogenic lineages, which was otherwise impaired following irradiation, as demonstrated by little or no increase in their expression of differentiation markers (Fig. 2E, 2F, n = 3). Together these data suggest that ZOL enhances DNA repair in hMSC after irradiation and protects their ability to proliferate and differentiate.
Figure 2.
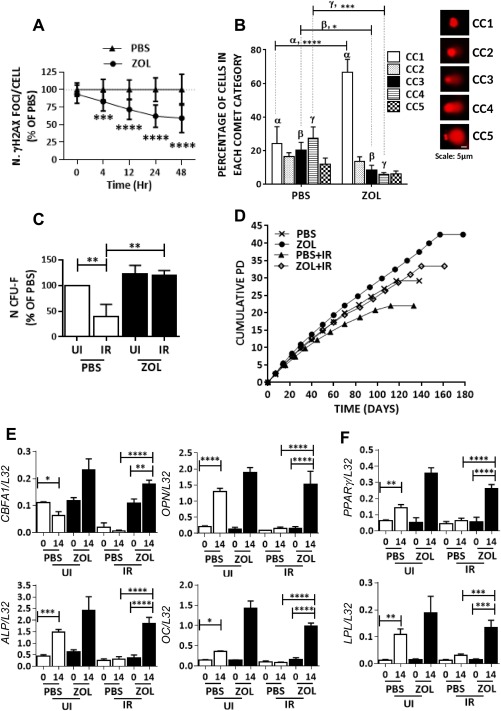
Zoledronate (ZOL) enhances DNA repair in human mesenchymal stem cell (hMSC) following irradiation and rescues their function. (A): Number of γH2AX foci enumerated in hMSC following irradiation (3Gy) in the presence or absence of ZOL (1 µM) and assessed at 0, 4, 12, 24, and 48 hours post irradiation (scored 25 cells from hMSC cultures from three independent donors). (B): Percentage of cells in different comet categories (CC) calculated based on increasing tail moments (CC1‐CC5) to determine cellular DNA damage (n = 3) 20×. (C): Number of colony forming unit fibroblast (CFU‐F) obtained from hMSC cultures exposed to irradiation (3 Gy) in the presence or absence of ZOL (n = 3). (D): Representative example of hMSC growth curve showing number of cumulative population doublings. Cultures were either left nonirradiated or irradiated at 3 Gy and grown in the presence or absence of ZOL for 3 days, and 12 hours later cultures were washed free from ZOL and expanded in hMSC medium (n = 3). (E, F): hMSC treated as described in (D): and at passage 9 exposed to osteogenic and adipogenic differentiation supplements, respectively. Cultures were assessed for the expression of osteogenic differentiation markers core‐binding factor subunit alpha, osteopontin, alkaline phosphatase, osteocalcin, and adipogenic differentiation markers lipoprotein lipase and peroxisome proliferator‐activated receptor γ. All markers were normalized to ribosomal protein L‐32 (n = 3). Data expressed as mean ± SD and analyzed by one way analysis of variance (ANOVA) and Bonferroni post hoc test for multiple comparisons, with the exception of data in panel A which was analyzed by two way ANOVA and Sidak's post‐test for multiple comparisons *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001. Abbreviations: ALP, alkaline phosphatase; CBFA, core‐binding factor subunit alpha; CFU, colony forming unit; IR, irradiation; LPL, lipoprotein lipase; OC, osteocalcin; OPN, osteopontin; PBS, phosphate buffer saline; PD, population doubling; PPAR γ, peroxisome proliferator‐activated receptor γ; UI, not irradiated; ZOL, zoledronate.
ZOL Repairs DNA Damage by Inhibiting the Mevalonate Pathway
To determine whether ZOL enhanced DNA repair through the inhibition of the mevalonate pathway (Fig. 3A), hMSC were treated with ZOL at increasing concentrations (100 nM–1 µM) for 3 days and expression of unprenylated Rap1A was assessed. A dose‐dependent increase in unprenylated RAP1A was observed (Fig. 3B; Supporting Information Fig. S3A) and this was mirrored by a dose‐dependent decrease in the number of γH2AX foci 4 hours after 1 Gy of irradiation (Fig. 3C; n = 3). More importantly, when the down‐stream metabolites, FOH and GGOH, were added to overcome the inhibition of FPPS, the DNA repair activity was abrogated (Fig. 3D, n = 3). Moreover, when we used two BP enantiomers with similar structure but different inhibitory activity on the FPPS enzyme, only the enantiomer with the higher potency, 1R,6S‐pyrindine bisphosphonate, PG‐1014491 (IC50: 15 nM) was able to enhance DNA repair (Fig. 3E, n = 3), while the less potent enantiomer, 1S,6R‐pyrindine bisphosphonate, PG‐1014493 (IC50: 359nM) 20 did not (Fig. 3F). These data suggest that the DNA repair response induced by ZOL depends on the inhibition of FPPS and the mevalonate pathway.
Figure 3.
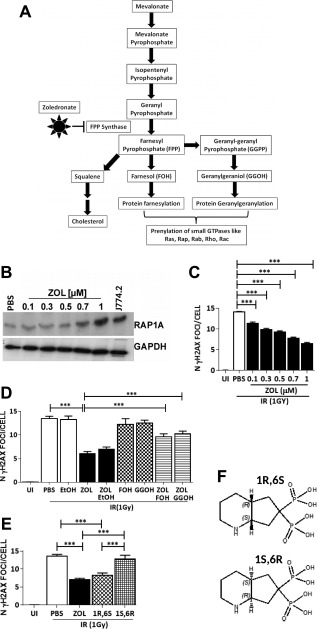
Zoledronate (ZOL) enhances DNA repair by inhibition of the mevalonate pathway. (A): A schematic representation of the mevalonate pathway. (B): A representative example of expression of unprenylated Rap1A (top panel) and glyceraldehyde 3‐phosphate dehydrogenase (GAPDH; bottom panel) in human mesenchymal stem cell (hMSC) following treatment with increasing concentrations of ZOL. (C): Number of γH2AX foci in response to different doses of ZOL following irradiation at 1Gy. (D): Number of γH2AX foci in hMSC not irradiated or exposed to 1 Gy of irradiation in the presence or absence of ZOL (1 µM) and with the addition of farnesol (FOH; 33 µM) or geranylgeraniol (GGOH; 33 µM) (n = 3). Ethanol was added as control with ZOL in the same amount used to dissolve GGOH and FOH. (E): Number of γH2AX foci in hMSC exposed to irradiation (1 Gy) in the presence of the octahydropyrindine‐BP enantiomeric pair (F) 1R,6S (PG‐1014491) is the stronger inhibitor of farnesyl diphosphate synthase with an IC‐50 = 15 nM, whereas the 1S,6R enantiomer (PG‐1014493) is a weaker inhibitor with an IC‐50 = 359 nM) (n = 3). Data are expressed as mean ± SD and were analyzed by one way analysis of variance and Bonferroni post hoc test for multiple comparisons *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001. Abbreviations: FOH, farnesol; FPPS, farnesyl diphosphate synthase; GGOH, geranylgeraniol; GGPP, gernyl‐geranyl phospahate; IR, irradiation; PBS, phosphate buffer saline; ZOL, zoledronate.
ZOL Mediates Its DNA Damage Response via the mTOR Signaling Pathway
To determine whether the action of ZOL might be mediated by inhibition of mTOR signaling (Fig. 4A) downstream of the mevalonate pathway, hMSC (n = 3) were cultured in the presence or absence of ZOL for 3 days and analyzed for protein expression of Ribosomal Protein S6 Kinase (P70S6K), AKT, mTOR and FOXO3A and their phosphorylated forms. As expected no changes in expression of mTOR, AKT, and P70S6K (Fig. 4B; Supporting Information Fig. S3C, n = 3) was observed in hMSC, whereas p‐mTOR (Ser2448), p‐AKT (Ser473), and p‐P70S6K (Thr421/Ser424) and p‐FOXO3A (Ser318/321) were significantly reduced on treatment with ZOL (Fig. 4B; Supporting Information Fig. S3B). Moreover, when the effects of inhibition of the FPPS enzyme was reversed by adding back the downstream metabolites, FOH and GGOH, no significant reduction in the expression of these phosphorylated proteins was observed (Fig. 4B; Supporting Information Fig. S3B), suggesting that ZOL abrogated both TORC1 and TORC2 mediated mTOR signaling and this was via inhibition of the mevalonate pathway.
Figure 4.
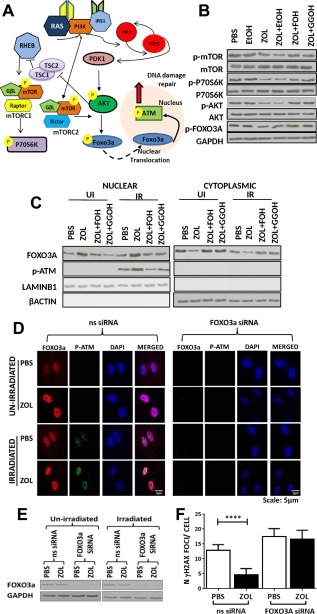
Zoledronate (ZOL) mediates an enhanced DNA damage response via inhibition of mTOR signaling. (A): A schematic representation of the mTOR pathway. (B): A representative example of expression of p‐mTOR, mTOR, p‐P70S6K, P70S6K, p‐AKT, AKT, and p‐FOXO3A by Western blot in human mesenchymal stem cell (hMSC) exposed to ZOL alone or in combination with farnesol (FOH) or geranylgeraniol (GGOH). Phosphate buffer saline was added in the same amount than ZOL, and ethanol in the same amount than GGOH and FOH. Protein expression was normalized to the expression of glyceraldehyde 3‐phosphate dehydrogenase (GAPDH). (C): A representative example of expression of nuclear and cytosolic FOXO3A and p‐ATM in nonirradiated hMSC and in hMSC 10 minutes after irradiation (IR) in the presence or absence of ZOL normalized to expression levels of LAMINB1 and β‐actin, respectively. (D): A representative example of hMSC transfected with a nonsilencing RNA (ns siRNA) or a siRNA specifically designed to knock down FOXO3a (FOXO3a siRNA) and analyzed by confocal microscopy for the expression of FOXO3a and phosphorylated ATM (p‐ATM); 63×. (E): A representative example of Western blot performed for the analysis of the expression of FOXO3a in hMSC tranfected with ns siRNA or FOXO3a siRNA and cultured in the presence or absence of ZOL and/or irradiation (1 Gy); (F) Number of γH2AX foci in hMSC transfected with ns siRNA or FOXO3a siRNA (n = 3). Data are expressed as mean ± SD and were analyzed by one way analysis of variance and Bonferroni post hoc test for multiple comparisons *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001. Abbreviations: DAPI, 4′,6‐diamidino‐2‐phenylindole; EtOH, ethanol; GGOH, geranylgeraniol; IR, irradiation; PBS, phosphate buffer saline; UI, not irradiated; ZOL, zoledronate.
To determine whether inhibition of mTORC2 signaling resulted in an enhanced DNA damage response, we determined expression levels of FOXO3A, p‐ATM (Ser1981) in hMSC, which were not irradiated or 10 minutes after IR (1 Gy). Nuclear levels of FOXO3A were significantly increased in ZOL‐treated hMSC regardless of irradiation (Fig. 4C; Supporting Information Fig. S3D, n = 3). In contrast, p‐ATM showed increased nuclear expression in ZOL‐treated cultures only following irradiation (Fig. 4C; Supporting Information Fig. S3E). On addition of FOH and GGOH, the levels of both FOXO3A and p‐ATM reverted to control levels confirming that inhibition of mTOR signaling is downstream of the mevalonate pathway (Fig. 4C; Supporting Information Fig. S3D, S3E). To determine whether translocation to the nuclei of FOXO3A was required to enhance DNA damage repair in response to ZOL treatment, expression of FOXO3A was knocked down and expression was verified both by confocal microscopy and by Western blotting (Fig. 4D, 4E, n = 3). As expected when FOXO3A was knocked down no expression of FOXO3A was detected by Western blot and by confocal microscopy. This also resulted in abrogation of nuclear expression of p‐ATM (Fig. 4D) and no decrease in the number of DNA damage foci in ZOL‐treated cultured 12 hours after irradiation (Fig. 4F, n = 3). This was in contrast to cultures treated with ZOL and transfected with a nonsilencing siRNA, where the expected significant decrease in the number of DNA damage foci was observed (Fig. 4F).
To investigate the type of DNA repair mechanism induced by ZOL, we quantified Ku70 and Ku80, which are components of the nonhomologous end joining (NHEJ) mechanism and RAD51 recombinase protein expression, which is increased in presence of homologous recombination activity. Expression of Ku80 was present but remained unchanged following irradiation or ZOL treatment (Supporting Information Fig. S4A, S4B). In addition, an increase in Ku70 was observed in both untreated and ZOL‐treated cultures following irradiation with no significant differences in expression between the two cultures (Supporting Information Fig. S4A, S4B). In contrast, a significant increase in RAD51 was observed after IR in ZOL‐treated MSC (Supporting Information Fig. S4A, S4B). No significant difference was found in the number of ZOL‐treated MSC in the S/G2 phase of the cell cycle (Supporting Information Fig. S4C), suggesting that any change in expression of RAD51 was not due to a change in cell cycle profile. These data suggest that ZOL increases DNA repair by HR.
In addition to determine whether this effect was seen with other mTOR inhibitors, we exposed hMSC to the inhibitor of Phosphoinositol‐3 kinase Ly294002 and to the inhibitor of mTOR KU0063794, both resulting in inhibition of both TORC1 and TORC2. Moreover, we also exposed the cells to rapamycin known to inhibit mainly TORC1 upon short term exposure 22. As expected, expression of the downstream targets of TORC1 and TORC2 p‐p70S6K, p‐AKT, and p‐FOXO3a was inhibited by Ly294002 and KU0063794 (Fig. 5A, n = 3). In contrast, only the expression of p‐p70S6K, but not p‐AKT and p‐FOXO3a, was decreased when cells were exposed to Rapamycin (Fig. 5A), in agreement with the notion that rapamycin inhibits only TORC1. Similarly, increased nuclear expression of FOXO3a and p‐ATM was seen only in cells exposed to Ly294002 and KU0063794 but not to rapamycin (Fig. 5B) and this resulted in a significant decrease in the number of γH2AX DNA damage foci (to similar levels to ZOL) only in cells treated with Ly294002 and KU0063794 but not with rapamycin (Fig. 5C). Taken together, these results suggest that abrogation of mTORC2 signaling by ZOL results in low levels of p‐AKT and increased nuclear translocation of FOXO3A, which in turn increases phosphorylation of ATM in the presence of DNA damage, resulting in an activated response to DNA damage.
Figure 5.
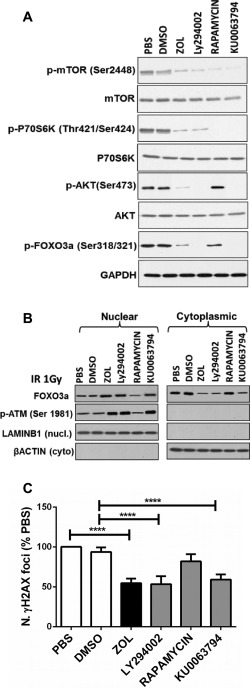
Inhibitors of both TORC1 and TORC2, but not rapamycin, promote enhanced DNA repair similar to ZOL in human mesenchymal stem cell (hMSC). (A): A representative example of expression of p‐mTOR, mTOR, p‐P70S6K, P70S6K, p‐AKT, AKT, and p‐FOXO3A by Western blot analysis in hMSC exposed to ZOL at 1 µM, inhibitor of phosphoinositol‐3 kinase Ly294002 (10 µM), inhibitor of mTOR KU0063794 (10 nM) and rapamycin (10 nM). Phosphate buffer saline was added in the same amount as used for ZOL, and dimethyl sulfoxide was added in the same amount as used for the other inhibitors as controls. Protein expression was normalized to the expression of glyceraldehyde 3‐phosphate dehydrogenase (GAPDH). (B): A representative example of expression of nuclear and cytosolic FOXO3A and p‐ATM in hMSC 10 minutes after irradiation (1 Gy) in the presence or absence of ZOL, Ly294002, KU0063794, and rapamycin normalized to expression levels of LaminB1 and β‐actin, respectively. (C): Number of γH2AX foci in response to ZOL, Ly294002, KU0063794 and rapamycin following irradiation at 1Gy. Data are expressed as mean ± SD and were analyzed by one way analysis of variance and Bonferroni post hoc test for multiple comparisons *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001. Abbreviations: DMSO, dimethyl sulfoxide; PBS, phosphate buffer saline; ZOL, zoledronate.
ZOL Protects MSCs from Radiation Induced Damage In Vivo
To determine whether ZOL was able to protect MSC from radiation‐induced damage in vivo, C57Bl/6 mice (n = 6) were exposed to ZOL for 3 days before total body irradiation and culled 24hours after IR. A significant increase in the number of γH2AX DNA damage foci was seen in cortical bone and bone marrow following IR, which was significantly reduced by pretreatment with ZOL (Fig. 6A, 6B). Irradiation reduced the number of surviving bone marrow CFU‐F and CFU‐O, used as measure of mesenchymal progenitors, but mice exposed to ZOL had a significantly higher number of progenitors compared with untreated controls (Fig. 6C).
Figure 6.
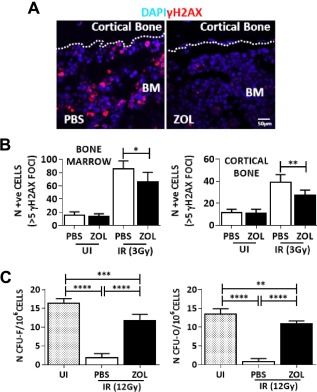
Zoledronate (ZOL) enhances protection of mesenchymal progenitors from radiation induced damage in vivo. (A): Representative example of bone marrow (BM) from a mouse 24 hours after exposure to irradiation (3Gy) and stained for γH2AX foci. Dotted lines indicate endosteal surface. (B): Number of cells (identified by 4′,6‐diamidino‐2‐phenylindole staining) with more than five γH2AX DNA damage foci (in red) in the bone marrow (left panel) or adjacent to the cortical bone (right panel). (C): The number of BM colony forming unit fibroblast (CFU‐F) and CFU‐osteoblast (CFU‐O) following the same treatment. Data are expressed as mean ± SD and were analyzed by one way analysis of variance and Bonferroni post hoc test for multiple comparisons *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001. Abbreviations: BM, bone marrow; CFU, colony forming unit; DAPI, 4′,6‐diamidino‐2‐phenylindole; IR, irradiation; PBS, phosphate buffer saline; UI, not irradiated; ZOL, zoledronate.
Discussion
In this study, we were able to demonstrate that ZOL attenuates accumulation of DNA damage in MSCs during ex vivo expansion or to irradiation and preserves their function. In previous studies ZOL has been shown to have cytotoxic effects and lead to loss of stemness when administered to MSC in vitro 23, 24. However, the concentrations used were 12 to 126 times higher than in our in vitro studies. Although BPs do not enter cells readily, we showed that our dose of 1 µM was sufficient to obtain the effects we have described in vitro and the dose equivalent to that already used in patients affected by cancer‐associated bone loss was appropriate to produce similar results in vivo 25. Our studies were performed in MSC derived from bone marrow of young donors. Further studies will be required to confirm that similar effects are observed in MSC obtained from the bone marrow of older donors.
The effects appeared to be mediated via inhibition of the mevalonate pathway as happens in other BP responsive cells such as osteoclasts and macrophages 26. The evidence that this is the case is strongly supported by the reversal achieved by adding back appropriate isoprenoid metabolites (e.g., FOH or GGOH) downstream of FPPS. Furthermore, the differential effects of the octahydropyrindine enantiomeric pair of N‐containing BPs were related to their known potencies as inhibitors of FPPS.
We were also able to identify TORC2‐FOXO3A‐ATM signaling as the molecular mechanism through which ZOL leads to enhanced DNA repair, downstream of the mevalonate pathway. Knockdown of FOXO3A, downstream of TORC2, abrogated the enhanced DNA damage response promoted by ZOL. Moreover, the same effect could be obtained using other mTOR inhibitors capable of inhibiting the TORC2 complex. Indeed short incubation with the TORC1 inhibitor rapamycin (3 days), induced little or no significant enhancement of DNA damage repair. This appears in contrast with the results of Gharibi et al. 27, who showed inhibition by rapamycin of the PI3K/Akt/mTOR pathway and lower levels of DNA damage in MSCs. The discrepancy may lie in the time of incubation. In the study by Gharibi et al. 27, cells were incubated in the presence of rapamycin for prolonged times (5 weeks), not the 3 days of treatment we used. Indeed, it is known that chronic administration of rapamycin leads to inhibition of TORC2 as well as TORC1 28.
It is unknown whether there is a direct link between the TORC2‐FOXO3A‐ATM signaling cascade and the reduced prenylation of progerin suggested as mechanism of action in the study by Varela et al. 15. The combination of ZOL and statins has been shown to prolong lifespan in a mouse model of HGPS and to prevent DNA damage 15. Accumulation of progerin, the abnormally splice variants of lamin A, which causes HGPS, has been shown to accumulate during cellular aging 29 and to be associated with accumulation of DNA damage 30. These evidence suggest that inhibition of prenylation of progerin may also be a player in the DNA repair activity of ZOL during normal aging. Therefore, the relationship between the TORC2‐FOXO3A‐ATM pathway and reduced prenylation of progerin in promoting the DNA repair activity warrants further investigation.
The involvement of the mTOR–FOXO3A–pATM signaling suggests that ZOL may attenuate DNA damage accumulation in other stem cell types as these molecules have been shown to be central to the survival and function of several stem cell types. Loss of the DNA damage sensor ATM depleted HSC 31, exacerbated the loss of melanocyte stem cells in response to low dose radiation 32, and promoted the loss of undifferentiated spermatogonia 33. Depletion of FOXO3A led to depletion of HSC and neural stem cells 34, 35, 36. Phosphorylation of ATM is central to initiate a number of DNA repair mechanisms including nonhomologous end joining or homologous recombination (HR) 37. Our data show that there is a NHEJ response in MSC regardless of ZOL. This is in agreement with the work of Oliver et al. 38, which shows that MSC have a robust NHEJ response to irradiation. In contrast, ZOL seems to increase the HR repair activity. Although more in depth work is required this is an important finding which has implication with regard to the level of fidelity obtained during the repair following ZOL.
This study has potentially important clinical implications in tissue engineering applications and in the treatment of cancer patients when radiation therapy is required. ZOL can be used during ex vivo expansion of MSC before transplantation to delay cellular aging and preserve their progenitor cell function. In our previous work, we have shown that treatment of MSCs with a molecule targeting prion protein prevented accumulation of DNA damage during ex vivo expansion and led to a 10‐fold increase in engraftment levels in bone marrow 4. Whether ZOL will result in a similar outcome will need to be confirmed.
Radiation therapy is one of the most common treatments for cancer patients and ZOL has already been used in cancer patients to reduce bone loss. However, it is usually administered long after radiation therapy when signs of severe bone loss appears. Here, we propose that administration of ZOL should be given before irradiation to prevent the loss of mesenchymal progenitor and their ability to differentiate to osteoblasts rather than exploit the anti‐resorptive properties when bone loss ensues.
One concern is that ZOL may enhance survival of cancer associated fibroblast in a similar way to MSCs and this may promote tumor growth. However, this is unlikely. ZOL has been given to cancer patients including patients affected by breast and prostate cancer with no evidence of reduced efficacy of any cancer treatment. This also points to a differential effect of its action in normal and cancer cells 39 and is in line with published work showing that reduced mTOR/AKT signaling mediates differential protection of normal cells from cancer cells 40. The signaling pathways mTOR has been shown to be hyper‐active in certain cancer types such as breast, prostate, and hematological malignancies due to loss of function mutations in tumor suppressor genes such as PTEN or gain of function mutations in genes such as RAS and PI3K. This renders cancer cells insensitive to such inhibitory signals. Considerable effort has been devoted to developing dual inhibitors of TORC1 and TORC2 in the hope of stopping the proliferation of cancer cells 41, 42. Here, we show that ZOL targets both components of the mTOR signaling pathway supporting further the view that ZOL may exert a differential action protecting normal cells and having cytostatic effects on cancer cells.
Conclusion
We have shown that ZOL protects normal MSCs from the accumulation of DNA damage following expansion and irradiation via mTOR signaling, a pathway which is central to longevity and cancer survival. Our data opens up exciting new potential applications for the protection of MSCs from the side effects of radiotherapy in cancer patients and during ex vivo expansion. Given the good safety profile of ZOL, these therapeutic opportunities may be readily exploited.
Author Contributions
J.M.: conception and design of the study, collection and assembly of data, data analysis and interpretation, manuscript writing final approval of the manuscript; S.T.M.: collection and assembly of data, data analysis, final approval of the manuscript; S.M., J.A.F., and F.H.E.: study design, provision of study material and final approval of the manuscript; R.G.G.R.: conception and study design, financial support, provision of material, final approval of the manuscript; I.B.: conception and study design, data analysis and interpretation, manuscript writing and final approval.
Disclosure of Potential Conflicts of Interest
R.G.G.R. received research support from Warner Chilcott and received income (legal work) from Novartis and Lilly. F.H.E. serves as a consultant for Terpenoid Therapeutics Inc. and BioVinc LLC. All other authors indicate no potential conflicts of interest.
Supporting information
Additional Supporting Information may be found in the online version of this article
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Acknowledgments
We thank Susan Smith, Healthcare Gateway, for helpful discussions. The enantiomeric pair of pyrindine bisphosphonates was kindly provided by Prof. Robert Boeckman, University of Rochester, Rochester, NY. This work was supported by the proof of concept fund a studentship from the University of Sheffield (to J.M.) and by the MRC and Arthritis Research UK as part of the MRC–Arthritis Research UK Centre for Integrated research into Musculoskeletal Ageing (CIMA).
References
- 1. Alves H, Munoz‐Najar U, De Wit J et al. A link between the accumulation of DNA damage and loss of multi‐potency of human mesenchymal stromal cells. J Cell Mol Med 2010;14:2729–2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cao X, Wu X, Frassica D et al. Irradiation induces bone injury by damaging bone marrow microenvironment for stem cells. Proc Natl Acad Sci USA 2011;108:1609–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cmielova J, Havelek R, Soukup T et al. Gamma radiation induces senescence in human adult mesenchymal stem cells from bone marrow and periodontal ligaments. Int J Radiat Biol 2012;88:393–404. [DOI] [PubMed] [Google Scholar]
- 4. Mohanty ST, Cairney CJ, Chantry AD et al. A small molecule modulator of prion protein increases human mesenchymal stem cell lifespan, ex vivo expansion, and engraftment to bone marrow in NOD/SCID mice. Stem Cells 2012;30:1134–1143. [DOI] [PubMed] [Google Scholar]
- 5. Baxter MA, Wynn RF, Jowitt SN et al. Study of telomere length reveals rapid aging of human marrow stromal cells following in vitro expansion. Stem Cells 2004;22:675–682. [DOI] [PubMed] [Google Scholar]
- 6. Zhang X, Xiang L, Ran Q et al. Crif1 promotes adipogenic differentiation of bone marrow mesenchymal stem cells after irradiation by modulating the PKA/CREB signaling pathway. Stem Cells 2015;33:1915–1926. [DOI] [PubMed] [Google Scholar]
- 7. Grayson WL, Bunnell BA, Martin E et al. Stromal cells and stem cells in clinical bone regeneration. Nat rev Endocrinol 2015;11:140–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Stolzing A, Jones E, McGonagle D et al. Age‐related changes in human bone marrow‐derived mesenchymal stem cells: Consequences for cell therapies. Mech Age Dev 2008;129:163–173. [DOI] [PubMed] [Google Scholar]
- 9. Costantino PD, Friedman CD, Steinberg MJ. Irradiated bone and its management. Otolaryngol Clin North Am 1995;28:1021–1038. [PubMed] [Google Scholar]
- 10. Jegoux F, Malard O, Goyenvalle E et al. Radiation effects on bone healing and reconstruction: Interpretation of the literature. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 2010;109:173–184. [DOI] [PubMed] [Google Scholar]
- 11. Baxter NN, Habermann EB, Tepper JE et al. Risk of pelvic fractures in older women following pelvic irradiation. JAMA 2005;294:2587–2593. [DOI] [PubMed] [Google Scholar]
- 12. Naveiras O, Nardi V, Wenzel PL et al. Bone‐marrow adipocytes as negative regulators of the haematopoietic microenvironment. Nature 2009;460:259–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Russell RGG. Bisphosphonates: The first 40 years. Bone 2011;49:2–19. [DOI] [PubMed] [Google Scholar]
- 14. Russell RGG, Watts NB, Ebetino FH et al. Mechanisms of action of bisphosphonates: Similarities and differences and their potential influence on clinical efficacy. Osteoporos Int 2008;19:733–759. [DOI] [PubMed] [Google Scholar]
- 15. Varela I, Pereira S, Ugalde AP et al. Combined treatment with statins and aminobisphosphonates extends longevity in a mouse model of human premature aging. Nature Med 2008;14:767–772. [DOI] [PubMed] [Google Scholar]
- 16. Lyles KW, Colon‐Emeric CS, Magaziner JS et al. Zoledronic acid in reducing clinical fracture and mortality after hip fracture. New Engl J Med 2007;357:nihpa40967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Colon‐Emeric CS, Mesenbrink P, Lyles KW et al. Potential mediators of the mortality reduction with zoledronic acid after hip fracture. J Bone Miner Res 2010;25:91–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Johnson SC, Rabinovitch PS, Kaeberlein M. mTOR is a key modulator of ageing and age‐related disease. Nature 2013;493:338–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tsai WB, Chung YM, Takahashi Y et al. Functional interaction between FOXO3a and ATM regulates DNA damage response. Nat Cell Biol 2008;10:460–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ebetino FH, Hogan AM, Sun S et al. The relationship between the chemistry and biological activity of the bisphosphonates. Bone 2011;49:20–33. [DOI] [PubMed] [Google Scholar]
- 21. Wu X, Peters JM, Gonzalez FJ et al. Frequency of stromal lineage colony forming units in bone marrow of peroxisome proliferator‐activated receptor‐alpha‐null mice. Bone 2000;26:21–26. [DOI] [PubMed] [Google Scholar]
- 22. Loewith R, Jacinto E, Wullschleger S et al. Two TOR complexes, only one of which is rapamycin sensitive, have distinct roles in cell growth control. Mol Cell 2002;10:457–468. [DOI] [PubMed] [Google Scholar]
- 23. Ebert R, Zeck S, Krug R et al. Pulse treatment with zoledronic acid causes sustained commitment of bone marrow derived mesenchymal stem cells for osteogenic differentiation. Bone 2009;44:858–864. [DOI] [PubMed] [Google Scholar]
- 24. Otto S, Pautke C, Opelz C et al. Osteonecrosis of the jaw: Effect of bisphosphonate type, local concentration, and acidic milieu on the pathomechanism. J Oral Maxillofac Surg 2010;68:2837–2845. [DOI] [PubMed] [Google Scholar]
- 25. Coleman R, Cameron D, Dodwell D et al. Adjuvant zoledronic acid in patients with early breast cancer: Final efficacy analysis of the AZURE (BIG 01/04) randomised open‐label phase 3 trial. Lancet Oncol 2014;15:997–1006. [DOI] [PubMed] [Google Scholar]
- 26. Rogers MJ, Crockett JC, Coxon FP et al. Biochemical and molecular mechanisms of action of bisphosphonates. Bone 2011;49:34–41. [DOI] [PubMed] [Google Scholar]
- 27. Gharibi B, Farzadi S, Ghuman M et al. Inhibition of Akt/mTOR attenuates age‐related changes in mesenchymal stem cells. Stem Cells 2014;32:2256–2266. [DOI] [PubMed] [Google Scholar]
- 28. Sarbassov DD, Ali SM, Sengupta S et al. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell 2006;22:159–168. [DOI] [PubMed] [Google Scholar]
- 29. Scaffidi P, Misteli T. Lamin A‐dependent nuclear defects in human aging. Science 2006;312:1059–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Liu B, Wang J, Chan KM et al. Genomic instability in laminopathy‐based premature aging. Nat Med 2005;11:780–785. [DOI] [PubMed] [Google Scholar]
- 31. Ito K, Hirao A, Arai F et al. Regulation of oxidative stress by ATM is required for self‐renewal of haematopoietic stem cells. Nature 2004;431:997–1002. [DOI] [PubMed] [Google Scholar]
- 32. Inomata K, Aoto T, Binh NT et al. Genotoxic stress abrogates renewal of melanocyte stem cells by triggering their differentiation. Cell 2009;137:1088–1099. [DOI] [PubMed] [Google Scholar]
- 33. Takubo K, Ohmura M, Azuma M et al. Stem cell defects in ATM‐deficient undifferentiated spermatogonia through DNA damage‐induced cell‐cycle arrest. Cell Stem Cell 2008;2:170–182. [DOI] [PubMed] [Google Scholar]
- 34. Miyamoto K, Miyamoto T, Kato R et al. FoxO3a regulates hematopoietic homeostasis through a negative feedback pathway in conditions of stress or aging. Blood 2008;112:4485–4493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Paik J‐h, Ding Z, Narurkar R et al. FoxOs cooperatively regulate diverse pathways governing neural stem cell homeostasis. Cell Stem Cell 2009;5:540–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Renault VM, Rafalski VA, Morgan AA et al. FoxO3 regulates neural stem cell homeostasis. Cell Stem Cell 2009;5:527–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chowdhury D, Choi YE, Brault ME. Charity begins at home: Non‐coding RNA functions in DNA repair. Nat Rev Mol Cell Biol 2013;14:181–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Oliver L, Hue E, Séry Q et al. Differentiation‐related response to DNA breaks in human mesenchymal stem cells. Stem Cells 2013;31:800–807. [DOI] [PubMed] [Google Scholar]
- 39. Coleman R, Cook R, Hirsh V et al. Zoledronic acid use in cancer patients. Cancer 2011;117:11–23. [DOI] [PubMed] [Google Scholar]
- 40. Lee C, Safdie FM, Raffaghello L et al. Reduced levels of IGF‐I mediate differential protection of normal and cancer cells in response to fasting and improve chemotherapeutic index. Cancer Res 2010;70:1564–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Xue Q, Hopkins B, Perruzzi C et al. Palomid 529, a novel small‐molecule drug, is a TORC1/TORC2 inhibitor that reduces tumor growth, tumor angiogenesis, and vascular permeability. Cancer Res 2008;68:9551–9557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wallin JJ, Edgar KA, Guan J et al. GDC‐0980 is a novel class I PI3K/mTOR kinase inhibitor with robust activity in cancer models driven by the PI3K pathway. Mol Cancer Ther 2011;10:2426–2436. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found in the online version of this article
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information