

Abstract
BACKGROUND
Prostate cancer incidence and mortality rates are significantly increased in African–American men, but limited studies have been performed within Sub–Saharan African populations. As mitochondria control energy metabolism and apoptosis we speculate that somatic mutations within mitochondrial genomes are candidate drivers of aggressive prostate carcinogenesis.
METHODS
We used matched blood and prostate tissue samples from 87 South African men (77 with African ancestry) to perform deep sequencing of complete mitochondrial genomes. Clinical presentation was biased toward aggressive disease (Gleason score >7, 64%), and compared with men without prostate cancer either with or without benign prostatic hyperplasia.
RESULTS
We identified 144 somatic mtDNA single nucleotide variants (SNVs), of which 80 were observed in 39 men presenting with aggressive disease. Both the number and frequency of somatic mtDNA SNVs were associated with higher pathological stage.
CONCLUSIONS
Besides doubling the total number of somatic PCa‐associated mitochondrial genome mutations identified to date, we associate mutational load with aggressive prostate cancer status in men of African ancestry. Prostate 76:349–358, 2016. © 2015 The Authors. The Prostate published by Wiley Periodicals, Inc.
Keywords: African ancestry, mitochondria, outcomes, prostate cancer, variation
INTRODUCTION
Although prostate cancer (PCa) is the most commonly diagnosed male cancer and second highest contributor to cancer mortality in Western countries 1, 2, the prevalence of PCa throughout Africa is largely unknown. The significance of African ancestry in PCa risk and outcomes has been driven by studies comparing incidence and mortality rates, as well as reported younger age at diagnosis, in African–Americans compared with European or Asian ancestral Americans 3, 4. We recently reported more aggressive histopathologically defined PCa and elevated prostate specific antigen (PSA) levels in Black men from South Africa 5, and postulate that genetics may be driving this ethnic‐based disparity.
PCa is the most heritable of the adult cancers 6, with carcinogenesis often driven by a complex series of acquired genetic events 7. Although genome‐wide association studies (GWAS) have identified 76 variants associated with PCa risk in predominantly European ancestral populations 8, more than half of these variants failed validation within a Black South African cohort 9. Additionally, GWAS and PCa genome profiling studies have been biased towards interrogation of the nuclear genome, largely ignoring the maternally inherited mitochondrial genome. Evidence for the significance of maternal inheritance in driving PCa risk and outcomes has been shown in a number of pivotal studies 10, 11, 12.
Unlike nuclear DNA, multiple copies of the mitochondrial (mt)DNA are present within the 100–1,000 mitochondria per cell 13. Lacking introns, over 93% of mtDNA codes for one of 37 genes including 13 proteins, 22 tRNAs, and 2 rRNAs, that are critical for energy production and apoptosis. Together with its high mutation rate 14, it is not surprising that mtDNA mutations have been implicated in human aging, disease, and cancer 15, 16, 17. Heteroplasmy and mutational load of acquired mtDNA mutations, will further impact cancer progression 18 contributing to extreme clinical heterogeneity. An analysis of somatic mtDNA mutations in 1,675 cancers from 31 tissue types, including 80 examples of PCa, demonstrated that functionally deleterious mtDNA mutations were more likely to be heteroplasmic possibly as a result of negative selection 19.
The role of acquired functionally relevant mtDNA mutations driving carcinogenesis has been supported by the observation that cancer cells switch from aerobic glycolysis in mitochondria to lactic acid fermentation in the cytosol, known as the Warburg effect 20. In PCa, acquired mtDNA mutations have been shown to; (i) accumulate in PCa tissue 21, 22 at a rate around 55‐times greater than nuclear DNA mutations 23, (ii) are most frequent in PCa, after gastric and hepatocellular cancers, than any other human cancer 19, (iii) increase PCa tumorigenesis, including indications as early events 24, 25, 26, (iv) associate with biochemical indicators of aggressive PCa 27, and (v) enhance PCa bone metastasis 28.
The aim of this study was to determine the spectrum and associated clinical relevance of mitochondrial genome variation within a unique cohort of predominantly Black South African men presenting with PCa. Biased towards histopathologically aggressive disease, complete mitochondrial genomes were deep sequenced in a set of matched blood and prostate tissue derived DNA from 87 patients. Among the most diverse, earliest diverged, mitochondrial genomes 29, 30, we identify a broad spectrum of somatic mutations associated with advanced PCa and elevated PSA levels at diagnosis.
MATERIALS AND METHODS
Study Design: Clinical and Pathological Data
A study sample of 87 men was recruited at the Steve Biko Academic Hospital in Pretoria, South Africa. Patients were consented as per the requirements of the University of Pretoria Faculty of Health Sciences Research Ethic Committee (#43/2010) and additional approvals obtained from the J. Craig Venter Institute Institutional Review Board (#2010–129) and Human Research Ethics Committee at the University of New South Wales (#HREC08244). Individuals self‐identified ethno‐linguistically as Black/Southern Bantu (n = 70), Coloured (n = 7), or European (n = 10). Further discussion of ancestral history and terminology can be found in supplementary material. All tissue core biopsies were blindly rescored by a single histopathologist and confirmed as presenting with high‐risk PCa defined as a Gleason score (GS) > 7 (n = 39), intermediate‐risk PCa with a GS = 7 (n = 12), low‐risk PCa with a GS < 7 (n = 10), or no PCa (n = 26) either with or without benign prostate hyperplasia (BPH). Matched blood and fresh tissue core biopsies were obtained, DNA isolated (Qiagen) and clinical information relating to age, serum PSA levels (ng/ml) and GS at diagnosis, were made available (Table I).
Table I.
Clinical and Pathological Characteristics
| Total, n = 87 | Southern Bantu, n = 70 | Coloured, n = 7 | White, n = 10 | |||||
|---|---|---|---|---|---|---|---|---|
| Patient characteristic at presentation | median (range) | |||||||
| Age (years) | 70 | (50–99) | 70 | (50–99) | 67 | (54–76) | 73.5 | (63–80) |
| Serum PSA (ng/ml) | 26 | (0.34–3459) | 26.7 | (4–2194) | 59 | (5.5–3459) | 7.85 | (0.34–1210) |
| Mitochondrial haplogroup | n | (%) | ||||||
| L0 | 37 | (43%) | 32 | (46%) | 5 | (71%) | 0 | |
| L1 | 7 | (8%) | 7 | (10%) | 0 | 0 | ||
| L2 | 19 | (22%) | 17 | (24%) | 2 | (29%) | 0 | |
| L3 | 14 | (16%) | 14 | (20%) | 0 | 0 | ||
| R/L4 | 10 | (11%) | 0 | 0 | 10 | (100%) | ||
| Diagnosis and PCa grade group | n | (%) | ||||||
| PCa GG = 5 (GS > 8) | 20 | (23%) | 17 | (24%) | 2 | (29%) | 1 | (10%) |
| PCa GG = 4 (GS = 8) | 19 | (22%) | 16 | (23%) | 2 | (29%) | 1 | (10%) |
| PCa GG = 3 (GS = 4 + 3) | 7 | (8%) | 6 | (9%) | 0 | 1 | (10%) | |
| PCa GG = 2 (GS = 3 + 4) | 5 | (6%) | 4 | (6%) | 1 | (14%) | 0 | |
| PCa GG = 1 (GS < 7) | 10 | (11%) | 8 | (11%) | 1 | (14%) | 1 | (10%) |
| No PCa, possible BPH | 26 | (30%) | 19 | (27%) | 1 | (14%) | 6 | (60%) |
Ancestral Contributions: Autosomal Genotyping
The 87 patients underwent genotyping using the Illumina Infinium HumanCore Beadchip (>250 K markers), with variant inclusion dependent on a GenTrain score > = 0.5 (Illumina GenomeStudio 1.9.4). The SNP and Variation Suite version 8.3.1 (Golden Helix; www.goldenhelix.com) was used to facilitate merging of genotype data of study participants with ancestral populations with (i) a previously published dataset 31 that includes 19 Ju/'hoan representing the earliest‐diverged population and (ii) the Illumina iControl database (www.illumina.com) that includes the more recent‐diverged Yoruba (n = 20), Han Chinese (n = 20), and European (n = 20) populations. Markers were excluded that were monomorphic, indels, or had linkage disequilibrium (LD) with r2 > = 0.2 within a 50‐variant sliding window. A total of 103,670 SNP markers were used to determine ancestral substructure using STRUCTURE 2.3.3 32, with the most appropriate K = 4 determined by the ΔK method 33.
Data Generation: Mitochondrial Genome Sequencing
The complete mitochondrial genome was captured via long‐range PCR amplification using the Platinum® Taq DNA Polymerase High Fidelity kit (Invitrogen) and previously published primer sets creating two overlapping amplicons of ∼7.2 kilobases (kb) and ∼9.7 kb. Pre‐amplification allowed for mtDNA specificity, eliminating mtDNA‐derived pseudogenes in the nuclear genome (NuMTs). Amplified products were quantified using the Quant‐iT™ PicoGreen® dsDNA Assay Kit (Invitrogen) prior to library preparation using the Ion Xpress™ Plus Fragment Library Kit. The 200 bp libraries were barcoded (Ion Xpress™ Barcode Adapters 1–16 Kit), quantified and sized (Agilent High Sensitivity DNA Kit), template prepared (Ion OneTouch™ 2 System), and sequenced on the Ion Personal Genome Machine (PGM™) System using the Ion 318™ Chip. Each chip was run with eight matched blood and prostate tissue samples generating at least 600 Mb of data with an average coverage depth of almost 3,000 × per sample.
Data Inferences: Statistical Analyses
All Ion Torrent sequence data were quality trimmed (Phred‐scale Q20, see Supplementary Table SII). Reads were aligned to the rCRS mtDNA reference genome using BWA MEM alignment 34. Variant analysis of somatic mutations was done using Varscan 2.0 35 from read pileups generated by Samtools 36 based on the BWA alignments. The Varscan somatic program was run with strand bias filtering, and tumor purity allowed as low as 10%. SNVs from matched blood and prostate tissue samples were further filtered for a difference of tumor above blood frequency exceeding 2.0%.
Statistical analyses were performed using R (www.R-project.org). Linear regression (lm) and Generalized Linear Models (glm) were used for linear or categorical regression of PCa status. PCa categories were defined by the new grading group (GG) system 37 as follows: 1. GS < = 6, 2. GS = 3 + 4, 3. GS = 4 + 3, 4. GS = 8, 5. GS > = 9. The Welsh t‐test (t‐test) was used to compare means between two classes of data. The binomial exact test (binom.test) was used to compare observed to expected somatic mutation coverage of mtDNA genes and PCa categories. One‐way ANOVA (anova) was used to evaluate the significance of multi‐term linear models for association with GS.
Large Deletions: Targeted Sanger Sequencing
A large 3,379 bp mitochondrial genome deletion (junction 10744:14124) has previously been reported as a potential PCa biomarker 38. As detection of large deletions is problematic using short read next generation sequencing technology, together with the large long‐range amplicons described, we designed a targeted approach including amplification of a 3.8 kb region using primers 10534F: 5′‐AGGAGTATCCTGAGGCATGG‐3′ and 14447R: 5′‐ATCGCTCACACCTCATATCC‐3′. Deletion bands identified during agarose gel electrophoresis were excised from the gel and purified, followed by Sanger sequencing using both forward and reverse primers.
Sequence Data Validation: Illumina MiSeq
Data validation using an alternative sequencing platform was performed for a random subset of 12 tissue and matched blood samples from 12/87 men. Library preparation using the Illumina Nextera XT DNA Library Preparation Kit involved simultaneous fragmentation and adapter tagging, followed by the addition of sequencing indexes to the amplified tagged DNA. A total of 24 barcoded samples were pooled for a single 2 × 250 bp paired‐end sequencing run on the Illumina MiSeq system. Quality trimming was done at Q33 (Phred scale) resulting in 18 M reads and 4 Gb of sequence. The same procedure was used to quantify somatic SNVs from MiSeq data as were used for Ion Torrent data to validate the results across the two sequencing platforms. Although a number of somatic indels were found in the Ion Torrent data, all indels were invalidated by MiSeq data, yet all SNVs were validated. Only SNVs were used in subsequent mtDNA somatic mutation analyses.
Previous Disease Associations: Database Interrogation
All 113 somatic SNVs identified in this study that were associated with PCa were researched for previous association to diseases. The list of somatic mutations was downloaded from Mitomap (www.mitomap.org/mitomap) on May 15, 2015 and merged with results from searches of each mutation on Pubmed (www.ncbi.nlm.nih.gov/pubmed). See Table SI for the full list of SNVs associated with PCa in the 77 ancestral African men from this study. There were 17/113 SNVs found to be previously associated with a disease in Mitomap, and an additional three found in Pubmed searches. A single SNV, (m.G3357A) of the 113 presented here was previously implicated in PCa 24.
RESULTS
Ancestral Contributions of Study Participants
In order to clarify the extent of ancestral African versus non‐African contribution to the study participants, STRUCTURE (K = 4) analysis was performed for over 100,000 randomly distributed autosomal markers. In contrast to African–Americans with on average ∼20% reported European ancestral contribution 39, Black South Africans have negligible non‐African ancestral contributions (Fig. 1). The Coloured participants, however, show varying contributions of both African and non‐African ancestral fractions, concurring with previous population‐based studies 31, 40. In concordance with previous reports on South African populations 5, 31, the African ancestral contribution to Black and Coloured South Africans can be further classified as African–Bantu and African–Khoesan. We confirm a lack of African ancestral contribution to the 10 European South Africans.
Figure 1.
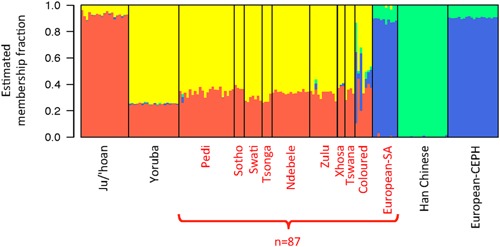
Cluster ancestral fractions for PCa participants in this study (red label) and reference populations (black label) for comparison. Ancestral fractions estimated by STRUCTURE with K = 4 clusters. The Orange cluster corresponds to African–Khoesan ancestry, Yellow to African–Bantu, Blue to European, and Green to Asian.
Association of mtDNA Haplogroup With Clinical Presentation
Significant maternal African–Khoesan contribution to the Southern Bantu and Coloured of South Africa is reflected in the notable representation of ancient modern human maternal lineages, specifically the L0 mitochondrial haplogroups (See Supplementary Fig. S1). Among the 87 patients, 37 (32 Black and 5 Coloured) carried L0 (Table I), including almost 50% with the earliest diverging L0d haplogroup recently shown to have emerged ∼172 thousand years ago (kya) (95%CI:149–199 kya) 30. Interestingly, these individuals presented with more aggressive PCa, or higher average GS (mean = 6.3), than those carrying recently diverging (non‐L0) haplogroups (mean = 4.9), P = 0.049, (Fig. 2A). We also observed a general trend towards elevated serum PSA levels at the time of clinical presentation (mean = 190 ng/ml) in individuals with earlier diverged maternal lineages (mean = 116 ng/ml), although these differences were not significant at the P = 0.05 Level, (Fig. 2B). Although it is important to note the potential susceptibility of earlier diverging haplogroups to PCa and extreme PSA levels, we must take caution as the effects of population structure and culture on the likelihood of an individual seeking clinical diagnosis are not always possible to mitigate.
Figure 2.
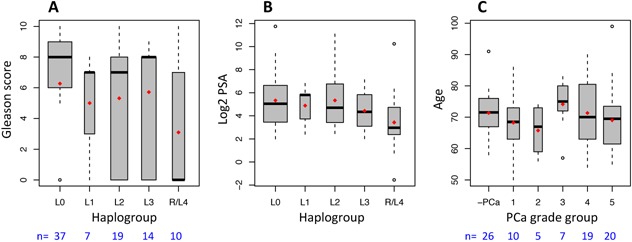
Correlations between (A) ancestral mtDNA‐haplogroups and clinical presentation by Gleason score, (B) haplogroups and serum PSA levels, and (C) PCa grade group categories of risk associated with age at presentation, including no PCa (‐PCa). The earliest lineage (L0) presented with significantly (P < 0.05) higher PCa risk than later diverging lineages. There was no significant association between age and PCa grade group. Red dots on box‐plots indicate mean values.
We find no significant association between PCa status and age at presentation (Fig. 2C). With a mean age of 70.4 years the ages of subjects are more evenly distributed among PCa risk categories for this study than may be present in general in the South African population. Age ranges also were not significantly different between ancestral haplogroups (Supplementary Fig. S2), minimizing confounding effects due to age.
Spectrum of mtDNA Somatic Mutations and Associated Clinical Presentation
Sequencing of matched blood and prostate tissue mtDNA samples were used to identify 144 somatic mutations. With a focus on African ancestry, we assessed the distribution of 134 somatic mutations identified in 53 of 77 patients with African ancestry (70 Southern Bantu, 7 Coloured), in 128 different positions across the mitochondrial genome. There were 34 in NADH dehydrogenase subunits, 33 in rRNAs, 27 in Cytochrome c oxidases, 26 in the control region, 7 in Cytochrome b, 5 in ATP synthase, and 2 in tRNAs. Although somatic mutations were distributed across the entire mitochondrial genome (Fig. 3, we observe significantly more than expected in the control region under a purely stochastic process (binomial P = 1.2e‐6). The rRNA regions were also enriched for somatic mutations, though to a lesser extent (P = 5.2e‐3), while the NADH dehydrogenases and tRNAs had fewer somatic mutations than expected (P = 1.8e‐3 and P = 7.0e‐4, respectively).
Figure 3.
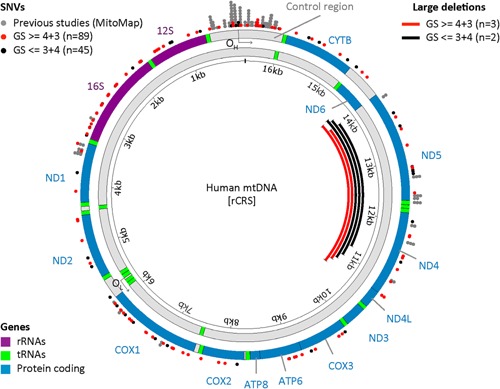
Distribution of somatic SNVs in aggressive PCa patients (red, this study GS > = 4 + 3), and no‐PCa or indolent PCa (black, this study GS < = 3 + 4), overlaid with previously reported PCa associated SNVs from Mitomap (gray). Large deletions detected by targeted amplification are depicted as red (GS > = 4 + 3) and black (GS < = 3 + 4) lines. The human mitochondrial genome is depicted as the heavy strand (outer) and light strand (inner), with location of genes, and origins of replication for each strand. The control region and rRNA genes contain a significant enrichment of SNVs compared to other regions.
The low number of somatic mutations found in tRNAs, is in contrast to the findings of Kloss–Brandstatter, 2010 27, where tRNA somatic mutations were found to be abundant in patients with PCa. The only tRNA mutations found here were in a patient presenting with indolent PCa (GG = 1, GS < 7) and one with no PCa. Of the 61 African‐ancestral patients with PCa, a total of 113 different SNVs were observed, of which 95 did not appear to have any previous PCa association (see Supplementary Table SI).
Unlike, the previous association of a large 3,379 bp mtDNA deletion mutation (34 mtΔ) identified in a European population as a significant biomarker for PCa 38, this mutation was absent within our study. However, analysis of the entire mitochondrial genome found this region to be the most likely to acquire a deletion variant at sufficient detectable frequency. Four patients (4/87, 4.6%) presented with a large deletion mutation including a single individual presenting with two. Deletions varied in length (from 2,879 to 3,769 bp), involved a repeat sequence at the donor and recipient junction (2–10 bases) removing most of ND4 and ND5, and the three intervening tRNAs (Supplementary Table SIII). These four patients represented all three population identifiers and carried one or multiple somatic SNVs, suggesting patient specific genomic instability.
We find only five mutations shared between multiple individuals (Fig. 3). Four of the five are in the control region, with two G‐> A transitions m.G103A and m.G16390A, a A‐> G transition m.A189G, shared between two individuals, and a C‐> T transition m.C16380T shared by three individuals. The fifth shared mutation is a synonymous T‐> C transition found in the ND4L subunit, m.T10756C, and both patients have aggressive high‐risk PCa (GG > 3, GS > 7). Most of these shared mutations involve at least one patient with aggressive PCa, with the exception of m.G103A, which is found in two men both with no PCa.
There is also a trend among these shared mutations that the individual with an appreciably larger variant frequency of the mutant allele also has the higher‐risk aggressive PCa. We examine further this association between PCa progression and the number of somatic mutations or the cumulative total of variant frequency in the population of heteroplasmic mitochondria.
Associating the extent of mutational load in mtDNA and African ancestral PCa clinical presentation, we make the following observations. There are 80/134 somatic mutations among the highest risk PCa patients (GS > 7), which is higher than would be expected at random (P = 7.3e‐3), and only 17/134 among individuals with no PCa, which is far fewer than we would expect given the number of subjects with no PCa (P = 2.4e‐4). As with serum PSA levels (Fig. 4A), we observe an accumulation of both the number of somatic mutations (Fig. 4B) and the frequency of these mutations (Fig. 4C) within the heteroplasmic mtDNA population that are associated with increased PCa risk.
Figure 4.
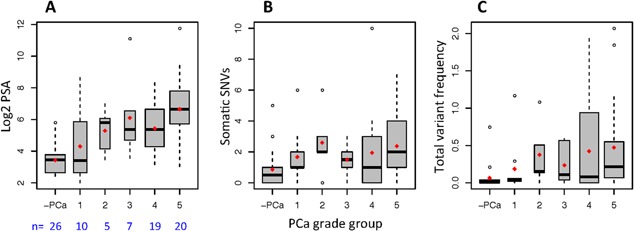
PCa grade groups, including no PCa (‐PCa), associated with (A) serum PSA levels, and mtDNA mutational load measured by (B) number of somatic SNVs, and (C) total variant frequency of somatic SNVs in South African men of African ancestry. Red dots on box‐plots indicate mean values.
Factors correlated with increasing GS and PCa risk include serum PSA levels, number of somatic variants (SNV), and the cumulative variant frequency of somatic mutations (VF). We use the log2 transform of PSA for correlations due to the extensive range of PSA values measured among the patients. We find the strongest association to increasing GS with log‐PSA (linear regression, P = 8.8e‐7), and categorically between PCa (GG > = 3 or GS > = 4 + 3, generalized linear model P = 9.6e‐5). Significant associations with GS were also apparent from linear regression with VF and SNVs, (P = 2.5e‐3 and P = 1.3e‐2, respectively). All three factors are positively correlated with each other and increasing GS, and provide predictive power for determining PCa aggression (Fig. 5). Multi‐term linear models were compared using ANOVA but no significant increase was found above the single term log‐PSA model. The strong interdependence of these factors limits the effectiveness of combining multiple predictors.
Figure 5.
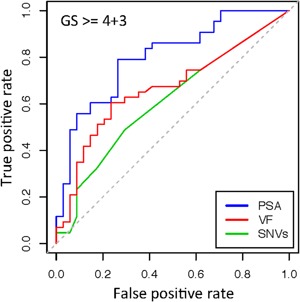
Receiver Operator Characteristic (ROC) curve for three factors associated with PCa aggression. Serum PSA levels, number of SNVs, and cumulative variant frequency (VF) calculated as the sum of variant frequency differences between tumor and blood samples that are above 2.0%. VF thus accounts for both the breadth and depth of mutation load within each population of heteroplasmic mtDNA.
DISCUSSION
In this study, we more than double the number of somatic PCa‐associated mitochondrial genome mutations identified to date by focusing on men with African ancestry biased towards more aggressive disease. Data presented here suggest elevated risk associated with somatic mtDNA mutations in the control region and rRNA subunits, and no correlation between synonymous and non‐synonymous exonic mutations. Other studies have found more risk‐associated mutations in tRNAs or with non‐synonymous mutations in specific genes. While it is possible our findings are unique to an ancestral African cohort, the limited concordance of previous studies continues to suggest that specific mutations are not as important as a non‐specific mutational load to the presentation and aggression of PCa in general.
Although the increase in mtDNA mutations is a factor associated with PCa aggression, there must be a limitation on how many mutations a cell can withstand among mitochondrial populations before it is no longer viable. We expect the cancer phenotype to be more apparent with increasing mutations and frequency of mitochondria with these mutations, but with upper limits on both. Our findings are consistent with this hypothesis of limited mutation range. There is also the possibility that mitochondrial mutations are elevated simply due to a background of higher mutagenesis or reduction in repair, and that they are not causal but rather are indicators of such a cellular environment. In either case, mutational load in mtDNA is an important indicator of PCa risk.
CONCLUSIONS
Among men with African ancestry presenting with extreme serum PSA levels and a bias towards histopathologically defined aggressive PCa, we find the breadth and depth of mutational load, measured as the number, and frequency of mtDNA somatic mutations respectively, to be associated with PCa aggression. We confirm that PSA screening is the most important indicator for determining PCa risk in ancestral African populations. These results provide evidence that PCa progression is more likely characterized by the accumulation of mitochondrial mutations across the entire genome and in greater frequency within heteroplasmic cellular populations than it is by specific mutations. However, it remains important to continue cataloging common PCa‐associated mutations in different populations as the effects of specific mutations, especially within varied genetic backgrounds, cannot be assumed to be consistent. The mutations provided here will further help with the quantification of risk across the mitochondrial genome, while with increasing precision and decreasing cost of next‐generation sequencing we hope more studies will follow that focus on PCa within understudied populations.
Supporting information
Additional supporting information may be found in the online version of this article at the publisher's web‐site.
Supplementary Table SI.
Supporting Information.
ACKNOWLEDGMENTS
The authors would like to acknowledge the following for their help and funding of this study: NIH R21‐CA170081, Australian Prostate Cancer Research Centre NSW, the J. Craig Venter Institute, the Garvan Institute, the Petre Foundation, Australia, the Cancer Association of South Africa (CANSA), and all the study participants.
Conflicts of interest: The authors have no conflict of interest related to this work.
REFERENCES
- 1. Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics, 2009. CA Cancer J Clin 2009; 59(4):225–249. [DOI] [PubMed] [Google Scholar]
- 2. La Vecchia C, Bosetti C, Lucchini F, Bertuccio P, Negri E, Boyle P, Levi F. Cancer mortality in Europe, 2000‐2004, and an overview of trends since 1975. Ann Oncol 2010; 21(6):1323–1360. [DOI] [PubMed] [Google Scholar]
- 3. Bigler SA, Pound CR, Zhou X. A retrospective study on pathologic features and racial disparities in prostate cancer. Prostate Cancer 2011; 2011:239460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hsing AW, Tsao L, Devesa SS. International trends and patterns of prostate cancer incidence and mortality. Int J Cancer 2000; 85(1):60–67. [DOI] [PubMed] [Google Scholar]
- 5. Tindall EA, Monare LR, Petersen DC, van Zyl S, Hardie RA, Segone AM, Venter PA, Bornman MS, Hayes VM. Clinical presentation of prostate cancer in black South Africans. Prostate 2014; 74(8):880–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hjelmborg JB, Scheike T, Holst K, Skytthe A, Penney KL, Graff RE, Pukkala E, Christensen K, Adami HO, Holm NV, Nuttall E, Hansen S, Hartman M, Czene K, Harris JR, Kaprio J, Mucci LA. The heritability of prostate cancer in the Nordic Twin Study of Cancer. Cancer Epidemiol Biomarkers Prev 2014; 23(11):2303–2310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Barbieri CE, Bangma CH, Bjartell A, Catto JW, Culig Z, Grönberg H, Luo J, Visakorpi T, Rubin MA. The mutational landscape of prostate cancer. Eur Urol 2013; 64(4):567–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Al Olama AA, Kote‐Jarai Z, Berndt SI, Conti DV, Schumacher F, Han Y, Benlloch S, Hazelett DJ, Wang Z, Saunders E, Leongamornlert D, Lindstrom S, Jugurnauth‐Little S, Dadaev T, Tymrakiewicz M, Stram DO, Rand K, Wan P, Stram A, Sheng X, Pooler LC, Park K, Xia L, Tyrer J, Kolonel LN, Le Marchand L, Hoover RN, Machiela MJ, Yeager M, Burdette L, Chung CC, Hutchinson A, Yu K, Goh C, Ahmed M, Govindasami K, Guy M, Tammela TL, Auvinen A, Wahlfors T, Schleutker J, Visakorpi T, Leinonen KA, Xu J, Aly M, Donovan J, Travis RC, Key TJ, Siddiq A, Canzian F, Khaw KT, Takahashi A, Kubo M, Pharoah P, Pashayan N, Weischer M, Nordestgaard BG, Nielsen SF, Klarskov P, Røder MA, Iversen P, Thibodeau SN, McDonnell SK, Schaid DJ, Stanford JL, Kolb S, Holt S, Knudsen B, Coll AH, Gapstur SM, Diver WR, Stevens VL, Maier C, Luedeke M, Herkommer K, Rinckleb AE, Strom SS, Pettaway C, Yeboah ED, Tettey Y, Biritwum RB, Adjei AA, Tay E, Truelove A, Niwa S, Chokkalingam AP, Cannon‐Albright L, Cybulski C, Wokołorczyk D, Kluźniak W, Park J, Sellers T, Lin HY, Isaacs WB, Partin AW, Brenner H, Dieffenbach AK, Stegmaier C, Chen C, Giovannucci EL, Ma J, Stampfer M, Penney KL, Mucci L, John EM, Ingles SA, Kittles RA, Murphy AB, Pandha H, Michael A, Kierzek AM, Blot W, Signorello LB, Zheng W, Albanes D, Virtamo J, Weinstein S, Nemesure B, Carpten J, Leske C, Wu SY, Hennis A, Kibel AS, Rybicki BA, Neslund‐Dudas C, Hsing AW, Chu L, Goodman PJ, Klein EA, Zheng SL, Batra J, Clements J, Spurdle A, Teixeira MR, Paulo P, Maia S, Slavov C, Kaneva R, Mitev V, Witte JS, Casey G, Gillanders EM, Seminara D, Riboli E, Hamdy FC, Coetzee GA, Li Q, Freedman ML, Hunter DJ, Muir K, Gronberg H, Neal DE, Southey M, Giles GG, Severi G, Cook MB, Nakagawa H, Wiklund F, Kraft P, Chanock SJ, Henderson BE, Easton DF, Eeles RA, Haiman CA, (BPC3) BaPCCC, Consortium PPCAGtIC‐AAitG, Consortium CCOG‐eS, Consortium G‐OE. A meta‐analysis of 87,040 individuals identifies 23 new susceptibility loci for prostate cancer. Nat Genet 2014; 46(10):1103–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tindall EA, Bornman MS, van Zyl S, Segone AM, Monare LR, Venter PA, Hayes VM. Addressing the contribution of previously described genetic and epidemiological risk factors associated with increased prostate cancer risk and aggressive disease within men from South Africa. BMC Urol 2013; 13:74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kiciński M, Vangronsveld J, Nawrot TS. An epidemiological reappraisal of the familial aggregation of prostate cancer: A meta‐analysis. PLoS ONE 2011; 6(10):e27130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hemminki K. Familial risk and familial survival in prostate cancer. World J Urol 2012; 30(2):143–148. [DOI] [PubMed] [Google Scholar]
- 12. Brandt A, Sundquist J, Hemminki K. Risk for incident and fatal prostate cancer in men with a family history of any incident and fatal cancer. Ann Oncol 2012; 23(1):251–256. [DOI] [PubMed] [Google Scholar]
- 13. Legros F, Malka F, Frachon P, Lombès A, Rojo M. Organization and dynamics of human mitochondrial DNA. J Cell Sci 2004; 117(Pt 13):2653–2662. [DOI] [PubMed] [Google Scholar]
- 14. Soares P, Ermini L, Thomson N, Mormina M, Rito T, Röhl A, Salas A, Oppenheimer S, Macaulay V, Richards MB. Correcting for purifying selection: An improved human mitochondrial molecular clock. Am J Hum Genet 2009; 84(6):740–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wallace DC. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: A dawn for evolutionary medicine. Annu Rev Genet 2005; 39:359–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lu J, Sharma LK, Bai Y. Implications of mitochondrial DNA mutations and mitochondrial dysfunction in tumorigenesis. Cell Res 2009; 19(7):802–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Schon EA, DiMauro S, Hirano M. Human mitochondrial DNA: Roles of inherited and somatic mutations. Nat Rev Genet 2012; 13(12):878–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. He Y, Wu J, Dressman DC, Iacobuzio‐Donahue C, Markowitz SD, Velculescu VE, Diaz LA, Kinzler KW, Vogelstein B, Papadopoulos N. Heteroplasmic mitochondrial DNA mutations in normal and tumour cells. Nature 2010; 464(7288):610–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ju YS, Alexandrov LB, Gerstung M, Martincorena I, Nik‐Zainal S, Ramakrishna M, Davies HR, Papaemmanuil E, Gundem G, Shlien A, Bolli N, Behjati S, Tarpey PS, Nangalia J, Massie CE, Butler AP, Teague JW, Vassiliou GS, Green AR, Du MQ, Unnikrishnan A, Pimanda JE, Teh BT, Munshi N, Greaves M, Vyas P, El‐Naggar AK, Santarius T, Collins VP, Grundy R, Taylor JA, Hayes DN, Malkin D, Foster CS, Warren AY, Whitaker HC, Brewer D, Eeles R, Cooper C, Neal D, Visakorpi T, Isaacs WB, Bova GS, Flanagan AM, Futreal PA, Lynch AG, Chinnery PF, McDermott U, Stratton MR, Campbell PJ, Group IBC, Group ICMD, Group IPC. Origins and functional consequences of somatic mitochondrial DNA mutations in human cancer. Elife 2014; 3:e02935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Koppenol WH, Bounds PL, Dang CV. Otto Warburg's contributions to current concepts of cancer metabolism. Nat Rev Cancer 2011; 11(5):325–337. [DOI] [PubMed] [Google Scholar]
- 21. Gómez‐Zaera M, Abril J, González L, Aguiló F, Condom E, Nadal M, Nunes V. Identification of somatic and germline mitochondrial DNA sequence variants in prostate cancer patients. Mutat Res 2006; 595(1–2):42–51. [DOI] [PubMed] [Google Scholar]
- 22. Chen JZ, Gokden N, Greene GF, Mukunyadzi P, Kadlubar FF. Extensive somatic mitochondrial mutations in primary prostate cancer using laser capture microdissection. Cancer Res 2002; 62(22):6470–6474. [PubMed] [Google Scholar]
- 23. Lindberg J, Mills IG, Klevebring D, Liu W, Neiman M, Xu J, Wikström P, Wiklund P, Wiklund F, Egevad L, Grönberg H. The mitochondrial and autosomal mutation landscapes of prostate cancer. Eur Urol 2013; 63(4):702–708. [DOI] [PubMed] [Google Scholar]
- 24. Jerónimo C, Nomoto S, Caballero OL, Usadel H, Henrique R, Varzim G, Oliveira J, Lopes C, Fliss MS, Sidransky D. Mitochondrial mutations in early stage prostate cancer and bodily fluids. Oncogene 2001; 20(37):5195–5198. [DOI] [PubMed] [Google Scholar]
- 25. Petros JA, Baumann AK, Ruiz‐Pesini E, Amin MB, Sun CQ, Hall J, Lim S, Issa MM, Flanders WD, Hosseini SH, Marshall FF, Wallace DC. MtDNA mutations increase tumorigenicity in prostate cancer. Proc Natl Acad Sci USA 2005; 102(3):719–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Parr RL, Dakubo GD, Crandall KA, Maki J, Reguly B, Aguirre A, Wittock R, Robinson K, Alexander JS, Birch‐Machin MA, Abdel‐Malak M, Froberg MK, Diamandis EP, Thayer RE. Somatic mitochondrial DNA mutations in prostate cancer and normal appearing adjacent glands in comparison to age‐matched prostate samples without malignant histology. J Mol Diagn 2006; 8(3):312–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kloss‐Brandstätter A, Schäfer G, Erhart G, Hüttenhofer A, Coassin S, Seifarth C, Summerer M, Bektic J, Klocker H, Kronenberg F. Somatic mutations throughout the entire mitochondrial genome are associated with elevated PSA levels in prostate cancer patients. Am J Hum Genet 2010; 87(6):802–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Arnold RS, Sun CQ, Richards JC, Grigoriev G, Coleman IM, Nelson PS, Hsieh CL, Lee JK, Xu Z, Rogatko A, Osunkoya AO, Zayzafoon M, Chung L, Petros JA. Mitochondrial DNA mutation stimulates prostate cancer growth in bone stromal environment. Prostate 2009; 69(1):1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Morris AG, Heinze A, Chan EK, Smith AB, Hayes VM. First ancient mitochondrial human genome from a prepastoralist southern African. Genome Biol Evol 2014; 6(10):2647–2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chan EK, Hardie RA, Petersen DC, Beeson K, Bornman RM, Smith AB, Hayes VM. Revised timeline and distribution of the earliest diverged human maternal lineages in southern Africa. PLoS ONE 2015; 10(3):e0121223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Petersen DC, Libiger O, Tindall EA, Hardie RA, Hannick LI, Glashoff RH, Mukerji M, Fernandez P, Haacke W, Schork NJ, Hayes VM, Consortium IGV. Complex patterns of genomic admixture within southern Africa. PLoS Genet 2013; 9(3):e1003309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics 2000; 155(2):945–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Evanno G, Regnaut S, Goudet J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol Ecol 2005; 14(8):2611–2620. [DOI] [PubMed] [Google Scholar]
- 34. Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA‐MEM. arXiv:13033997 [q‐bioGN] 2013.
- 35. Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, Miller CA, Mardis ER, Ding L, Wilson RK. VarScan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res 2012; 22(3):568–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, Subgroup GPDP. The sequence alignment/map format and SAMtools. Bioinformatics 2009; 25(16):2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Epstein JI, Zelefsky MJ, Sjoberg DD, Nelson JB, Egevad L, Magi‐Galluzzi C, Vickers AJ, Parwani AV, Reuter VE, Fine SW, Eastham JA, Wiklund P, Han M, Reddy CA, Ciezki JP, Nyberg T, Klein EA. A Contemporary prostate cancer grading system: A validated alternative to the gleason score. Eur Urol 2015. [ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Maki J, Robinson K, Reguly B, Alexander J, Wittock R, Aguirre A, Diamandis EP, Escott N, Skehan A, Prowse O, Thayer RE, Froberg MK, Wilson MJ, Maragh S, Jakupciak JP, Wagner PD, Srivastava S, Dakubo GD, Parr RL. Mitochondrial genome deletion aids in the identification of false‐and true‐negative prostate needle core biopsy specimens. Am J Clin Pathol 2008; 129(1):57–66. [DOI] [PubMed] [Google Scholar]
- 39. Bryc K, Durand EY, Macpherson JM, Reich D, Mountain JL. The genetic ancestry of African Americans, Latinos, and European Americans across the United States. Am J Hum Genet 2015; 96(1):37–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Patterson N, Petersen DC, van der Ross RE, Sudoyo H, Glashoff RH, Marzuki S, Reich D, Hayes VM. Genetic structure of a unique admixed population: Implications for medical research. Hum Mol Genet 2010; 19(3):411–419. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional supporting information may be found in the online version of this article at the publisher's web‐site.
Supplementary Table SI.
Supporting Information.