

Abstract
Small cell lung cancers (SCLCs) and extrapulmonary small cell cancers (SCCs) are very aggressive tumors arising de novo as primary small cell cancer with characteristic genetic lesions in RB1 and TP53. Based on murine models, neuroendocrine stem cells of the terminal bronchioli have been postulated as the cellular origin of primary SCLC. However, both in lung and many other organs, combined small cell/non‐small cell tumors and secondary transitions from non‐small cell carcinomas upon cancer therapy to neuroendocrine and small cell tumors occur. We define features of “small cell‐ness” based on neuroendocrine markers, characteristic RB1 and TP53 mutations and small cell morphology. Furthermore, here we identify a pathway driving the pathogenesis of secondary SCLC involving inactivating NOTCH mutations, activation of the NOTCH target ASCL1 and canonical WNT‐signaling in the context of mutual bi‐allelic RB1 and TP53 lesions. Additionaly, we explored ASCL1 dependent RB inactivation by phosphorylation, which is reversible by CDK5 inhibition. We experimentally verify the NOTCH‐ASCL1‐RB‐p53 signaling axis in vitro and validate its activation by genetic alterations in vivo. We analyzed clinical tumor samples including SCLC, SCC and pulmonary large cell neuroendocrine carcinomas and adenocarcinomas using amplicon‐based Next Generation Sequencing, immunohistochemistry and fluorescence in situ hybridization. In conclusion, we identified a novel pathway underlying rare secondary SCLC which may drive small cell carcinomas in organs other than lung, as well.
Keywords: Lung cancer, small cell lung cancer, achaete‐scute homolog 1, neurogenic locus notch homolog, retinoblastoma protein
Short abstract
What's new?
Using next generation sequencing and establishing features of ‘small cell‐ness’, we identified a NOTCH‐ASCL1‐RB1‐TP53 signaling axis driving small cell cancers. In contrast to the previously described bi‐allelic RB1/TP53 loss in neuroendocrine stem cells as origin of primary small cell neuroendocrine cancers, the NOTCH‐ASCL1 mediated signaling defines an alternative pathway driving secondary small cell neuroendocrine cancers arising from non‐small cell cancers. Moreover, we show a preclinical rational for therapeutically testing WNT‐inhibitors in small cell cancers.
Abbreviations
- AdC
(pulmonary) adenocarcinoma
- ASCL1
Achaete‐scute homolog 1
- CDK
cylin dependent kinase
- DKK1
Dickkopf 1
- eV
empty vector
- FISH
fluorescence in situ hybridization
- FFPE
formalin‐fixed paraffin‐embedded
- IF
Immunofluorescence
- IHC
immunohistochemistry
- LCNEC
(pulmonary) large cell neuroendocrine carcinoma
- LRP6
low density lipoprotein receptor‐related protein‐6
- NE
neuroendocrine
- NGS
next generation sequencing
- NOTCH
neurogenic locus notch homolog
- NSCLC
non‐small cell lung cancer
- p53
tumor protein 53
- RB
retinoblastoma protein
- SCC
small cell cancer
- SCLC
small cell lung cancer
- SqCC
(pulmonary) squamous cell carcinoma
- WNT
wingless‐type
The current WHO classification of lung cancer discriminates small cell lung cancer (SCLC) from non‐small cell lung cancer (NSCLC) comprising the entities adenocarcinoma (AdC), squamous cell carcinoma (SqCC), a few rare subtypes of NSCLC, large cell neuroendocrine carcinoma (LCNEC), and finally typical and atypical carcinoids. A novel genomics‐based taxonomy of lung tumors proposed by the worldwide initiative of the Clinical Lung Cancer Genome Project (CLCGP) and the Network Genomic Medicine (NGM) suggests that a combination of histological and genomic denominators will redefine the classification into SCLC/LCNEC, AdC, SqCC and carcinoids.1
SCLC has distinct pathological and clinical features. Tumor cells have round, spindled nuclei with finely granulated chromatin, inconspicuous nucleoli, scant cytoplasm, and frequently shows nuclear moulding. SCLCs have high mitotic rates (>60 mitoses per 2 mm2) and frequently a neuroendocrine (NE) phenotype. All small cell carcinomas (SCCs), however representing a rare tumor entity, share a very aggressive biology with early systemic spread, irrespective of organ of origin.2, 3, 4, 5 Therefore, it is likely that general molecular mechanisms drive “small cell‐ness” with cancer stem cell‐related features.
We and others showed that mutual bi‐allelic TP53 and RB1 alterations are central events in SCLC biology.6 Bi‐allelic loss of TP53 and RB1 is sufficient to induce a SCC phenotype in murine lung tumors.7 Nevertheless, combined lung carcinoma phenotypes and relapses with a changed phenotype upon cancer therapy occur in patients. Thus, we suggest that NE SCCs may not only arise as primary lesions or as a synchronous combined carcinoma but also arise as secondary lesions in form of relapses originating from non‐small cell carcinomas induced by cancer therapy.
Achaete‐scute homolog 1 (ASCL1) is a basic‐helix‐loop‐helix transcription factor pivotal for NE differentiation and expressed in pulmonary NE cells and in SCLC.8 Moreover, ASCL1 promotes more aggressive AdC growth in vivo and may interact with the central “retinoblastoma protein‐tumor protein 53” (RB‐p53) axis in the carcinogenesis of NE lung cancers.9 ASCL1 contributes to enhanced proliferation and migration in lung cancer cells in vitro by targeting cyclin‐dependent kinase 5 (CDK5).10
ASCL1 expression is regulated downstream of neurogenic locus notch homolog (NOTCH) signaling mediated through four different receptors which causes polyubiquitination‐mediated ASCL1 degradation.11, 12 Altered NOTCH‐signaling by receptor mutations is frequently found in cancer. Thereby the mutated domain determines the functionality, for example, activating mutations located in the Proline Glutamic acid Serine Threonine rich (PEST) domain12 or inactivating mutations in the EGF‐like13 and ankyrin (ANK) repeats.14
We defined features of “small cell‐ness” and investigated signaling via the NOTCH‐ and ASCL1‐dependent pathway in vitro. We then performed amplicon‐based Next Generation Sequencing (NGS) of different tumor sample cohorts to identify NOTCH mutations. Large parts of the genomic Guanine Cytosine (GC) rich NOTCH1‐4 loci are difficult to sequence and hence, data from whole genome sequencing and The Cancer Genome Atlas (TCGA) are not fully informative.
Taken together, our data suggest that there are two oncogenic pathways for NE SCCs. Primary SCLC originates from NE stem cells with mutual bi‐allelic TP53 and RB1 alteration in contrast to secondary SCLC developing from NOTCH‐defective NSCLC that already harbor TP53 mutations and acquire additional RB inactivation.
Material and Methods
Cell culture and reagents
The cell lines A549, PC9, H1975, H441, H460, GLC1, GLC2, GLC8, N417, DMS114 and SW1271 were kindly provided by Roman Thomas (University of Cologne, Germany), from American Type Culture Collection (ATCC) or Lou de Leij. Cells were authenticated by NGS.
Jerry Crabtree (Stanford, USA) donated pTight‐hASCL1‐N174 (ASCL1 expression plasmid), published by Yoo et al. in Nature15 and delivered by Addgene (plasmid ID: 31876, Cambridge, USA). The empty Vector (eV) control was generated by SpeI/XhoI digest. 100 nM siRNAs were applied using Lipofectamine 2000 (Life Technologies): RB (New England Biolabs), ASCL1, NOTCH1 and NOTCH2 (Santa Cruz). The CDK5 inhibitor Roscovitine16 (Absource Diagnostics) and the wingless‐type (WNT)‐pathway inhibitor IWP‐2 (Sigma‐Aldrich) were used as indicated. Proteins were blotted onto nitrocellulose membranes (BioRad). Quantitative Real‐Time PCR (qRT‐PCR) was performed with SYBR Green (Qiagen). Used antibodies and primers are listed in Supporting Information Tables S1 and S2.
Flow cytometry
Cells were washed in PBS, prestained with fixable viability dye (eBioscience), fixed in 4% PBS‐buffered formalin, permeabilized using 0.2% Saponin and stained with antibodies in PBS. Flow cytometry was performed by FACSCanto I (Becton Dickinson) and data were analyzed using FlowJo (Tree Star).
Immunofluorescence (IF) and Immunohistochemistry (IHC)
For IF cells were plated on cover slips, fixed in 4% PBS‐buffered formalin and pre‐treated with 0.25% TritonX. Staining was performed for 30 min in a humidified chamber at RT. Images were taken by an inverted microscope fitted with an ApoTome (Zeiss). IHC stain was performed as previously described.17
Fluorescence in situ hybridization (FISH)
FISH was performed as previously described.18 RB1 probe (red) (artificial BAC clone: RP11‐893E5, Life Technologies) and chromosome 13 centromeric probe (green) (Empire Genomics) were used. Evaluation of RB1 deletions in 100 tumor cells was performed by fluorescence microscopy using 60× magnification (Zeiss).
Amplicon‐based NGS of formalin‐fixed paraffin‐embedded tumor samples
Formalin‐fixed paraffin‐embedded (FFPE) tumor samples were obtained from our routine diagnositics with approval of the local ethics committee (Ref Number: 10‐242). Ion AmpliSeq™ Custom DNA Panels (Life Technologies) were designed (Supporting Information Table S3) and used and analyzed according to manufactureŕs instructions with modifications.19
Statistics
Statistics were calculated using Excel (Microsoft), Graph Pad Prism (STATCON) and SPSS (Armonk). We used two‐sided Students t test. If normal distribution and similar variance in an experiment were not applicable, Kruskal‐Wallis‐Test was used. Error bars indicate standard error of the mean (SEM).
Results
Establishment of features of “small cell‐ness” according to lung cancer cell lines
Pathological and clinical features of SCLC were described in patients based on IHC20 and integrative genome analysis.6 We adapted criteria of markers, mutations and morphology characteristic for SCLC to establish features of “small cell‐ness,” especially for in vitro studies (Fig. 1).
Figure 1.
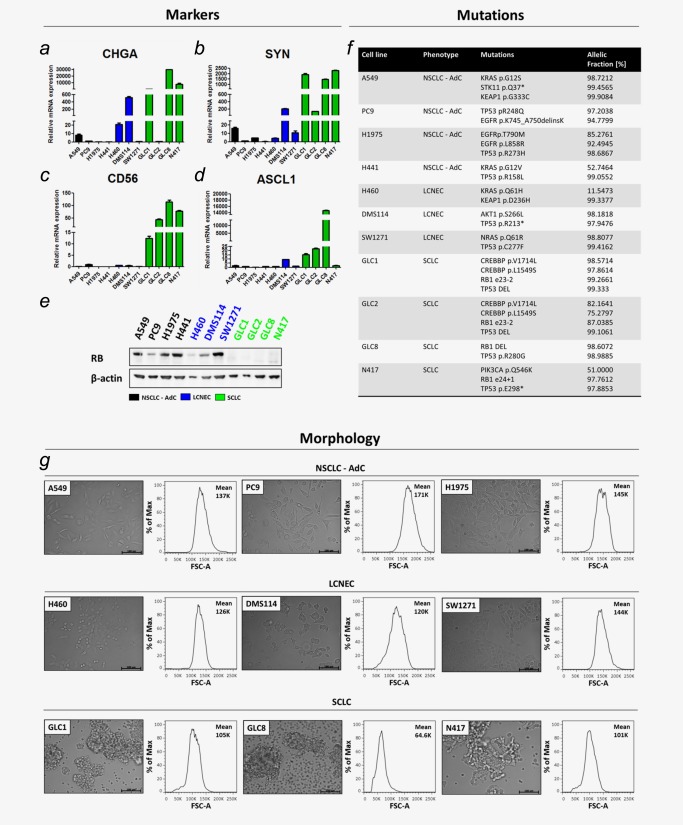
Establishment of “small cell‐ness” features for lung carcinoma cell lines analyzing markers, mutations and morphology. NSCLC‐AdC (black), LCNEC (blue) and SCLC (green) were compared. NE marker expressions of (a) Chromogranin A (CHGA), (b) Synaptophysin (SYN), (c) CD56 and (d) ASCL1 were determined by qRT‐PCR and calculated by ΔΔCT‐method. (e) RB protein expression determined by Western Blot. (f) Mutations identified by amplicon‐based NGS. Allelic fraction was listed in %. (g) Cell morphologies determined in monolayer cell culture by microscopy. Bars indicate 100 μm. Cell size determined by forward scatter (FSC‐A) properties measured by flow cytometry.
However, intermediate forms of lung carcinomas with large cells but NE differentiation such as LCNECs were of special interest. Finally, we categorized the two cell lines, DMS114 and SW1271, declared as SCLC, as LCNEC, since they did not completely meet the features of “small cell‐ness.”
First, we analyzed expression of classical NE markers Chromogranin A, Synaptophysin and CD56 (Figs. 1 a–1 c). We also included ASCL1 (Fig. 1 d), since reanalysis of previously published expression‐array data of NSCLC and SCLC samples21, 22 identified significant expression of ASCL1 in SCLC. Additionally, we determined RB protein expression (Fig. 1 e).
At least one NE marker was expressed by cell lines categorized as SCLC (green) and LCNEC (blue). RB protein was expressed in AdC (black) and LCNEC cell lines but nor on SCLC cell lines.
We used amplicon‐based NGS to identify mutations on RB1 and TP53 (Fig. 1 f). Mutual RB1 and TP53 mutations were only identified in SCLC cell lines. Thereby RB1 mutations correlated with the lack of RB protein expression. Using different amplicon‐based panels, we identified other oncogenic mutations, for example EGFR mutations in PC9 and H1975.
To determine cell morphology, we used monolayer cell culture and flow cytometry measuring cell size by forward scatter (FSC‐A) (Fig. 1 g).
The three analyzed AdC cell lines (A549, PC9 and H1975) grew adherently, had cells with spindle shape, abundant cytoplasm and a mean FSC‐A of 151K referring to large cell size. LCNEC cell lines (H460, DMS114 and SW1271) grew adherently, had round to spindle formed or irregular shaped cells with intermediate cytoplasm. Mean FSC‐A was 130K. SCLC cell lines (GLC1, GLC8 and N417) grew in cell clusters in suspension, had round cells with scant cytoplasm and a mean FSC‐A of 90.2K referring to small cell size.
Taken together, the features of “small cell‐ness” clearly describe the most prominent characteristics of SCLC and may represent a model to pre‐categorize SCLC and LCNEC, especially for in vitro experiments. Thus, we postulate a general phenotype also of extrapulmonary small cell carcinomas (SCCs) that comprises NE marker expression including ASCL1, mutual RB1 and TP53 mutations and small round clustered cells.
ASCL1 overexpression induced a small cell carcinoma phenotype and canonical WNT‐signaling
ASCL1 is a NE master regulator and expressed in multipotent stem cells of NE lineage. Lineage decisions in lung development are triggered by NOTCH‐signaling upstream of ASCL1 down‐regulation.23 Thus, we hypothesized that ASCL1 may be a key‐factor in SCC development.
We transfected PC9 cells (AdC) with an expression plasmid containing ASCL1 or vector control (Fig. 2). PC9 cells harbor an activating EGFR mutation and an inactivating TP53 mutation and may therefore represent a model system for secondary SCLC, as clinically observed after intensive treatment AdCs relapse as SCLC.
Figure 2.
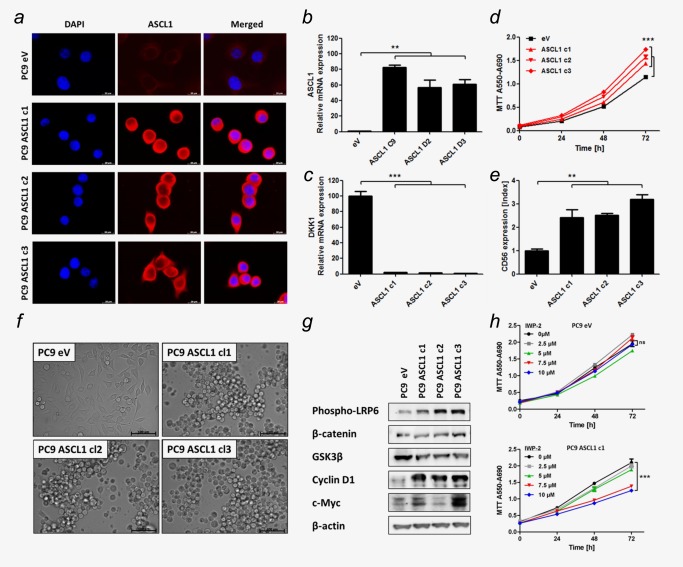
ASCL1 expression induced “small cell‐ness” and canonical WNT‐signaling. (a) IF of PC9 transfected with ASCL1 expression plasmid (ASCL1) or empty vector (eV). Bars indicate 20 μm. (b+c) ASCL1 and DKK1 mRNA expression determined using qRT‐PCR. (d) Cell proliferation measured by MTT assay. (e) CD56 expression determined by flow cytometry. Mean fluorescence intensity of CD56‐PE‐Cy7 was normalized on IgG‐PE‐Cy7 (Index). (f) Cell morphologies determined in monolayer cell culture by microscopy. Bars indicate 100 μm. (g) Protein levels of members of WNT‐signaling pathway determined by Western blot. (h) eV transfected PC9 and an ASCL1 expressing representative clone c1 were treated with IWP‐2 WNT‐pathway inhibitor. Cell proliferation measured by MTT assay. Analysis was done using ΔΔCT‐method. Data are presented as mean ± SEM (n = 5). Statistical significance was calculated using a Student's t test, two‐sided, * p < 0.05, ** p < 0.01, *** p < 0.001.
Stable transfectants only harboring the empty vector (eV) and three clones overexpressing ASCL1 (c1‐c3) were derived (Fig. 2 a). We analyzed ASCL1 clones for features of “small cell‐ness.” Prior to experiments we controlled clonal origin by microsatellite analysis referring to the parental PC9 cell line (Supporting Information Fig. S1).
ASCL1 clones showed at least 50‐fold increased ASCL1 expression (Fig. 2 b). Interestingly, Dickkopf 1 (DKK1) expression was significantly decreased upon ASCL1 expression (Fig. 2 c). Moreover, cell proliferation was significantly enhanced in ASCL1 clones compared to eV (Fig. 2 d) and they expressed CD56 (Fig. 2 e). Remarkably, ASCL1 overexpression directly induced a switch towards SCLC‐like cell morphology (Fig. 2 f).
We hypothesized that ASCL1 activates pro‐proliferative WNT‐signaling because ASCL1 directly represses transcription of DKK1, a negative WNT‐signaling pathway regulator.24 DKK1 acts as a core repressor of low density lipoprotein receptor‐related protein‐6 (LRP6), a co‐receptor recruited by Frizzled to canonically transduce WNT‐signals into the cell.25 Thus, we analyzed WNT‐signaling and the phosphorylation of LRP6 (Fig. 2 g).
We found robust induction of phospho‐LRP6 (pLRP6) and the WNT targets CyclinD1 and c‐Myc and moderate reduction of Glykogen synthase kinase 3β (GSK3β) in ASCL1 clones compared to eV.
Effects of WNT‐pathway inhibition in cancers are tested in phase I clinical trials, for example in previously treated NSCLC patients (ClinicalTrials.gov Identifier: NCT01957007, Bayer) or in patients with other malignancies (ClinicalTrials.gov Identifier: NCT01351103, Novartis).
Consistently, upon treatment with WNT‐inhibitor IWP‐2 the ASCL1 clone showed significantly reduced cell proliferation in a dose‐dependent manner whereas the eV control remained unaffected with respect to cell growth (Fig. 2 h). Inhibition of WNT‐signaling was controlled by qRT‐PCR of Cyclin D1 and Axin2 (Supporting Information Fig. S2).
Moreover, we showed induction of apoptosis by 7AAD and AnnexinV stain upon WNT‐pathway inhibition in two SCLC cell lines, GLC1 and GLC2, in a dose‐dependent manner (Supporting Information Fig. S3). Thus, WNT‐signaling provided a potential therapeutic target in SCLC.
In conclusion, overexpression of ASCL1 was sufficient to trigger canonical WNT‐signaling via phosphorylation of the co‐receptor LRP6, to induce NE differentiation and to mediate the phenotypic switch towards “small cell‐ness,” which did not results from an accidental de novo mutation in RB1 (data not shown).
ASCL1 triggered phosphorylation of RB by CDK5
ASCL1 clones presented a SCC phenotype and fulfilled the criteria of “small cell‐ness” apart from mutual RB1 and TP53 mutation, since de novo mutations in RB1 were not acquired.
In addition to direct genetic inactivation, RB can be inactivated by phosphorylation.26 Thus, we performed Western blot analysis to determine total RB protein and phosphorylation status. All three ASCL1 clones showed higher expression of phosphorylated RB at Serine (Ser) 780, Ser795, Ser807/811 than PC9 eV cells (Fig. 3 a). Total RB protein expression, however, remained unchanged. Thus, we concluded that ASCL1 overexpression caused inactivation of RB by phosphorylation. Phosphorylation of RB is triggered by CDKs. We found upregulated CDK5 in ASCL1 clones compared to eV control. In contrast, CDK2 protein levels were not altered by ASCL1 overexpression and CDK4 and CDK6 expression was reduced (Fig. 3 a).
Figure 3.
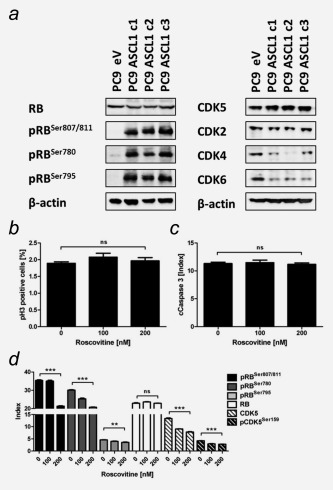
ASCL1 mediated phosphorylation of RB triggered by CDK5. (a) RB and CDK protein levels determined by Western blot. (b+c) Mitotic cell number (pHistoneH3 – pH3) and induction of apoptosis (cleaved Caspase 3 – cCaspase3) determined by flow cytometry upon CDK5 inhibition by Roscovitine for 48 hrs. (d) RB and CDK5 protein levels measured by flow cytometry. Mean fluorescence intensity was normalized on secondary antibody control (Index). Data are presented as mean ± SEM of analysis in triplicates. Statistical significance was calculated using the Student's t test, two‐sided, * p < 0.05, ** p < 0.01, *** p < 0.001.
Based on these data, we further validated CDK5 as driver of RB phosphorylation in ASCL1 clones using Roscovitine to selectively inhibit CDK5 activity (Figs. 3b–3d). CDK5 inhibition did not affect mitotic cell count and also did not induce cleaved Caspase 3 referring to apotosis. Analysis of RB, pRBSer780, pRBSer795, pRBSer807/811, CDK5 and pCDK5Ser159 expression showed significantly decreased levels of CDK5 and pCDK5Ser159 and RB phosphorylation. To further elucidate the connection between ASCL1, CDK5 and pRB, we performed siRNA mediated knock‐down of ASCL1 in a SCLC cell line (GLC8) and a LCNEC cell line (DMS114). Consistently, ASCL1 knock‐down reduced protein levels of CDK5 and of pRBSer780 (Supporting Information Fig. S4).
Since ASCL1 is targeted by NOTCH‐signaling, we performed siRNA mediated knock‐down of NOTCH1 and NOTCH2 in PC9 cells (Supporting Information Fig. S5). We observed significantly increased ASCL1 and CD56 expression. Flow cytometry revealed stable RB protein expression and significantly increased RB phosphorylation at Ser780, but not as evident as in ASCL1 clones.
Nevertheless, NOTCH1 and NOTCH2 knock‐down were not potent enough to cause a phenotypic switch towards SCC and cell proliferation was even reduced, since PC9 cells originally harbored intact NOTCH‐signaling (Supporting Information Fig. S5).
To prove direct effects of RB inactivation on SCC growth, we performed siRNA mediated knock‐down of RB in A549 and PC9. A549 cells (p53 wild‐type) showed reduced number of mitotic cells positive for phospho‐histone H3 (pH3) and increased apoptosis indicated by elevated cleaved Caspase 3 and cleaved PARP. PC9 cells (p53 mutated) were protected from both and proliferated faster upon RB knock‐down than A549 cells (Supporting Information Fig. S6).
Thus, we propose that ASCL1 overexpression induced CDK5 upregulation and thereby RB inactivation by phosphorylation and that p53 mutated cells had a selective advantage when RB was inactivated.
Conclusively, ASCL1 assists the central RB‐p53 signaling axis in the establishment of a SCC phenotype.
Mutational patterns of RB1, TP53 and NOTCH genetic alterations in SCLC, SCC, LCNEC and AdC
We hypothesized a central signaling axis of NOTCH inactivation, ASCL1/CDK5 activation and mutual RB/p53 inactivation. Since isolated NOTCH knock‐down was not potent enough, we suggested that genetic lesions may be driver events in this signaling axis in vivo.
We examined mutations in all four NOTCH genes (NOTCH1‐4), RB1 and TP53 by NGS in 35 SCLCs, 28 extrapulmonary SCCs, 19 pulmonary LCNECs and 33 pulmonary AdCs. All samples underwent routine IHC based diagnostics to determine cell morphology by HE stain, a pulmonary tumor origin by Thyroid Transcription Factor‐1 (TTF1) and cytokeratin 7/8 (CK7/8) stain and a proliferation score by Ki‐67 stain. Additonally, we included ASCL1 and DKK1 IHC for further characterization (Figs. 4 and 5).
Figure 4.
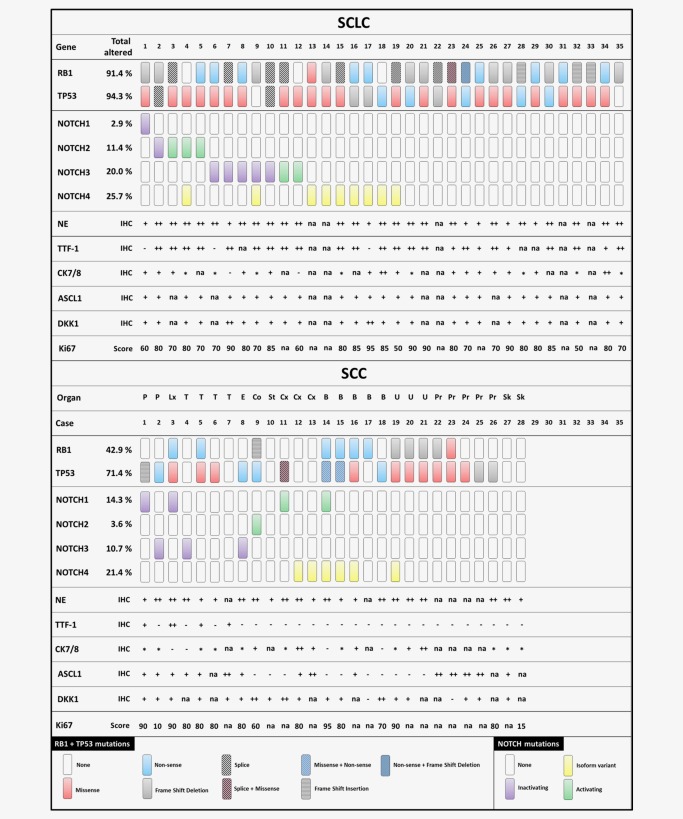
Distribution of genetic lesions in different small cell carcinoma entities. Mutations in RB1 and TP53 shown in the upper panel. Missense mutations occurring in all four NOTCH genes (NOTCH1‐4) shown in the lower panel. DNA of 35 small cell lung carcinomas (SCLC) and 28 extrapulmonary small cell carcinomas (SCC) was analyzed by NGS. Organ of origin: P–Parotis; Lx–Larynx; T–Trachea; E–Esophagus; St–Stomach; Cx– Cervix; B–Bladder; U–Urothel; Pr–Prostate; Sk–Skin. – not expressed; + expressed; ++ strongly expressed; * dot‐like expressed; na – not available.
Figure 5.
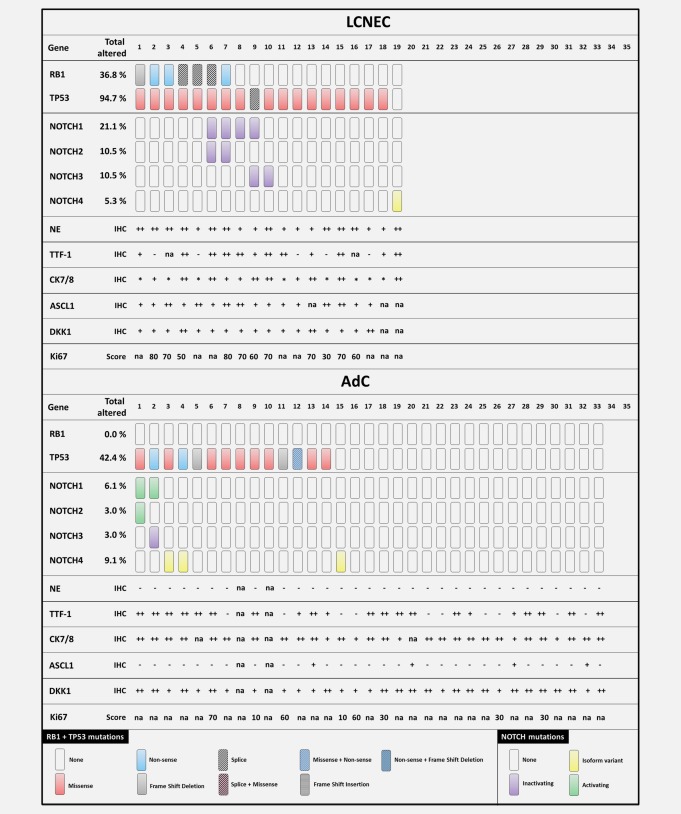
Distribution of genetic lesions in different lung carcinoma entities. Mutations in RB1 and TP53 shown in the upper panel. Missense mutations occurring in all four NOTCH genes (NOTCH1‐4) shown in the lower panel. DNA of 19 pulmonary large cell neuroendocrine carcinomas (LCNEC) and 33 pulmonary adenocarcinomas (AdC) was analyzed by NGS. – not expressed; + expressed; ++ strongly expressed; * dot‐like expressed; na – not available.
A comprehensive list of mutations according to carcinoma subtype is given in Supporting Information Table S4. Since the activation status of NOTCH is highly dependent on the specific mutation, an additional list stating the effect of the mutation on the specific NOTCH receptor domain and available COSMIC ID is given in Supporting Information Table S5. We excluded known single nucleotide polymorphisms (SNPs) by screening SNP databases of dbSNP (NCBI) and the NHLBI GO Exome Sequencing Project (ESP) EPS5400. We included NOTCH4 isoform variants identified by our routine diagnostics pipeline.
Genetic alterations in RB1 and TP53 were characteristic for SCLCs (RB1 91.4%, TP53 94.3%, combined 85.7%) and frequently found in SCCs (RB1 42.9%, TP53 71.4%, combined 39.3%) and LCNECs (RB1 36.8%, TP53 94.7%, combined 36.8%).
Importantly, the AdC cohort did not harbor any RB1 mutation. 42.4% of AdCs harbored a TP53 mutation. Thus, we reconfirm that mutual inactivation of both RB1 alleles and both TP53 alleles is a hallmark of SCLC.
Only 12.1% of AdCs expressed ASCL1 whereas all SCLCs and LCNECs were positively stained. 54.5% of AdCs showed a strong expression of DKK1 which we observed only in 8.5% of SCLC, SCC and LCNEC cases.
Inactivating NOTCH mutations in NOTCH1, NOTCH2 and NOTCH3 occurred in SCLC, SCC and LCNEC. The most remarkable cohort was LCNEC where no activating NOTCH mutation occurred. Thus, although NOTCH mutations were a rare genetic event, we postulate that they occur predominantly in NE lesions and that they are a hallmark of a representative subgroup of NE differentiated neoplasms including secondary SCLC that relapsed from NSCLC induced by cancer therapy (Fig. 6 a).
Figure 6.
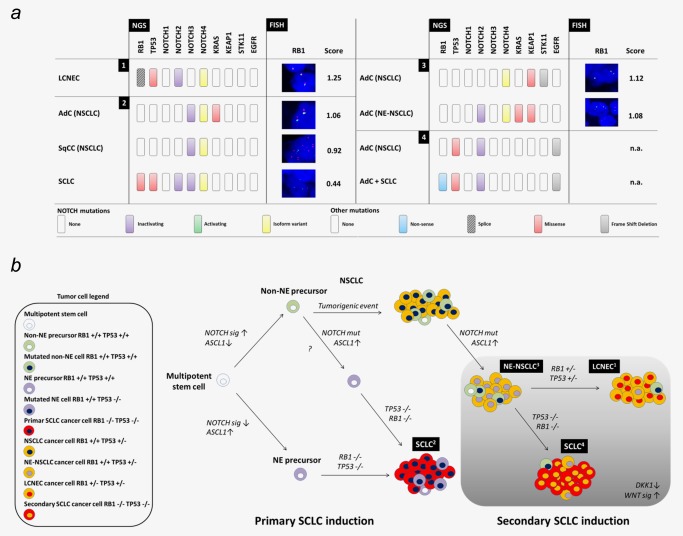
Primary and secondary SCLC within the network of NE lung carcinomas. (a) Four special lung carcinoma cases were selected for comprehensive NGS, IHC and FISH analysis. Case 1 – LCNEC, Case 2 – primary SCLC with combined AdC and SqCC, Case 3 – NE AdC combined with non‐NE AdC, Case 4 – secondary SCLC with combined AdC as relapse of TKI‐treated AdC. Corresponding IHC data are presented in Supporting Information Figure S7. (b) Primary SCLC (2) is suggested to arise from cancer stem cells out of a NE niche upon bi‐allelic loss of TP53 and RB1. Secondary SCLC (4) may originate from non‐NE cancer stem cells that acquire NE differentiation through inactivating NOTCH mutations and additionally a bi‐allelic loss of TP53 and RB1. Intermediate tumor stages as NE differentiated NSCLC (3) or LCNEC (1) depend on the mutation status of TP53 and RB1.
Primary and secondary SCLC within the network of pulmonary NE lesions
Finally, we analyzed representative cases of four NE lung carcinoma categories, namely LCNEC, primary combined SCLC, NE NSCLC and secondary SCLC (Fig. 6). We performed NGS, IHC and FISH analysis if applicable (Fig. 6a and Supporting Information Fig. S7). Material of such specimens is often limited because most patients with aggressive NE lung cancers receive radio‐chemotherapies and are diagnosed by small biopsies.
Case 1 was a LCNEC with a high Ki67 index (70%) comparable to that of SCLCs. NGS revealed a NOTCH4 isoform (SNP), an inactivating NOTCH2 mutation and alterations in RB1 and TP53. Interestingly, the RB1/chromosome 13 quotient was 1.25, referring to no allelic RB1 deletion (Fig. 6 a and Supporting Information Fig. S7).
Case 2 harbored combined synchronous AdC, SqCC and SCLC which were discriminated by characteristic IHC stain pattern (Supporting Information Fig. S7). The AdC harbored a KRAS mutation. All three tumor entities revealed the same NOTCH4 isoform variant and the same inactivating NOTCH3 mutation (SNPs). Importantly, only the SCLC showed an additional inactivating NOTCH2 mutation, and additional RB1 and TP53 missense mutations. RB1/chromosome 13 ratio determined in AdC and SqCC with 1.06 and 0.92, respectively, referred to no allelic RB1 deletion. In the SCLC, the ratio of 0.44 indicated heterozygous RB1 deletion. Combined FISH and NGS results indicated a complete loss of both RB1 alleles. Furthermore, we detected bi‐allelic TP53 inactivation by positive p53 IHC stain and TP53 mutation with an excessively high allelic frequency of [mt]80% (Supporting Information Table S4) suggesting presence of the mutated p53 allele and allelic loss affecting the wt allele (Fig. 6 a and Supporting Information S7).
These results indicate a different tumor origin of the SCLC compontent compared to the AdC and the SqCC, thus representing a primary SCLC as part of a combined lung carcinoma.
Case 3 harbored two distinct but synchronous AdCs, one AdC harbored STK11 and KEAP1 mutations, the other AdC KRAS and KEAP1 mutations. The two KEAP1 mutations were not identical. Both tumors showed the same NOTCH4 isoform variant (SNP). The KRAS mutated AdC showed NE differentiation and harbored an additional inactivating NOTCH2 mutation. RB1/chromosome 13 ratio was 1.12 and 1.08, respectively, indicating no allelic RB1 deletion (Fig. 6 a and Supporting Information Fig. S7). Since this NE differentiated lung carcinoma did not provide the typical rosette structure of LCNECs (Fig. S7b), this case represents a further NE lung carcinoma category of NE NSCLC.
Case 4 was defined as combined AdC with SCLC. However, this combined carcinoma was a relapse from TKI‐treated AdC. We received extracts from both specimens. NGS analysis revealed an EGFR mutated AdC as primary tumor. Furthermore, this AdC harbored a TP53 mutation and an inactivating NOTCH2 mutation. Unfortunately, FFPE material was so limited that no further IHC and FISH analysis could be performed to determine NE marker expression. In the relapsed combined AdC‐SCLC specimen we identified the same EGFR mutation and the same TP53 and NOTCH2 mutation. In addition, the combined AdC SCLC harbored a RB1 splice mutant.
Allelic fractions of the TP53 and the RB1 mutation were > 90%. Thus, we postulate mutual bi‐allelic alteration of both genes defined as a prerequisite for SCLC formation. As all other mutations were identical in the primary tumor and the relapse we conclude that the SCLC fraction represented a small cell outgrowth of a transdifferentiated NSCLC.
Small cell outgrowths as secondary NSCLC relapse or as primary synchronous combined carcinomas were often observed in combination with LCNEC (Supporting Information Fig. S8). For secondary SCLC, bi‐allelic TP53 mutations in the non‐small cell precursor may be a prerequisite, which was more frequent in SqCCs than in AdCs (Supporting Information Fig. S9). However, an independent, primary SCLC origin or a NSCLC‐dependent secondary SCLC origin can only be determined by NGS.
Taken together we here established features of “small cell‐ness” and confirmed the central signaling axis of NOTCH‐ASCL1‐RB‐p53. Finally, our results provide evidence for a signaling pathway based on inactivating NOTCH mutations that drive the development of NE neoplasms including secondary SCLC (Fig. 6 b).
Discussion
In this study, we comprehensively investigated features of “small cell‐ness” and genetic alterations underlying the NE and SCC phenotype. We used NGS and in vitro assays to map an oncogenic pathway along the central driving axis of RB‐p53 in SCC development, especially in lung cancer.
Previous hypotheses that cytoskeletal alterations drive “small cell‐ness” and epithelial to mesenchymal transition (EMT) were not confirmed, and we rather showed that these were secondary events in SCC pathology27 which may be controlled by NOTCH‐signaling.28 Consistent to our features of “small cell‐ness,” inactivation of all four alleles of RB1 and TP53 were described to be causative for SCC, including SCLC.7, 29 We confirmed that this mechanism was likely for independent primary SCLC, and suggest an alternate pathway for secondary SCLC relapsing from NSCLC with a central driving axis including NOTCH‐ASCL1‐RB‐p53.
A multipotent epithelial precursor was discussed as tumor cell of origin 2003 by Meuwissen et al.7 However, in 2011 NE precursor cells were described as the predominant origin of SCLC by Sutherland et al.29
Experimentally, we here show that ASCL1 overexpression leads to SCC morphology in vitro, as previously described by Osada et al.24 We found activated canonical WNT‐signaling upon ASCL1 expression, already evaluated as therapeutical target in SCLC and LCNEC,30 and responsible for enhanced proliferation and invasion in lung cancer.31 In accordance to our data, ASCL1 drives WNT‐signaling by inhibition of DKK1 in glioblastoma.32
Bi‐allelic RB inactivation was frequently found in patients suffering from SCLC or extrapulmonary SCC.6, 33, 34 Importantly, we found RB inactivation by extensive phosphorylation at three different serine positions in ASCL1 clones in vitro. Upon RB inactivation E2F1 is liberated and cells are directed towards p53 mediated apoptosis. Therefore, the Ser795 phosphorylation is functionally important, since it is the most potent site inhibiting E2F1 binding to the RB pocket.35 Tumor cells can evade this mechanism by acquiring mutations in the TP53 gene.36, 37
During cell cycle, CDKs mediate RB phosphorylation.36 We found upregulated CDK5 upon ASCL1 overexpression. A direct interaction of ASCL1 and CDK5 was shown in lung cancer cells where ASCL1 stimulated migration by activated CDK5.10 It is known that CDK5 is able to phosphorylate RB at the same residues as CDK4 and CDK2 in postmitotic neurons and enables cell cycle re‐entry and proliferation.38 Furthermore, CDK5 mediated phosphorylation of RB at Ser807/811 is essential for tumorigenesis and tumor progression in NE thyroid cancer.39
We found that ASCL1 overexpression leads to CD56 expression in vitro. In vivo ASCL1 expression induced NE differentiation of murine lung tumors and enhanced tumorigenesis. Nevertheless, ASCL1 expression alone was not sufficient to induce a full SCC phenotype but it was reported that ASCL1 may cooperate with RB and p53 loss when forming SCLC.9
SCLCs occur as pure carcinomas or as combined carcinomas with non‐small cell components in up to 30% of cases40 and also mixed phenotypes were reported after chemotherapy of primary SCLC.41 However, clinical observations also suggest that SCCs may arise as secondary neoplasms from non‐small cell cancer background in form of relapses after genotoxic chemotherapies or targeted therapies.42, 43, 44
The complex patterns of inactivating NOTCH mutations in context of mutual RB1 and TP53 alteration in our clinical collection of tumors with NE differentiation indeed suggests that some NE neoplasm may represent a NSCLC‐dependent secondary tumor origin overgrowing their non‐small cell origin.
Therefore, it may be interesting to analyze NOTCH mutation status also in other representative subgroups such as the recently identified subgroup of ASCL1 and RET expressing pulmonary AdC.45
Our results suggested one inactivating NOTCH mutation to be sufficient to induce NE differentiation from non‐NE tumor cells or tumor precursors. Nevertheless, it may be possible that inactivation of more than one NOTCH receptor increases the likelihood of primary SCLC. Morimoto et al. found a significantly increased number of NE bodies (NEBs) which represent the niche for pulmonary NE stem cells – the likely origin of primary SCLC – in NOTCH double knock‐out mice.46
ASCL1 is targeted by NOTCH‐pathway inhibition which is involved in cell‐fate decisions in the lung47 and our in vitro data strongly suggest ASCL1 assisting signaling via the RB‐p53 axis in SCC development. Furthermore, the findings of Viatour et al. strengthen our hypothesis of secondary SCLC origin. They showed that NOTCH‐inhibition enhanced tumor growth in RB family member‐depleted hepatocellular carcinomas.48
Finally, reactivating NOTCH‐signaling may represent a therapy option for SCLC patients.28, 49
In summary, we here mapped a comprehensive NOTCH‐ASCL1‐RB‐p53 signaling pathway driving and maintaining the phenotype of SCCs and thereby “small cell‐ness.” We found that mutual genetic alterations in RB1 and TP53 were characteristic for a SCC phenotype. RB1 lesions were directly associated to elevated proliferation and inactivating NOTCH mutations to NE differentiation.
Likewise, our research proposes to further explore therapies interfering with the NE or “small cell‐ness” signaling molecules, such as WNT‐inhibitors. Recently published data of Hassan et al. postulated reactivation of NOTCH1 in SCLC as a therapeutic target, since this promoted cell adhesion and reduced metastasis formation by blocking EMT.28 These recent findings also strengthen our results concerning inactivating NOTCH mutations as an important genetic event in secondary SCLC.
Supporting information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Acknowledgements
We acknowledge our clinical collaborators and patients supporting the Network Genomic Medicine (www.lungcancergroup.de). K.K. is now affiliated with Labor Dr Quade & Kollegen GmbH (Cologne, Germany). L.C.H. is now affiliated with NEO New Oncology AG (Cologne, Germany). R.B. is cofounder and scientific coordinator of Targos Molecular Pathology GmbH (Kassel, Germany). R.B., L.C.H., K.K. and all other authors declare no conflict of interest.
References
- 1. Clinical Lung Cancer Genome Project (CLCGP); Network Genomic Medicine (NGM) . A genomics‐based classification of human lung tumors. Sci Transl Med 2013;5:209ra153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Epstein JI, Amin MB, Beltran H, et al. Proposed morphologic classification of prostate cancer with neuroendocrine differentiation. Am J Surg Pathol 2014;38:756–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Foulkes WD, Clarke BA, Hasselblatt M, et al. No small surprise—small‐cell carcinoma of the ovary, hypercalcaemic type is a malignant rhabdoid tumour. J Pathol 2014;233:209–14. [DOI] [PubMed] [Google Scholar]
- 4. Tudor, J , Cantley, RL , Jain, S. Primary small cell carcinoma arising from a bladder diverticulum. J Urol 2014;192:236–7. [DOI] [PubMed] [Google Scholar]
- 5. Pavithra V, Sai Shalini CN, Priya S, et al. Small cell neuroendocrine carcinoma of the cervix: a rare entity. J Clin Diagn Res 2014;8:147–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Peifer M, Fernández‐Cuesta L, Sos ML, et al. Integrative genome analyses identify key somatic driver mutations of small‐cell lung cancer. Nat Genetics 2012;44:1104–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Meuwissen R, Linn SC, Linnoila RI, et al. Induction of small cell lung cancer by somatic inactivation of both Trp53 and Rb1 in a conditional mouse model. Cancer Cell 2003; 4:181–189. [DOI] [PubMed] [Google Scholar]
- 8. Borges M, Linnoila RI, van de Velde HJ, et al. An achaete‐scute homologue essential for neuroendocrine differentiation in the lung. Nature 1997;386:852–5. [DOI] [PubMed] [Google Scholar]
- 9. Linnoila RI, Zhao B, DeMayo JL, et al. Constitutive achaete‐scute homologue‐1 promotes airway dysplasia and lung neuroendocrine tumors in transgenic mice. Cancer Res 2000;60:4005–9. [PubMed] [Google Scholar]
- 10. Demelash A, Rudrabhatla P, Pant HC, et al. Achaete‐scute homologue‐1 (ASH1) stimulates migration of lung cancer cells through Cdk5/p35 pathway. Mol Biol Cell 2012;23:2856–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sriuranpong V, Borges MW, Strock CL, et al. Notch signaling induces rapid degradation of achaete‐scute homolog 1. Mol Cellular Biol 2002;22:3129–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. South AP, Cho RJ, Aster JC. The double‐edged sword of Notch signaling in cancer. Semin Cell Dev Biol 2012;23:458–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang NJ, Sanborn Z, Arnett KL, et al. Loss‐of‐function mutations in Notch receptors in cutaneous and lung squamous cell carcinoma. Proc Natl Acad Sci USA 2011;108:17761–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Deregowski V, Gazzerro E, Priest L, et al. Role of the RAM domain and ankyrin repeats on notch signaling and activity in cells of osteoblastic lineage. J Bone Mineral Res 2006;21:1317–26. [DOI] [PubMed] [Google Scholar]
- 15. Yoo AS, Sun AX, Li L, et al. MicroRNA‐mediated conversion of human fibroblasts to neurons. Nature 2011;476:228–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Goodyear S, Sharma MC. Roscovitine regulates invasive breast cancer cell (MDA‐MB231) proliferation and survival through cell cycle regulatory protein cdk5. Exp Mol Pathol 2007;82:25–32. [DOI] [PubMed] [Google Scholar]
- 17. Pauls K, Schorle H, Jeske W, et al. Spatial expression of germ cell markers during maturation of human fetal male gonads: an immunohistochemical study. Hum Reprod 2006;21:397–404. [DOI] [PubMed] [Google Scholar]
- 18. Boehm D, von Massenhausen A, Perner S. Analysis of receptor tyrosine kinase gene amplification on the example of FGFR1. Methods Mol Biol 2015;1233:67–79. [DOI] [PubMed] [Google Scholar]
- 19. König K, Peifer M, Kröger C, et al. Implementation of amplicon parallel sequencing leads to improvement of diagnosis and therapy of lung cancer patients. J Thorac Oncol 2015;10:1049–57. [DOI] [PubMed] [Google Scholar]
- 20. Travis WD. Classification of lung cancer. Semin Roentgenol 2011;46:178–86. [DOI] [PubMed] [Google Scholar]
- 21. Beer DG, Kardia SL, Huang CC, et al. Gene‐expression profiles predict survival of patients with lung adenocarcinoma. Nat Med 2002;8:816–24. [DOI] [PubMed] [Google Scholar]
- 22. Kaderali L, Zander T, Faigle U, et al. CASPAR: a hierarchical bayesian approach to predict survival times in cancer from gene expression data. Bioinformatics 2006;22:1495–502. [DOI] [PubMed] [Google Scholar]
- 23. Linnoila RI. Functional facets of the pulmonary neuroendocrine system. Lab Invest 2006;86:425–44. [DOI] [PubMed] [Google Scholar]
- 24. Osada H, Tomida S, Yatabe Y, et al. Roles of achaete‐scute homologue 1 in DKK1 and E‐cadherin repression and neuroendocrine differentiation in lung cancer. Cancer Res 2008;68:1647–55. [DOI] [PubMed] [Google Scholar]
- 25. Anastas JN, Moon RT. WNT signalling pathways as therapeutic targets in cancer. Nat Rev Cancer 2013;13:11–26. [DOI] [PubMed] [Google Scholar]
- 26. Chau BN, Wang JY. Coordinated regulation of life and death by RB. Nat Rev Cancer 2003;3:130–8. [DOI] [PubMed] [Google Scholar]
- 27. König K, Meder L, Kröger C, et al. Loss of the keratin cytoskeleton is not sufficient to induce epithelial mesenchymal transition in a novel KRAS driven sporadic lung cancer mouse model. PLoS One 2013;8:e57996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hassan WA, Yoshida R, Kudoh S, et al. Notch1 controls cell invasion and metastasis in small cell lung carcinoma cell lines. Lung Cancer 2014;86:304–10. [DOI] [PubMed] [Google Scholar]
- 29. Sutherland KD, Proost N, Brouns I, et al. Cell of origin of small cell lung cancer: inactivation of Trp53 and Rb1 in distinct cell types of adult mouse lung. Cancer Cell 2011;19:754–64. [DOI] [PubMed] [Google Scholar]
- 30. Paripati A, Kingsley C, Weiss GJ. Pathway targets to explore in the treatment of small cell and large cell lung cancers. J Thorac Oncol 2009;4:1313–1321. [DOI] [PubMed] [Google Scholar]
- 31. Gao Y, Song C, Hui L, et al. Overexpression of RNF146 in non‐small cell lung cancer enhances proliferation and invasion of tumors through the Wnt/beta‐catenin signaling pathway. PLoS One 2014;9:e85377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rheinbay E, Suvà ML, Gillespie SM, et al. An aberrant transcription factor network essential for Wnt signaling and stem cell maintenance in glioblastoma. Cell Rep 2013;3:1567–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tan HL, Sood A, Rahimi HA, et al. Rb loss is characteristic of prostatic small cell neuroendocrine carcinoma. Clin Cancer Res 2014;20:890–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sahi H, Savola S, Sihto H, et al. RB1 gene in Merkel cell carcinoma: hypermethylation in all tumors and concurrent heterozygous deletions in the polyomavirus‐negative subgroup. APMIS 2014;122:1157–66. [DOI] [PubMed] [Google Scholar]
- 35. Burke JR, Liban TJ, Restrepo T, et al. Multiple mechanisms for E2F binding inhibition by phosphorylation of the retinoblastoma protein C‐terminal domain. J Mol Biol 2014;426:245–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Burkhart DL, Sage J. Cellular mechanisms of tumour suppression by the retinoblastoma gene. Nat Rev Cancer 2008;8:671–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature 2000;408:307–10. [DOI] [PubMed] [Google Scholar]
- 38. Futatsugi A, Utreras F, Rudrabhatla P, et al. Cyclin‐dependent kinase 5 regulates E2F transcription factor through phosphorylation of Rb protein in neurons. Cell Cycle 2012;11:1603–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pozo K, Castro‐Rivera E, Tan C, et al. The role of Cdk5 in neuroendocrine thyroid cancer. Cancer Cell 2013;24:499–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wagner PL, Kitabayashi N, Chen YT, et al. Combined small cell lung carcinomas: genotypic and immunophenotypic analysis of the separate morphologic components. Am J Clin Pathol 2009;131:376–82. [DOI] [PubMed] [Google Scholar]
- 41. Brambilla E, Moro D, Gazzeri S, et al. Cytotoxic chemotherapy induces cell differentiation in small‐cell lung carcinoma. J Clin Oncol 1991;9:50–61. [DOI] [PubMed] [Google Scholar]
- 42. D'Angelo SP, Janjigian YY, Ahye N, et al. Distinct clinical course of EGFR‐mutant resected lung cancers: results of testing of 1118 surgical specimens and effects of adjuvant gefitinib and erlotinib. J Thorac Oncol 2012;7:1815–22. [DOI] [PubMed] [Google Scholar]
- 43. Sequist LV, Waltman BA, Dias‐Santagata D, et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med 2011;3:75ra26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Alam N, Gustafson KS, Ladanyi M, et al. Small‐cell carcinoma with an epidermal growth factor receptor mutation in a never‐smoker with gefitinib‐responsive adenocarcinoma of the lung. Clin Lung Cancer 2010;11:E1–4. [DOI] [PubMed] [Google Scholar]
- 45. Kosari F, Ida CM, Aubry MC, et al. ASCL1 and RET expression defines a clinically relevant subgroup of lung adenocarcinoma characterized by neuroendocrine differentiation. Oncogene 2014;33:3776–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Morimoto M, Nishinakamura R, Saga Y, et al. Different assemblies of Notch receptors coordinate the distribution of the major bronchial Clara, ciliated and neuroendocrine cells. Development 2012;139:4365–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Collins BJ, Kleeberger W, Ball DW. Notch in lung development and lung cancer. Semin Cancer Biol 2004;14:357–64. [DOI] [PubMed] [Google Scholar]
- 48. Viatour P, Ehmer U, Saddic LA, et al. Notch signaling inhibits hepatocellular carcinoma following inactivation of the RB pathway. J Exp Med 2011;208:1963–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wael H, Yoshida R, Kudoh S, et al. Notch1 signaling controls cell proliferation, apoptosis and differentiation in lung carcinoma. Lung Cancer 2014;85:131–40. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information