

ABSTRACT
Weaver syndrome (WS) is a rare congenital disorder characterized by generalized overgrowth, macrocephaly, specific facial features, accelerated bone age, intellectual disability, and susceptibility to cancers. De novo mutations in the enhancer of zeste homolog 2 (EZH2) have been shown to cause WS. EZH2 is a histone methyltransferase that acts as the catalytic agent of the polycomb‐repressive complex 2 (PRC2) to maintain gene repression via methylation of lysine 27 on histone H3 (H3K27). Functional studies investigating histone methyltransferase activity of mutant EZH2 from various cancers have been reported, whereas WS‐associated mutations remain poorly characterized. To investigate the role of EZH2 in WS, we performed functional studies using artificially assembled PRC2 complexes containing mutagenized human EZH2 that reflected the codon changes predicted from patients with WS. We found that WS‐associated amino acid alterations reduce the histone methyltransferase function of EZH2 in this in vitro assay. Our results support the hypothesis that WS is caused by constitutional mutations in EZH2 that alter the histone methyltransferase function of PRC2. However, histone methyltransferase activities of different EZH2 variants do not appear to correlate directly with the phenotypic variability between WS patients and individuals with a common c.553G>C (p.Asp185His) polymorphism in EZH2.
Keywords: EZH2, histone methyltransferase, H3K27, Weaver syndrome, childhood cancer
Introduction
The histone methyltransferase EZH2 (enhancer of zeste homolog 2) (MIM #601573) is a key epigenetic regulator in mammals. This protein forms the catalytic subunit of the polycomb‐repressive complex 2 (PRC2) [Kuzmichev et al., 2002], and is thought to suppress gene transcription epigenetically by adding up to three methyl groups onto lysine residue 27 of histone H3 (H3K27). Epigenetic silencing via EZH2‐mediated histone methylation is further supported by the fact that some mutations at the level of H3K27 can inhibit the activity of PRC2 [Lewis et al., 2013; Pengelly et al., 2013].
Somatic mutations of EZH2 in circulating white blood cells have been shown to be extremely common in hematological malignancies [Lund et al., 2014]. Sequencing of human diffuse large B‐cell and non‐Hodgkin lymphomas revealed recurrent somatic mutations at positions Tyr646, Ala682, and Ala692 (or Tyr641, Ala677, and Ala687 in the shorter EZH2 isoform as referenced in the original publications) [Morin et al., 2010; Majer et al., 2012; McCabe et al., 2012a]. The most frequently mutated residue, tyrosine at position 646, has been reported as mutated to phenylalanine (p.Tyr646Phe), asparagine (p.Tyr646Asn), histidine (p.Tyr646His), and serine (p.Tyr646Ser) [Morin et al., 2010]. All of these heterozygous single amino acid substitutions have been shown to change the substrate specificity and favor trimethylation of H3K27 (H3K27me2 →H3K27me3) over monomethylation (H3K27me0→ H3K27me1) and dimethylation (H3K27me1→ H3K27me2). When combined with the activity of the wild‐type (WT) EZH2 copy, which has high affinity for unmethylated H3K27me0 and medium affinity for monomethylated H3K27me1, this results in an overall gain‐of‐function of EZH2 [Sneeringer et al., 2010; Yap et al., 2011]. Unlike for Tyr646, mutations at the other two sites have only been reported as specific amino acid substitutions, p.Ala682Gly [McCabe et al., 2012a] and p.Ala692Val [Majer et al., 2012].These two alterations also show gain‐of‐function activity but appear to favor different substrates. p.Ala682Gly promotes methyltransferase activity of nearly equal efficiency for all three substrates (H3K27me0/me1/me2), thereby resulting in hypertrimethylation of H3K27 [McCabe et al., 2012a]. By contrast, p,Ala692Val reduces monomethylation and enhances dimethylation of H3K27 while leaving trimethylation virtually unchanged in vitro [Majer et al., 2012], although global levels of H3K27me3 were found to be increased in a tumor‐derived p.Ala692Val mutant cell line, or when this mutant was transiently expressed in a cell line with an EZH2 WT background [Ott et al., 2014]. Given that additional data support the increased activity of EZH2 in other cancers, possibly independent of its assembly into the PRC2 complex [Xu et al., 2012], there is intense interest in developing EZH2 inhibitors as potential chemotherapeutic agents [Knutson et al., 2012; McCabe et al., 2012b; Qi et al., 2012; Knutson et al., 2012; Tan et al., 2014; Van Aller et al., 2014]. Targeting of other proteins in the complex, such as EED and SUZ12, may also become a useful therapeutic strategy [Tan et al., 2014], as may disruption of proper PRC2 complex assembly [Kim et al., 2013a]. Notably, though, inactivating mutations have been found at multiple sites throughout the EZH2 gene in myeloid disorders and acute lymphoblastic leukemia [Ernst et al., 2010; Makishima et al., 2010; Guglielmelli et al., 2011; Ntziachristos et al., 2012; Score et al., 2012; Zhang et al., 2012; The Cancer Genome Atlas Research Network, 2013], suggesting that some neoplasms may not respond well to EZH2 inhibition.
Recently, we and others have shown that de novo germline mutations in EZH2 cause Weaver syndrome (WS; MIM #277590), a rare but well‐described developmental disorder of prenatal onset that features intellectual disability, tall stature, macrocephaly, accelerated bone growth and maturation, and a susceptibility to cancers including hematological malignancies [Tatton‐Brown et al., 2011; Gibson et al., 2012; Tatton‐Brown et al., 2013; Tatton‐Brown and Rahman, 2013]. Aspects of this phenotype can be explained by the role of Ezh2 in craniofacial skeleton formation [Schwarz et al., 2014]. Though cerebral malformations were not part of the original description of WS [Weaver et al., 1974], recent clinical reports have documented the presence of neuronal migration disorders in association with physical features of WS [Freeman et al., 1999; Al‐Salem et al., 2013; Tatton‐Brown et al., 2013]. In the case reported by Al‐Salem et al. (2013), polymicrogyria was proven to be associated with a de novo mutation in EZH2, confirming the diagnosis of WS in this patient. In the more recent case reported by Tatton‐Brown et al. (2013), pachy‐ and polymicrogyria were associated with a truncating variant in EZH2, classified as likely pathogenic because parental samples were unavailable.
Given that both gain‐of‐function and loss‐of‐function mutations in EZH2 have been associated with human neoplastic disease when acquired during life in somatic cells [Ernst et al., 2010; Yap et al., 2011; McCabe et al., 2012a; Ntziachristos et al., 2012; Lund et al., 2014], we hypothesized that germline de novo mutations causative of WS would alter EZH2 activity within the PRC2 complex. We further hypothesized that more severe clinical features of WS (such as cerebral migration defects, or the development of leukemia) might be specifically associated with more significant changes in PRC2‐mediated methyltransferase activity conferred by individual mutations in EZH2.
Materials and Methods
To test our hypothesis, we designed recombinant human EZH2 proteins, had them preassembled into PRC2 complexes (BPS Bioscience, San Diego CA), and tested their activity in vitro using a well‐accepted in vitro assay [Ernst et al., 2010; Yap et al., 2011; Score et al., 2012]. Mutant EZH2–PRC2 complexes were selected for study based on the rare de novo variants observed in our original three patients [Gibson et al., 2012] and among other patients with WS identified since. Two other variants that had been observed in patients with both WS and neoplastic disease [Tatton‐Brown et al., 2011] were also selected.
Patients
Clinical data presented here were collected by the physicians who referred the patients to our study. Participating families provided informed consent, and this study was approved by the joint Clinical Research Ethics Board of the University of British Columbia and British Columbia Children's Hospital.
Sequencing
Sanger sequencing was performed on PCR products from genomic DNA. PCR primers were used to amplify the 19 coding exons of EZH2 (exons 2–20; primers available upon request) [Gibson et al., 2012]. All primer pairs were confirmed to be specific to the EZH2 gene on chromosome 7 (NC_000007.13: 148504464–148581441) rather than the pseudogene on chromosome 21 (NC_000021.8: 36971977–36972553) using the BLAST function on NCBI at http://www.ncbi.nlm.nih.gov//blast/Blast.cgi (GRCh37.p13 assembly). NCBI Reference sequence NM_004456.4 is available from GenBank. This corresponds to the longest isoform of EZH2. All nomenclature of described sequence variants is based on this sequence and follows the guidelines described in http://www.hgvs.org/mutnomen/. Population frequencies for each variant were investigated in the dbSNP database, searchable at http://www.ncbi.nlm.nih.gov/projects/SNP/, and the exome variant server, found at http://evs.gs.washington.edu/EVS/. Functional predictions for previously undescribed variants were done using the PROVEAN and SIFT tools, available at http://provean.jcvi.org/index.php (PROVEAN v1.1.3). Detailed prediction scores are provided in Supp. Table S1. All described pathogenic variants have been submitted to the LOVD locus‐specific database and can be found at http://www.lovd.nl/EZH2.
Histone Methyltransferase Assay
Core histones were purchased from EMD Millipore (13‐107; Billerica MA) and used as methyl acceptors in most of our in vitro reactions. This substrate contains a mix of all core histones and H3 peptides at different methylation states, thereby recapitulating the heterogeneity of endogenous nucleosomes. In an alternative assay, biotinylated peptides (mimicking the H3 tail, H3[21–44]) that had been unmethylated (H3K27me0), monomethylated (H3K27me1) or dimethylated (H3K27me2) were used as substrate (Supp. Fig. S1). We purchased PRC2 complexes containing WT EZH2 (51004) or mutant EZH2 from BPS Bioscience [Morin et al., 2010]. Methyltransferase assays were done using a commonly used kit (17–330; EMD Millipore) [Ernst et al., 2010; Yap et al., 2011; Score et al., 2012] as per manufacturer's instructions. We incubated 250 ng of individual HMTase complexes separately with 0.67 μM 3H‐S‐adenosyl‐methionine (3H‐SAM) (Perkin Elmer, Waltham MA) and 2 μg core histones (or 1 μM peptide), in 50 mM Tris–HCl, pH 9.0, and 0.5 mM DTT for 30 min at 30°C in a 10 μl volume. Excess volumes of 3H‐SAM, core histones, and a longer incubation were also tested (Supp. Fig. S2). In all reactions, five or eight microliters were spotted on a P81 square paper (Millipore), washed (three times with 10% tricholoracetic acid and once with 95% ethanol) to remove unincorporated 3H‐Met, air‐dried overnight, placed in a glass scintillation vial with 3 ml of scintillation fluid (ScintiSafe Econo1 SX20‐5 or Scintisafe 30% SX23‐5; Fisher Chemical, Waltham MA), and counted on a 1900TR Liquid Scintillation Analyzer (Perkin Elmer) or LS6500 Multi‐Purpose Scintillation Counter (Beckman Coulter, Brea CA). Normalized counts represent the subtraction of background counts (i.e., control tubes with no enzyme added) from total counts.
Results
Probands
Probands 1, 2, and 3 were described previously [Gibson et al., 2012]. The mutations identified in EZH2 were c.457_459delTAT (p.Tyr153del), c.2080C>T (p.His694Tyr), and c.394C>T (p.Pro132Ser), respectively.
Proband 4 was originally published in 2001 [Huffman et al., 2001]. More detailed clinical features are described in Supp. Table S2. Using Sanger sequencing, we identified a c.394C>T (p.Pro132Ser) mutation in EZH2, an alteration previously described in proband 3 from our cohort [Gibson et al., 2012] and not detected in any of the parents of probands 3 or 4. Proband 4 had a stage 4S neuroblastoma, which subsequently underwent spontaneous resolution, as was commonly observed for this type of tumor [Huffman et al., 2001]. At this time, a predisposition for neoplasm development in WS and other overgrowth syndromes was already recognized [Huffman et al., 2001]. This patient also presented with congenital heart defects. Recently, de novo mutations in histone‐modifying genes have been implicated in many cases of congenital heart disease [Zaidi et al., 2013], further supporting a role for EZH2 in this individual's phenotype.
The clinical features of proband 5 are summarized in Supp. Table S2. The most striking and unusual feature is polymicrogyria. Photographs and MRI brain images are presented in Figure 1. A clinical summary is provided in the Supp. Text along with fetal ultrasound results documenting prenatal onset of excessive growth (Supp. Table S3). We identified a c.2050C>T (p.Arg684Cys) mutation in EZH2, confirmed to be de novo by trio‐based testing at the BC Children's Hospital Clinical Molecular Genetics Lab. This mutation was previously described in four other WS patients [Tatton‐Brown et al., 2011], and thus appears to be a relatively frequent cause of WS.
Figure 1.
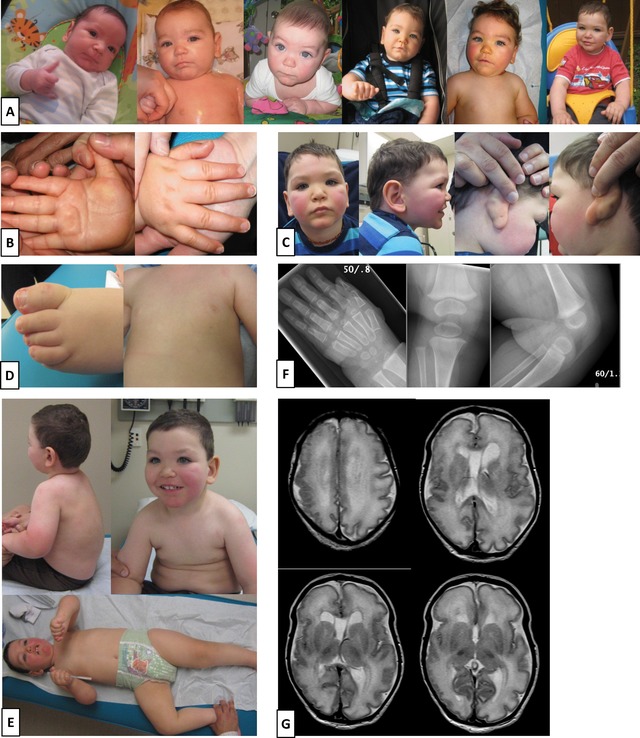
Weaver syndrome proband with polymicrogyria described in this study. A: Proband 5 is shown at 2, 4, 6, 8, 12, and 19 months. B: Both sides of the hand are shown at 12 months to illustrate the prominent palmar crease. C: At 27 months, face with prominent rosy cheeks, profile and ears are shown to confirm the dimple is only present behind the right ear. D: At 31 months, mild camptodactyly is seen on the toes and a third nipple is apparent. E: Full torso and full body are also shown at 31 months. F: X‐rays of the hand at 11½ months and the knee at 10½ months are indicative of advanced bone age. G: MRI done at 5 days of age illustrates asymmetric perisylvian polymicrogyria.
The clinical features of probands 6 and 7 are summarized in Supp. Table S2. Further clinical information is provided in the Supp. Text. In both unrelated patients, we identified a c.398A>G (p.Tyr133Cys) de novo mutation in EZH2. This mutation was predicted damaging by PROVEAN/SIFT (Supp. Table S1).
We also detected the c.553G>C (p.Asp185His) variant in EZH2 in individuals referred to our WS‐like cohort. This variant was predicted damaging by SIFT but not by PROVEAN (Supp. Table S1). Clinical features of carriers of this p.(Asp185His) variant (cases 15, 40, 53, 73, and 95) are summarized in Supp. Table S4 and further clinical information is provided in the Supp. Text. Other members of their families were tested at this locus and in all cases the variant was found to be inherited from one of the parents (Supp. Table S4). p.(Asp185His) is reported in dbSNP (rs2302427C>G) with minor allele frequencies of 8% in the 1000 Genomes phase 1 population, 6% in the Exome Variant Server, and 7.7% in a healthy ancestrally diverse cohort screened for common variants in cancer‐susceptibility genes [Bodian et al., 2014]. Because of its frequency in the general population, the p.(Asp185His) variant cannot in isolation be causative of WS.
EZH2 Mutations in WS Impair Histone Methyltransferase Activity In Vitro
In order to determine the functional impact of the EZH2 mutations observed in WS, EZH2 mutant proteins corresponding to the mutations observed in WS patients were expressed in vitro and then assembled together with other artificially expressed members of the PRC2 complex (EED/SUZ12/RbAp48 and AEBP2). Preassembly into the PRC2 complex was necessary because EZH2 requires other members of PRC2 for activity on nucleosomes [Kuzmichev et al., 2002]. The mutations studied included those identified within our cohort: p.(Tyr153del), p.(His694Tyr), p.(Pro132Ser), p.(Arg684Cys), and p.(Tyr133Cys). The p.(Ala682Thr) and p.(Glu745Lys) mutations were also of interest because of their associations with neuroblastoma, acute lymphoblastic leukemia, and lymphoma in WS patients [Tatton‐Brown et al., 2011]. As mentioned above, we also included the common variant p.(Asp185His). WT EZH2 was used as a positive control, and the methyltransferase‐inactive mutant EZH2 p.(Phe672Ile) (equivalent to the inactive fly mutant allele E(z)son 1 described in Joshi et al. [2008]) was used as a negative control. We then measured incorporation of tritiated methyl groups from 3H‐SAM onto mixed core histones in the presence of each EZH2‐PRC2 complex (Fig. 2; Supp. Fig. S3 and Supp. Table S5A). As expected, WT EZH2–PRC2 complex catalyzed the incorporation of 3H into core histones, consistent with the model whereby it uses 3H‐SAM as the methyl donor and nucleosomes as the recipient substrates for histone methylation [Kuzmichev et al., 2002]. In contrast, PRC2 complexes containing WS‐associated EZH2 mutants showed reduced histone methyltransferase activity in vitro (Fig. 2; Supp. Fig. S3 and Supp. Table S5A), suggesting that EZH2 mutations associated with WS are loss‐of‐function (hypomorphic) mutations.
Figure 2.
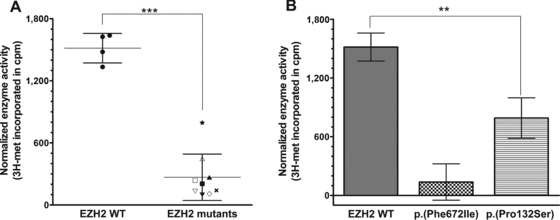
Weaver syndrome mutants are impaired in their histone methyltransferase activity in vitro. Histone methyltransferase reactions were performed using 2 μg purified core histones and 0.67 μM 3H‐S‐adenosyl‐methionine (3H‐SAM). Each reaction was incubated with 250 ng of either wild‐type (WT) or a mutant HMTase complex (or no enzyme controls). Histone methyltransferase activity was measured based on the incorporation of 3H‐labeled methyl groups, represented in scintillation counts per minute. Counts were normalized by subtracting background counts (i.e., no enzyme) from the total counts. A: Incorporation of tritiated methyl groups from 3H‐SAM onto core histones is shown for each complex: EZH2 WT •, p.(Phe672Ile) ×, p.(Pro132Ser) ★, p.(Tyr153del) △, p.(His694Tyr) ▽, p.(Glu745Lys) ▴, p.(Ala682Thr) ▾, p.(Arg684Cys) ▪, p.(Tyr133Cys) □, and p.(Asp185His) ◇. Error bars represent standard deviation (SD) within the groups “EZH2 WT” and “EZH2 mutants.” Unpaired t‐test showed statistically significant difference between the two groups (P value < 0.0001). B: Incorporation of tritiated methyl groups from 3H‐SAM onto core histones is shown for the positive control EZH2 WT, the negative control EZH2 (p.Phe672Ile), and the mutant complex with activity closest to WT, namely, EZH2 (p.Pro132Ser). Error bars represent SD of four independent replicates for the controls, and three independent replicates for the mutant EZH2 (p.Pro132Ser). One‐way ANOVA showed statistically significant difference between all groups (overall P value < 0.0001; P values between WT and p.(Phe672Ile), between p.(Phe672Ile) and p.(Pro132Ser), and between WT and p.(Pro132Ser) were all <0.05).
The Common p.(Asp185His) Variant Also Appears to Impair Histone Methyltransferase Activity In Vitro
Surprisingly, EZH2 p.(Asp185His) also showed impaired histone methyltransferase activity in this in vitro assay (Fig. 2A; Supp. Fig. S3). Based on the frequency of this variant and the rarity of WS, p.(Asp185His) cannot by itself be causative of WS. However, based on the number of replicates we performed under varied conditions (Supp. Fig. S2 and Supp. Tables S5B and C), we believe this result to be reproducible and to reflect accurately the activity of this enzyme variant under these artificial conditions.
Discussion
Expanding the Phenotype of WS to Include Neuronal Migration Disorders
Brain imaging in proband 5 (Fig. 1G) was consistent with that reported on two prior occasions in different children with WS [Freeman et al., 1999; Al‐Salem et al., 2013]. In each previous case, and in our case, there was asymmetric perisylvian polymicrogyria that appeared more severe on the right side, as well as mildly enlarged lateral ventricles. The report by Freeman et al. (1999) described pachygyria, but based on review of the published images (W. Dobyns), we believe the findings are more consistent with perisylvian polymicrogyria. The image shown in the report by Al‐Salem et al. (2013) demonstrates enlarged extra‐axial fluid spaces over the brain, similar in appearance to the polymicrogyria observed among megalencephaly syndromes associated with PI3K‐AKT pathway mutations [Mirzaa et al., 2012]. However, neither hydrocephalus nor Chiari malformations were seen in these WS patients. Tatton‐Brown et al. (2013) also reported a case with pachy‐ and polymicrogyria, but no images were published.
Our proband 5 has the recurrent p.(Arg684Cys) de novo mutation, the patient with polymicrogyria and WS described by Al‐Salem et al. (2013) was shown to have a de novo p.(Glu745Lys) mutation, and the patient from Tatton‐Brown et al. (2013) had an EZH2 variant predicted to truncate the protein at position 732. The association of polymicrogyria with WS in four independent cases, two of which have molecular confirmation of de novo mutations in different exons of EZH2 and another of which has a truncating variant in the last exon, strongly supports a causal association between EZH2 mutations and neuronal migration defects in some patients with WS. EZH2 has been shown to control the decision between self‐renewal and differentiation in the cerebral cortex, and inhibition of PRC2 complex activity had been shown to shift the balance toward differentiation [Pereira et al., 2010]. Furthermore, EZH2 has also been shown to orchestrate neuronal migration in the cortico‐ponto‐cerebellar pathway in mice [Di Meglio et al., 2013]. The possibility that diminished PRC2 complex activity could lead to premature neuronal differentiation, possibly at ectopic sites along the normal migration pathway, offers a plausible explanation for the cortical patterning defects seen in patients with WS and pachy‐ or polymicrogyria.
Thus, in addition to their known risk for neoplastic disease, patients with WS should be considered to be at risk for neuronal migration disorders, and physicians should have a low threshold for ordering cranial imaging studies. Similarly, children with overgrowth and cerebral migration disorders could be tested for rare variants in EZH2, and physicians performing prenatal diagnosis in the context of a fetus with polymicrogyria should consider the possibility of WS.
Given the large number of individuals now being studied with high‐throughput next‐generation sequencing, rare variants in EZH2 that are discovered through targeted sequencing panels, exome sequencing, or whole‐genome sequencing should be considered carefully in the context of clinical findings such as overgrowth phenotypes, cerebral malformations, and neoplastic disease. For highly heterogeneous disorders such as overgrowth syndromes, techniques such as exome sequencing are becoming cost‐effective for diagnosis at an early stage of the workup. An estimate of diagnostic costs incurred during the workup of proband 5 is presented in Supp. Table S6, for theoretical comparison to early exome sequencing (though in his particular case, EZH2 was selected on a candidate gene basis).
WS Mutations and Neoplastic Disease
With somatic mutations in EZH2 having been associated to both gain and loss of histone methyltransferase function, it was important to investigate mutations found in WS patients who had also developed malignancies. To date, none of the three cases from our original report have been diagnosed with neoplastic disease. However, proband 4 had a nonmetastatic stage 4S neuroblastoma in his left adrenal gland. Our functional analysis of the EZH2 p.(Pro132Ser) mutant complex suggested a loss‐of‐function effect, consistent with a previous report of this variant in the context of myeloid disorders [Guglielmelli et al., 2011]. Proband 6 had a prenatal neuroblastoma in the right adrenal gland that was successfully removed surgically shortly after birth, and the EZH2 p.(Tyr133Cys) mutant complex also appeared to be loss‐of‐function in our assay. The other two mutant complexes containing mutations found in patients with malignancies, EZH2 p.(Arg682Thr), and p.(Glu745Lys) [Tatton‐Brown et al., 2011], also showed loss‐of‐function in vitro, suggesting that the mechanism driving cancer in WS patients resembles that of myeloid disorders and acute lymphoblastic leukemia rather than that of diffuse large B‐cell and non‐Hodgkin lymphomas. Furthermore, the p.(Arg684Cys) mutation reported in several independent cases [Tatton‐Brown et al., 2011] and identified in proband 5, which has not yet been associated with malignancy development in WS but appears to be a true recurrent mutation, had already been described as likely inactivating in myeloid disorders [Ernst et al., 2010]. Overall, all de novo WS‐associated EZH2 mutations showed impaired histone methyltransferase activity in vitro, particularly with reduced ability to monomethylate H3K27 (Supp. Fig. S1 and Supp. Table S5D). Impaired histone methyltransferase activity had previously been observed among NSD1 mutations causing Sotos syndrome (MIM #117550) [Qiao et al., 2011; Kudithipudi et al., 2014], which is another overgrowth syndrome that shares significant phenotypic overlap with WS [Tatton‐Brown and Rahman, 2013]. Based on these results, we suggest that EZH2 inhibitors currently being developed against various cancers [Knutson et al., 2012; McCabe et al., 2012b; Qi et al., 2012; Knutson et al., 2013] may not be of specific benefit in WS. Importantly, we did not assay PRC2‐independent functions of EZH2, so additional complexity in the functional effects of disease‐associated EZH2 mutations remains to be unraveled.
Methyltransferase Activity of p.(Asp185His)
We identified the p.(Asp185His) variant in five individuals who were referred for WS‐like features, including macrocephaly. This variant has been found in multiple healthy controls, as well as among individuals affected with generalized overgrowth or acute leukemia [Tatton‐Brown et al., 2011; Grossmann et al., 2012]. Importantly, recent genome‐wide association studies for human height and for infant head circumference have not identified this variant as a risk factor for either of these quantitative traits [Taal et al., 2012; Wood et al., 2014]. Thus, population genetic evidence supports the classification of the p.(Asp185His) variant as a common, benign SNP (single‐nucleotide polymorphism). In this context, the fact that our functional work suggests impaired histone methyltransferase activity for this mutant is notable. Based on careful repetition of our assays with different lot numbers of the WT protein and under varied conditions (Fig. 2; Supp. Figs. S2 and S3), we do believe our in vitro results to be reproducible. Given the reproducibility of our assay in our hands and the use of a similar assay by multiple other groups, we do not believe the low methyltransferase activity exhibited by the p.(Asp185His) protein variant in this assay is indicative of experimental error. Rather, we believe that our results reflect the true activity of this variant under this select set of experimental conditions. Nevertheless, given the lack of a specific phenotype for p.(Asp185His) carriers, these in vitro results may not reflect its true activity in vivo. On this basis, we must conclude that this particular in vitro assay cannot be used in isolation to assess the potential pathogenicity of novel EZH2 variants. Instead, pathogenicity should be assessed based on the sum total of available evidence from family‐specific cosegregation with disease phenotypes, population genetics, and, where available, other orthogonal lines of functional evidence. The possibility of “pseudodeficiency alleles” is an uncommon but known phenomenon whereby some protein variants manifest impaired activity by in vitro assays but have no demonstrable phenotypic effects in vivo [Coulter‐Mackie and Gagnier, 2003; Yasuda et al., 2003; Tomatsu et al., 2009]. To understand the discrepancy between the predicted (normal) activity of a common protein variant and its observed (deficient) activity more fully would require more definitive studies such as determination of the binding constant (K m) for substrates, assays over the linear portion of the product versus time curve, and careful substudies of the different enzymatic steps for a variety of different rare and common protein variants.
Histone Methyltransferase Activity Does Not Correlate with Phenotypic Severity
We chose to assay EZH2 protein variants that represented a wide variety of WS phenotypes. We had hypothesized that more severe clinical features of WS (such as cerebral migration defects, or the development of malignancy) might be associated with mutations in specific protein domains that were in turn associated with more striking alterations of histone methyltransferase activity. However, we observed no clear correlation between these parameters. We also observed no correlation between clinical severity and profiles of substrate specificity (Supp. Fig. S1). Our results are consistent with Guglielmelli et al. (2011) who observed no correlation between EZH2 mutational status and hematologic or clinical parameters in patients with myelofibrosis. This lack of phenotype/genotype correlation suggests that factors apart from histone methyltransferase function (such as the presence of modifier genes, other epigenetic modifications such as H3K4 methylation, or stochastic modifiers) might explain the phenotypic differences observed in WS patients. Activity of accessory proteins that are absent from our in vitro assay might also change the conformation of the PRC2 complex and influence the resulting phenotype. Such factors include the presence of PHF1 [Sarma et al., 2008], the activity of NF‐kB [Lee et al., 2011], WNT [Wang et al., 2010], or Akt, that modifies EZH2 post‐translationally [Cha et al., 2005]. Alternatively, the activity of WS mutants on nonhistone substrates, such as STAT3 [Kim et al., 2013b] or JARID2 [Sanulli et al., 2015], may be a more important determinant of the ultimate phenotype of WS patients.
Supporting information
Disclaimer: Supplementary materials have been peer‐reviewed but not copyedited.
Supporting Information
Acknowledgments
We would like to thank the families for agreeing to participate in our study. We are also extremely grateful for the expert comments from Dr. Gregg Morin regarding our enzymatic assay. Further, we gratefully acknowledge the assistance of Drs. Brett Casey and Tanya Nelson in the clinical testing of EZH2 mutations among proband 5 and his family members, as well as Dr. Kym M. Boycott and Rebecca L. Hood for testing of EZH2 in proband 6. This work was funded by the Government of Canada through the Canadian Institutes of Health Research (CIHR MOP‐119595). WTG received salary support via Clinician Scientist Awards from the CIHR and from the Child and Family Research Institute. ASAC is supported by a Doctoral Grant from the Fundação para a Ciência e a Tecnologia (Government of Portugal and European Union joint grant).
Contract grant sponsors: Canadian Institutes of Health Research (CIHR MOP‐119595).
Communicated by Mark H. Paalman
References
- Al‐Salem A, Alshammari MJ, Hassan H, Alazami AM, Alkuraya FS. 2013. Weaver syndrome and defective cortical development: a rare association. Am J Med Genet A 161A:225–227. [DOI] [PubMed] [Google Scholar]
- Bodian DL, McCutcheon JN, Kothiyal P, Huddleston KC, Iyer RK, Vockley JG, Niederhuber JE. 2014. Germline variation in cancer‐susceptibility genes in a healthy, ancestrally diverse cohort: implications for individual genome sequencing. PLoS One 9:e94554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha TL, Zhou BP, Xia W, Wu Y, Yang CC, Chen CT, Ping B, Otte AP, Hung MC. 2005. Akt‐mediated phosphorylation of EZH2 suppresses methylation of lysine 27 in histone H3. Science 310:306–310. [DOI] [PubMed] [Google Scholar]
- Coulter‐Mackie MB, Gagnier L. 2003. Spectrum of mutations in the arylsulfatase A gene in a Canadian DNA collection including two novel frameshift mutations, a new missense mutation (C488R) and an MLD mutation (R84Q) in cis with a pseudodeficiency allele. Mol Genet Metab 79:91–98. [DOI] [PubMed] [Google Scholar]
- Di Meglio T, Kratochwil CF, Vilain N, Loche A, Vitobello A, Yonehara K, Hrycaj SM, Roska B, Peters AHFM, Eichmann A, Wellik D, Ducret S, et al. 2013. Ezh2 orchestrates topographic migration and connectivity of mouse precerebellar neurons. Science 339:204–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst T, Chase AJ, Score J, Hidalgo‐Curtis CE, Bryant C, Jones AV, Waghorn K, Zoi K, Ross FM, Reiter A, Hochhaus A, Drexler HG, et al. 2010. Inactivating mutations of the histone methyltransferase gene EZH2 in myeloid disorders. Nat Genet 42:722–726. [DOI] [PubMed] [Google Scholar]
- Freeman BM, Hoon AH Jr, Breiter SN, Hamosh A. 1999. Pachygyria in Weaver syndrome. Am J Med Genet 86:395–397. [PubMed] [Google Scholar]
- Gibson WT, Hood RL, Zhan SH, Bulman DE, Fejes AP, Moore R, Mungall AJ, Eydoux P, Babul‐Hirji R, An J, Marra MA, FORGE Canada Consortium , et al. 2012. Mutations in EZH2 cause Weaver syndrome. Am J Hum Genet 90:110–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guglielmelli P, Biamonte F, Score J, Hidalgo‐Curtis C, Cervantes F, Maffioli M, Fanelli T, Ernst T, Winkelman N, Jones AV, Zoi K, Reiter A, et al. 2011. EZH2 mutational status predicts poor survival in myelofibrosis. Blood 118:5227–5234. [DOI] [PubMed] [Google Scholar]
- Grossmann V, Bacher U, Kohlmann A, Artusi V, Klein HU, Dugas M, Schnittger S, Alpermann T, Kern W, Haferlach T, Haferlach C. 2012. EZH2 mutations and their association with PICALM‐MLLT10 positive acute leukaemia. Br J Haematol 157:387–390. [DOI] [PubMed] [Google Scholar]
- Huffman C, McCandless D, Jasty R, Matloub J, Robinson HB, Weaver DD, Cohen MM Jr. 2001. Weaver syndrome with neuroblastoma and cardiovascular anomalies. Am J Med Genet 99:252–255. [DOI] [PubMed] [Google Scholar]
- Joshi P, Carrington EA, Wang L, Ketel CS, Miller EL, Jones RS, Simon JA. 2008. Dominant alleles identify SET domain residues required for histone methyltransferase of polycomb repressive complex 2. J Biol Chem 283:27757–27766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim W, Bird GH, Neff T, Guo G, Kerenyi MA, Walensky LD, Orkin SH. 2013a. Targeted disruption of the EZH2‐EED complex inhibits EZH2‐dependent cancer. Nat Chem Biol 9:643–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E, Kim M, Woo DH, Shin Y, Shin J, Chang N, Oh YT, Kim H, Rheey J, Nakano I, Lee C, Joo KM, et al. 2013b. Phosphorylation of EZH2 Activates STAT3 Signaling via STAT3 Methylation and Promotes Tumorigenicity of Glioblastoma Stem‐like Cells. Cancer Cell 23:839–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knutson SK, Wigle TJ, Warholic NM, Sneeringer CJ, Allain CJ, Klaus CR, Sacks JD, Raimondi A, Majer CR, Song J, Porter Scott M, Jin L, et al. 2012. A selective inhibitor of EZH2 blocks H3K27 methylation and kills mutant lymphoma cells. Nat Chem Biol 8:890–896. [DOI] [PubMed] [Google Scholar]
- Knutson SK, Warholic NM, Wigle TJ, Klaus CR, Allain CJ, Raimondi A, Porter Scott M, Chesworth R, Moyer MP, Copeland RA, Richon VM, Pollock RM, et al. 2013. Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc Natl Acad Sci USA 110:7922–7927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudithipudi S, Lungu C, Rathert P, Happel N, Jeltsch A. 2014. Substrate specificity analysis and novel substrates of the protein lysine methyltransferase NSD1. Chem Biol 21:226–237. [DOI] [PubMed] [Google Scholar]
- Kuzmichev A, Nishioka K, Erdjument‐Bromage H, Tempst P, Reinberg D. 2002. Histone methyltransferase activity associated with a human multiprotein complex containing the enhancer of Zeste protein. Genes Dev 16:2893–2905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee ST, Li Z, Wu Z, Aau M, Guan P, Karuturi RK, Liou YC, Yu Q. 2011. Context‐specific regulation of NF‐kappaB target gene expression by EZH2 in breast cancers. Mol Cell 43:798–810. [DOI] [PubMed] [Google Scholar]
- Lewis PW, Muller MM, Koletsky MS, Cordero F, Lin S, Banaszynski LA, Garcia BA, Muir TW, Becher OJ, Allis CD. 2013. Inhibition of PRC2 activity by a gain‐of‐function H3 mutation found in pediatric glioblastoma. Science 340:857–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund K, Adams PD, Copland M. 2014. EZH2 in normal and malignant hematopoiesis. Leukemia 28:44–49. [DOI] [PubMed] [Google Scholar]
- Majer CR, Jin L, Scott MP, Knutson SK, Kuntz KW, Keilhack H, Smith JJ, Moyer MP, Richon VM, Copeland RA, Wigle TJ. 2012. A687V EZH2 is a gain‐of‐function mutation found in lymphoma patients. FEBS Lett 586:3448–3451. [DOI] [PubMed] [Google Scholar]
- Makishima H, Jankowska AM, Tiu RV, Szpurka H, Sugimoto Y, Hu Z, Saunthararajah Y, Guinta K, Keddache MA, Putnam P, Sekeres MA, Moliterno AR, et al. 2010. Novel homo‐ and hemizygous mutations in EZH2 in myeloid malignancies. Leukemia 24:1799–1804. [DOI] [PubMed] [Google Scholar]
- McCabe MT, Graves AP, Ganji G, Diaz E, Halsey WS, Jiang Y, Smitheman KN, Ott HM, Pappalardi MB, Allen KE, Chen SB, Della Pietra A 3rd, et al. 2012a. Mutation of A677 in histone methyltransferase EZH2 in human B‐cell lymphoma promotes hypertrimethylation of histone H3 on lysine 27 (H3K27). Proc Natl Acad Sci USA 109:2989–2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCabe MT, Ott HM, Ganji G, Korenchuk S, Thompson C, Van Aller GS, Liu Y, Graves AP, Della Pietra A 3rd, Diaz E, LaFrance LV, Mellinger M, et al. 2012b. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2‐activating mutations. Nature 492:108–112. [DOI] [PubMed] [Google Scholar]
- Mirzaa GM, Conway RL, Gripp KW, Lerman‐Sagie T, Siegel DH, deVries LS, Lev D, Kramer N, Hopkins E, Graham JM Jr, Dobyns WB. 2012. Megalencephaly‐capillary malformation (MCAP) and megalencephaly‐polydactyly‐polymicrogyria‐hydrocephalus (MPPH) syndromes: two closely related disorders of brain overgrowth and abnormal brain and body morphogenesis. Am J Med Genet A 158A:269–291. [DOI] [PubMed] [Google Scholar]
- Morin RD, Johnson NA, Severson TM, Mungall AJ, An J, Goya R, Paul JE, Boyle M, Woolcock BW, Kuchenbauer F, Yap D, Humphries RK, et al. 2010. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B‐cell lymphomas of germinal‐center origin. Nat Genet 42:181–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ntziachristos P, Tsirigos A, Van Vlierberghe P, Nedjic J, Trimarchi T, Flaherty MS, Ferres‐Marco D, da Ros V, Tang Z, Siegle J, Asp P, Hadler M, et al. 2012. Genetic inactivation of the polycomb repressive complex 2 in T cell acute lymphoblastic leukemia. Nat Med 18:298–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott HM, Graves AP, Pappalardi MB, Huddleston M, Halsey WS, Hughes AM, Groy A, Dul E, Jiang Y, Bai Y, Annan R, Verma SK, et al. 2014. A687V EZH2 is a driver of histone H3 lysine 27 (H3K27) hypertrimethylation. Mol Cancer Ther 13:3062–3073. [DOI] [PubMed] [Google Scholar]
- Pengelly AR, Copur O, Jackle H, Herzig A, Muller J. 2013. A histone mutant reproduces the phenotype caused by loss of histone‐modifying factor Polycomb. Science 339:698–699. [DOI] [PubMed] [Google Scholar]
- Pereira JD, Sansom SN, Smith J, Dobenecker MW, Tarakhovsky A, Livesey FJ. 2010. Ezh2, the histone methyltransferase of PRC2, regulates the balance between self‐renewal and differentiation in the cerebral cortex. Proc Natl Acad Sci USA 107:15957–15962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi W, Chan H, Teng L, Li L, Chuai S, Zhang R, Zeng J, Li M, Fan H, Lin Y, Gu J, Ardayfio O, et al. 2012. Selective inhibition of Ezh2 by a small molecule inhibitor blocks tumor cells proliferation. Proc Natl Acad Sci USA 109:21360–21365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao Q, Li Y, Chen Z, Wang M, Reinberg D, Xu RM. 2011. The structure of NSD1 reveals an autoregulatory mechanism underlying histone H3K36 methylation. J Biol Chem 286:8361–8368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanulli S, Justin N, Teissandier A, Ancelin K, Portoso M, Caron M, Michaud A, Lombard B, da Rocha ST, Offer J, Loew D, Servant N, et al. 2015. Jarid2 methylation via the PRC2 complex regulates H3K27me3 deposition during cell differentiation. Mol Cell 57:769–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarma K, Margueron R, Ivanov A, Pirrotta V, Reinberg D. 2008. Ezh2 requires PHF1 to efficiently catalyze H3 lysine 27 trimethylation in vivo. Mol Cell Biol 28:2718–2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz D, Varum S, Zemke M, Schöler A, Baggiolini A, Draganova K, Koseki H, Schübeler D, Sommer L. 2014. Ezh2 is required for neural crest‐derived cartilage and bone formation. Development 141:867–877. [DOI] [PubMed] [Google Scholar]
- Score J, Hidalgo‐Curtis C, Jones AV, Winkelmann N, Skinner A, Ward D, Zoi K, Ernst T, Stegelmann F, Döhner K, Chase A, Cross NC. 2012. Inactivation of polycomb repressive complex 2 components in myeloproliferative and myelodysplastic/myeloproliferative neoplasms. Blood 119:1208–1213. [DOI] [PubMed] [Google Scholar]
- Sneeringer CJ, Scott MP, Kuntz KW, Knutson SK, Pollock RM, Richon VM, Copeland RA. 2010. Coordinated activities of wild‐type plus mutant EZH2 drive tumor‐associated hypertrimethylation of lysine 27 on histone H3 (H3K27) in human B‐cell lymphomas. Proc Natl Acad Sci USA 107:20980–20985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taal HR, St Pourcain B, Thiering E, Das S, Mook‐Kanamori DO, Warrington NM, Kaakinen M, Kreiner‐Møller E, Bradfield JP, Freathy RM, Geller F, Guxens M, et al. 2012. Common variants at 12q15 and 12q24 are associated with infant head circumference. Nat Genet 44:532–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan JZ, Yan Y, Wang XX, Jiang Y, Xu HE. 2014. EZH2: biology, disease, and structure‐based drug discovery. Acta Pharmacol Sin 35:161–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatton‐Brown K, Hanks S, Ruark E, Zachariou A, Duarte S del V, Ramsay E, Snape K, Murray A, Perdeaux ER, Seal S, Loveday, C , Banka S, et al. 2011. Germline mutations in the oncogene EZH2 cause Weaver syndrome and increased human height. Oncotarget 2:1127–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatton‐Brown K, Murray A, Hanks S, Douglas J, Armstrong R, Banka S, Bird LM, Clericuzio CL, Cormier‐Daire V, Cushing T, Flinter F, Jacquemont ML, et al. 2013. Weaver syndrome and EZH2 mutations: clarifying the clinical phenotype. Am J Med Genet A 161A:2972–2980. [DOI] [PubMed] [Google Scholar]
- Tatton‐Brown, K. , Rahman, N. 2013. The NSD1 and EZH2 overgrowth genes, similarities and differences. Am J Med Genet C 163C:86–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Research Network . 2013. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med 368:2059–2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomatsu S, Montaño AM, Dung VC, Grubb JH, Sly WS. 2009. Mutations and polymorphisms in GUSB gene in mucopolysaccharidosis VII (Sly Syndrome). Hum Mutat 30:511–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Aller GS, Pappalardi MB, Ott HM, Diaz E, Brandt M, Schwartz BJ, Miller WH, Dhanak D, McCabe MT, Verma SK, Creasy CL, Tummino PJ, et al. 2014. Long residence time inhibition of EZH2 in activated polycomb repressive complex 2. ACS Chem Biol 9:622–629. [DOI] [PubMed] [Google Scholar]
- Wang L, Jin Q, Lee JE, Su IH, Ge K. 2010. Histone H3K27 methyltransferase Ezh2 represses Wnt genes to facilitate adipogenesis. Proc Natl Acad Sci USA 107:7317–7322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver DD, Graham CB, Thomas IT, Smith DW. 1974. A new overgrowth syndrome with accelerated skeletal maturation, unusual facies, and camptodactyly. J Pediatr 84:547–552. [DOI] [PubMed] [Google Scholar]
- Wood AR, Esko T, Yang J, Vedantam S, Pers TH, Gustafsson S, Chu AY, Estrada K, Luan J, Kutalik Z, Amin N, Buchkovich ML, et al. 2014. Defining the role of common variation in the genomic and biological architecture of adult human height. Nat Genet 46:1173–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu K, Wu ZJ, Groner AC, He HH, Cai C, Lis RT, Wu X, Stack EC, Loda M, Liu T, Xu H, Cato L, et al. 2012. EZH2 oncogenic activity in castration‐resistant prostate cancer cells is polycomb‐independent. Science 338:1465–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap DB, Chu J, Berg T, Schapira M, Cheng SW, Moradian A, Morin RD, Mungall AJ, Meissner B, Boyle M, Marquez VE, Marra MA, et al. 2011. Somatic mutations at EZH2 Y641 act dominantly through a mechanism of selectively altered PRC2 catalytic activity, to increase H3K27 trimethylation. Blood 117:2451–2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda M, Shabbeer J, Benson SD, Maire I, Burnett RM, Desnick RJ. 2003. Fabry disease: characterization of alpha‐galactosidase A double mutations and the D313Y plasma enzyme pseudodeficiency allele. Hum Mutat 22:486–492. [DOI] [PubMed] [Google Scholar]
- Zaidi S, Choi M, Wakimoto H, Ma L, Jiang J, Overton JD, Romano‐Adesman A, Bjornson RD, Breitbart RE, Brown KK, Carriero NJ, Cheung, YH , et al. 2013. De novo mutations in histone‐modifying genes in congenital heart disease. Nature 498:220–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Ding L, Holmfeldt L, Wu G, Heatley SL, Payne‐Turner D, Easton J, Chen X, Wang J, Rusch M, Lu C, Chen SC, et al. 2012. The genetic basis of early T‐cell precursor acute lymphoblastic leukaemia. Nature 481:157–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Disclaimer: Supplementary materials have been peer‐reviewed but not copyedited.
Supporting Information