

Abstract
Cathepsin B is a cysteine proteinase that primarily functions as an endopeptidase within endolysosomal compartments in normal cells. However, during tumoral expansion, the regulation of cathepsin B can be altered at multiple levels, thereby resulting in its overexpression and export outside of the cell. This may suggest a possible role of cathepsin B in alterations leading to cancer progression. The aim of this study was to determine the contribution of intracellular and extracellular cathepsin B in growth, tumorigenesis, and invasion of colorectal cancer (CRC) cells. Results show that mRNA and activated levels of cathepsin B were both increased in human adenomas and in CRCs of all stages. Treatment of CRC cells with the highly selective and non‐permeant cathepsin B inhibitor Ca074 revealed that extracellular cathepsin B actively contributed to the invasiveness of human CRC cells while not essential for their growth in soft agar. Cathepsin B silencing by RNAi in human CRC cells inhibited their growth in soft agar, as well as their invasion capacity, tumoral expansion, and metastatic spread in immunodeficient mice. Higher levels of the cell cycle inhibitor p27Kip1 were observed in cathepsin B‐deficient tumors as well as an increase in cyclin B1. Finally, cathepsin B colocalized with p27Kip1 within the lysosomes and efficiently degraded the inhibitor. In conclusion, the present data demonstrate that cathepsin B is a significant factor in colorectal tumor development, invasion, and metastatic spreading and may, therefore, represent a potential pharmacological target for colorectal tumor therapy. © 2015 The Authors. Molecular Carcinogenesis, published by Wiley Periodicals, Inc.
Keywords: cathepsin B, colorectal cancers, invasion, intestinal tumorigenesis, p27Kip1
Abbreviations
- APC
adenomatous polyposis coli
- CDK
cyclin‐dependent kinase
- CHX
cycloheximide
- CRC
colorectal cancer
- CTSB
cathepsin B
- ERK
extracellular signal regulated kinase
- FCS
fetal calf serum
- HEK
human embryonic kidney
- IEC
intestinal epithelial cells
- Kip1
kinase inhibitory protein 1
- Kip2
kinase inhibitory protein 2
- MAPK
mitogen‐activated protein kinase
- Min
multiple intestinal neoplasia
- MMP
matrix metalloproteases
- RNAi
RNA interference
- uPA
urokinase‐type plasminogen activator
- Wnt
Wingless
INTRODUCTION
Colorectal cancer (CRC) is one of the major malignancies worldwide and the second leading cause of cancer death in North America. CRC develops through multiple steps, with the sequential acquisition of genetic alterations in key tumor suppressors and oncogenes. This knowledge has been largely derived from early research on CRC 1. The phenotypic hallmarks of cancer cells include the ability to invade and metastasize which depends on the action of proteases actively taking center stage in extracellular proteolysis during cancer progression 2. Of all the proteases, the cysteine protease cathepsin B is of significant importance as it is involved in various pathologies and oncogenic processes 3. Cathepsin B primarily functions as an endopeptidase within endolysosomal compartments in normal cells. However, during malignant transformation, the regulation of cathepsin B can be altered at multiple levels thereby resulting in its overproduction 3, 4. Mechanisms that increase cathepsin B expression in tumors and in tumor‐associated cells include genetic amplification or alternative splicing 4. Moreover, in tumors, cathepsin B can either be secreted, bound to the cell membrane or released by shedding vesicles 4. Expression and redistribution of active cathepsin B to the basal plasma membrane occurs in late colon adenomas 5, 6 coincident with the activation of KRAS 1. In line with these results, Cavallo‐Medved et al. 7 have demonstrated that trafficking of cathepsin B to caveolae and its secretion are regulated by active KRAS in CRC cells in culture. Accordingly, secretion of cathepsin B is increased in the extracellular environment of CRC 8, 9 where it is suspected to play an essential role in disrupting extracellular matrix barriers between tumors and surrounding tissue, thereby facilitating invasion and metastasis 10, 11, 12. Altogether, these data are consistent with the link between cathepsin B protein expression in colorectal carcinomas and shortened patient survival 6.
In a recent prospective cohort study of 558 men and women with colonic tumors accessible for immunohistochemical assessment, Chan et al. 13 found that 82% of patients had tumors that expressed cathepsin B, irrespective of stage, while the remaining 18% had tumors that did not express cathepsin B. Moreover, other studies have suggested that cathepsin B expression or activity may actually peak during early stage cancer and subsequently decline with advanced disease 14, 15. Thus, the above data point to a possible role of cathepsin B in both early and late alterations leading to tumor formation in the colon.
In light of the above, the present study used two strategies to specifically counteract the action of cathepsin B. The first involved the use of RNA interference (RNAi) to inhibit the expression of cathepsin B protein into CRC cells while the second approach employed the highly selective cathepsin inhibitor Ca074 to block extracellular cathepsin B activity. Results suggest that extracellular cathepsin B is involved in cell invasion whereas intracellular cathepsin B controls tumoral properties of CRC cells. Of further importance, biochemical analysis suggests that intracellular cathepsin B regulates tumorigenesis by degrading the p27Kip1 cell cycle inhibitor.
MATERIALS AND METHODS
Materials
Antibodies
Primary antibodies were obtained from the following sources: Na+/K+ ATPase‐α, calnexin, calpain 2, CDK2, cyclin B1, cyclin E, ERK2, lamin B, HA tag, p21, p27Kip1, p57Kip2, β‐tubulin from Santa Cruz Biotechnology (Santa Cruz, CA), β‐actin from Chemicon International (Billerica, MA), phospho‐AKT (Ser473), AKT, phosphorylated ERK1/2 (T202/Y204), and Lamp1 from Cell Signaling Technology (Danvers, MA) and Cathepsin B from R&D Systems (Mississauga, ON, Canada). Mouse and rabbit horseradish peroxidase antibodies were purchased from Amersham Biosciences (Pittsburg, PA), goat horseradish peroxidase antibody from Santa Cruz, and alkaline phosphatase‐conjugated antibodies were purchased from Promega (Madison, WI).
Inhibitor and protease
Cathepsin B inhibitor IV CA074 and recombinant human cathepsin B were obtained from EMD Millipore (Billerica).
Cell culture
Colorectal adenocarcinoma cell lines (except Caco‐2/15 cell line) and HEK293T cells were all obtained from ATCC (Manassas, VA). The cell line Caco‐2/15 was obtained from Dr A. Quaroni (Cornell University, Ithaca, NY) and cultured in DMEM containing 10% FCS (Wisent, St‐Bruno, QC, Canada). The colon carcinoma cell lines HCT116 (CCL‐247) and HT29 (HTB‐38) were cultured in McCoy's medium containing 10% FCS. The colon adenocarcinoma cell line Lovo (CCL 229) was cultured in Ham's F12 medium containing 10% FCS. SW480 (CCL 228), SW620 (CCL‐227), and HEK293T cell lines were maintained in DMEM containing 10% FCS. The colon adenocarcinoma cell lines DLD‐1 (CCL‐21) and Colo205 (CCL‐222) were cultured in RPMI medium containing 10% FCS 16.
Mice
CD1 nu/nu and Fox Chase SCID Beige mice were purchased from Charles River Laboratory (Wilmington, MA). Mice were housed in individually ventilated cages and sterilized food, water, bedding, and cages were used. Experiments were approved by the Animal Research Committee of the Faculty of Medicine and Health Sciences of the Université de Sherbrooke.
Human Colorectal Tissues
Sixty‐six samples of colon tumors and paired normal colon tissues (at least 10 cm from the tumor) were obtained from patients undergoing surgical resection. Patients did not receive neoadjuvant therapy. Tissues were collected after obtaining the patient's written informed consent, according to the protocol approved by the Institutional Human Subject Review Board of the Centre Hospitalier Universitaire de Sherbrooke. Paired tissues were frozen in liquid nitrogen within 15 min from resection as recommended by the Canadian Tumor Repository Network [http://www.ctrnet.ca] and stored in liquid nitrogen until total RNA or protein extraction. Total RNA was extracted using the Totally RNA kit (Invitrogen, Burlington, ON) and processed according to the manufacturer's instructions. For protein extraction, paired tissues were lysed in Triton sample buffer (100 mM NaCl, 5 mM EDTA [pH 8.0], 50 mM Tris–HCl [pH 7.5], 1% Triton X‐100, 5% glycerol, 1 mM PMSF, 0.2 mM orthovanadate, 40 mM β‐glycerophosphate, 50 mM NaF, and 2% protease inhibitor cocktail [Sigma–Aldrich]) and immunoblotted as described in the Western blot analysis section. Clinical and pathological data were obtained from medical records and are provided in supplementary Table S1. Adenoma samples were endoscopically unresectable and defined as advanced because of their size larger than 1 cm or by the presence of high‐grade dysplasia or villous component. Tumors were histologically classified and graded according to overall TNM staging criteria (based on Tumor‐, Lymph Node‐, and Metastatic‐status). Genomic DNA was extracted from formalin‐fixed paraffin‐embedded tissue using a FFPE DNA Isolation Kit for Cells and Tissues (Qiagen, Toronto, ON, Canada). APC (exon 15) and KRAS (exons 1 and 2) were amplified by PCR and the presence of mutations was detected by direct sequencing (Plateforme de Séquençage et de Génotypage des Génomes, Québec, QC, Canada).
Reverse Transcription PCR
For human colorectal tissues, reverse transcription was performed using AMV‐RT (Roche, Laval, QC, Canada) according to the manufacturer's instructions while Q‐PCR was performed by the RNomics Platform at the Université de Sherbrooke (QC, Canada). CTSB expression was normalized using the qBase quantification Framework software with three reference genes: MRLP19, PUM1, and RPL13A. For cultured cells, total RNA was extracted with the RNeasy plus mini kit (Qiagen). Reverse transcription was performed using AMV‐RT according to the manufacturer's instructions and Q‐PCR performed using a LightCycler® Carousel‐Based System (Roche). Experiments were run and analyzed with the LightCycler software 4.0 according to the manufacturer's recommendations (Roche). Synthesis of double‐stranded DNA during PCR was monitored using SYBR Green I according to the manufacturer's recommendations (QuantiTect SYBR Green PCR Kit; Qiagen). Target expression was quantified relative to PDGB expression. Primer sequences and conditions are available upon request.
Plasmid Constructions and Lentiviruses Production
The lentiviral shRNA expression vector (pLenti6‐U6) was constructed 16 and shRNA oligonucleotides against human cathepsin B were designed according to Ambion guidelines (technical bulletin #506). siRNA sequences were 5′‐GGATCACTGTGGAATGGAATC‐3′ (Cathepsin B) or 5′‐GAGCCATTAGGACGGTTAGAT‐3′ (Scrambled) and TTCAAGAGA as loop sequence. The oligonucleotide‐annealed products were subcloned into pLenti6‐U6 between the BamHI and XhoI sites. Lentiviruses produced in 293 T cells were used for infection according to Invitrogen recommendations (ViraPower Lentiviral Expression System). No induction of OAS1 gene expression was detected by Q‐PCR analysis in the experiments involving lentiviruses infection (data not shown). OAS1 is a classic interferon target gene recommended as a key test for interferon induction before attributing a particular response to the targeted gene 17. The full‐length human p27Kip1 cDNA was subcloned after PCR amplification in expression vector pCDNA3.1. The HA tag was inserted in frame between a Kozak sequence and the N‐terminal end by PCR using the following sequences: 5′–TCA CTA GGA TCC ACC ATG TCA AAC GTG CGA G –3′ and 5′–AGT GAT CTC GAG TTA CGG GAG GCT AGC ATA ATC AGG AAC ATC ATA CGT TTG ACG TCT TCT GAG GCC AGG CTT–3′. PCR products were next ligated in BamHI/EcoRI‐digested pCDNA3.1 vector. The p27Kip1 mutants R58A, R152A, Δ96–100, and Δ189–198 were generated by site‐directed mutagenesis and deletion mutagenesis using the following oligonucleotides, respectively: R58A: 5′‐GAA GAG GCG AGC CAG GCT AAG TGG AAT TTC‐3′ ; 5′‐GAA ATT CCA CTT AGC CTG GCT CGC CTC TTC‐3′; R152A: 5′‐TGC GCA GGA ATA CCA AAG CGA CCT GCA ACC‐3′ ; 5′‐GCT TGC AGG TCG CTT TGC TAT TCC TGC‐3′ ; Δ96–100: 5′‐CCC CCG CGG CCC CCC GTC CCG GCG‐3′ ; 5′‐CTC CTG CGC CGG CAC GGG GGG CCG‐3′ ; Δ198–198 : 5′‐TCA CTA GGA TCC ACC ATG TCA AAC GTC‐3′ ; 5′‐AGT GAT CTC GAG TTA GGG CGT CTG CTC CTC CAC AGA‐3′. PCR products were next digested and ligated in BamHI/EcoRI‐digested PCDNA3.1 vector.
Western Blot Analysis
Cells were washed with ice‐cold PBS and lysed in Laemmli sodium dodecyl sulfate–sample buffer (90 mM Tris‐HCl [pH 6.8], 2.5% sodium dodecyl sulfate, 15% glycerol). Samples were then boiled, sonicated, and protein concentrations were determined using the bicinchoninic acid (BCA) assay (Fisher Healthcare, Houston, TX) with bovine serum albumin as standard. β‐mercaptoethanol and bromophenol blue were then added to a final concentration of 1% and 0.005%, respectively. Proteins (5–50 µg) were separated by SDS–PAGE in 7.5% or 10% gels and were detected immunologically following electrotransfer onto PVDF membranes (PerkinElmer, Woodbridge, ON, Canada). PVDF membranes were stained with Ponceau Red to assure a correct transfer of proteins and molecular weight markers (Bio‐Rad, Mississauga, ON, Canada). Membranes were blocked in PBS containing 5% powdered milk and 0.05% Tween 20 for 1 h at 25°C. Membranes were then incubated overnight at 4°C with primary antibodies in blocking solution and thereafter with horseradish peroxidase or alkaline phosphatase‐conjugated IgG for 1 h. Blots were visualized using the Amersham ECL system (Amersham–Pharmacia Biotech) or Millipore Immobilon AP substrate (Millipore, Etobicoke, ON, Canada).
Expression of Extracellular Cathepsin B Protein
For culture medium analysis, subconfluent CRC cells were serum‐deprived during 24 h. Medium was collected and centrifuged 5 min at 4 500g to remove non‐adherent cells and possible cellular debris. Thereafter, 4 mL of the medium was collected and concentrated through an Amicon(r) Ultra‐10 kDa centrifugal filter unit (Millipore) after which Laemmli buffer was added to the retentate and boiled for 5 min. Cells were lysed in Laemmli sodium dodecyl sulfate–sample buffer and samples subsequently boiled and sonicated. Protein concentration in cell lysates was determined using the bicinchoninic acid (BCA) assay. Volumes of culture media proportional to the intracellular protein concentration were separated on 10% SDS–PAGE gels for Western blot analysis.
Cathepsin B Activity Assay
Purified human cathepsin B (Calbiochem) was activated by incubation at 37°C with 5 mM DTT in 50 mM Tris (pH 6.1). The activity of cathepsin B was then measured in presence or absence of 10 μM Ca074 using the Cathepsin B Activity Fluorometric Assay Kit from BioVision according to the manufacturer's instructions (BioVision, Milpitas, CA). Subconfluent HT29 and HEK293 cells were incubated with 10 μM Ca074 for 12 h. Cells were then washed twice with PBS, lysed in cell lysis buffer provided in the assay kit and the resulting activity measured according to the manufacturer's instructions.
Cell Proliferation Assays
Cell populations were plated for growth assay in 6‐well plates at a concentration of 2 × 105 (HT‐29) or 1.2 × 105 (DLD1) cells/well in 2% FCS‐containing medium. Proliferation was measured during 7–8 d using a Cell particle counter.
Soft Agarose
DMEM 2X medium without phenol red was prepared from powder (Wisent) according to the manufacturer's instructions. DMEM (DLD1 and SW480) and McCoy5A (HT‐29) media were subsequently supplemented with 20% FCS, 8 mM glutamine, 100 U/mL penicillin, 100 mg/mL streptomycin (Invitrogen), and 40 mM HEPES. Pre‐warmed media were mixed 1:1 with autoclaved 1.4% agarose type VII maintained at ≈42°C, and 6‐well dishes were pre‐coated with 1 mL/well. Cells were added to the medium‐agarose mix at a concentration of 5 × 104 (HT‐29), 3 × 104 (DLD1), and 7.5 × 103 cells/mL (SW480), and seeded at 1.5 mL/well. Plates were allowed to solidify under the hood and then placed at 37°C and 5% CO2. Fresh media supplemented with 10% FCS were added on the surface of the agarose every 1–2 d. After 2–3 wk, colonies were stained by adding 500 µL of PBS containing 0.5 mg/mL MTT on the surface of the agarose and incubated for 2 h at 37°C and 5% CO2. Images were acquired using an AlphaImager camera (Alpha Innotech Corporation) and colonies counted using ImageJ software.
Invasion Assays
Invasion assays were conducted using BD Matrigel(tm) Invasion Chamber 24‐well plates 8.0 micron according to the manufacturer's instructions. Briefly, plates were thawed at room temperature for 30 min after which Matrigel was humidified with serum‐free culture medium for at least 2 h at 37°C and 5% CO2. Thereafter, 3 × 104 HT29 cells, 1 × 104 DLD‐1 cells, or 4 × 104 SW480 cells in serum‐free medium were seeded into the upper chamber while culture medium containing 20% FCS was placed into the lower chamber as chemoattractant. Cells were allowed to migrate for the next 48 h in the presence of 2 mM hydroxyurea in both chambers to prevent HT29 and DLD1 proliferation. For SW480 cells, hydroxyurea was not added since these cells are particularly sensitive to this compound and do not proliferate significantly within 48 h under these conditions. Non‐migrating cells were removed with two cotton swabs, while migrating cells were fixed for 2 min with methanol and stained with DAPI for manual counting under the microscope. The number of invading cells was counted in seven fields per chamber.
Xenograft Assays in Mice
A total of 1 × 106 cells suspended in 100 µL McCoy 5 A medium were injected into the dorsal subcutaneous tissue of 6‐wk‐old female CD1 nu/nu mice. Mice were sacrificed after 23 d post‐injection. Tumors were excised, weighed, and lysed in RIPA buffer.
Experimental Tail Vein Assays in Mice
The tail‐vein of 5‐wk‐old female Fox Chase SCID mice was injected with 106 cells suspended in 100 µL McCoy 5 A medium. Mice were sacrificed after 28 d post‐injection. Lungs were maintained in Bouin's fixative for 24 h before visualization of surface‐visible metastases.
p27Kip1 Cleavage by Cathepsin B
293T cells were transfected by Lipofectamine (Invitrogen) with 3 μg/60 mm Petri dish of each expression vectors encoding for HA‐tagged wild‐type p27Kip1 or mutants described above. Thirty‐six hours after transfection, cells were washed twice with ice‐cold PBS then lysed in Triton lysis buffer. Protein concentrations were determined using the BCA assay. For the reaction step, 7.5 μg of each cellular extract was incubated with reaction buffer containing 50 mM Tris‐HCl (pH 6.0), 5 mM DTT, and 300 ng of purified human cathepsin B (Calbiochem) at 37°C. After different time points, 4X Laemmli buffer was added to the samples for gel analysis.
Confocal Microscopy
Cells grown on glass coverslips were fixed for 15 min at room temperature with 3.7% paraformaldehyde, permeabilized for 15 min with 0.1% Triton X‐100/PBS, and then blocked for 1 h with 2% BSA/PBS. To visualize lysosomes, cells were incubated for 1 h at 37°C with LysoTracker Red probe (DN99, Invitrogen). Cells were incubated overnight at 4°C with anti‐cathepsin B antibodies (AF‐953, R&D systems). Cathepsin B was visualized with donkey anti‐goat AlexaFluor®488 (Invitrogen). For double p27Kip1‐cathepsin B immunofluorescence, cells were incubated overnight at 4°C with anti‐p27Kip1 (C‐19, Santa Cruz) and anti‐cathepsin B. Cathepsin B and p27Kip1 were visualized using donkey anti‐goat AlexaFluor 594 and chicken anti‐rabbit AlexaFluor®488 (Invitrogen), respectively. In double immunofluorescence experiments, single staining of each protein was performed to ensure nonoverlapping fluorescence. Images were acquired on a Zeiss Axio Observer LSM700 confocal microscope with an x63 oil‐immersion objective (N.A. 1.4, Carl Zeiss Canada). Confocal microscope settings were adjusted to produce the optimum signal/noise ratio. The sequential mode was used in image acquisition to avoid any interference from overlapping fluorescence. Fluorescence analysis was performed using Zen Black edition software (Carl Zeiss).
Subcellular Fractionation (Differential Centrifugations)
Cells were washed twice with ice‐cold PBS and scraped in cell fractionation buffer: 50 mM Tris (pH 7.5), 5 mM MgCl2, 25 mM KCl, 250 mM sucrose, supplemented with protease inhibitors (0.1 mM phenylmethylsulfonyl fluoride [PMSF], 10 µg/mL leupeptin, 1 µg/mL pepstatin, 10 µg/mL aprotinin). Cell membranes were disrupted through a 25 G syringe (five strokes). Crude cellular extracts were centrifuged at 500g for 5 min. The pellet containing nuclei was homogenized in fractionation buffer, washed, and lysed by addition of 4X Laemmli buffer. After centrifugation of the supernatant at 20 000g for 15 min, the resulting pellet containing plasma membrane, mitochondria, lysosomes, and peroxisomes was washed and homogenized in fractionation buffer and lysed by addition of 4X Laemmli buffer. The 20 000g supernatant was then centrifuged at 105 000g for 60 min. The pellet containing microsomes was homogenized in fractionation buffer and lysed by addition of 4X Laemmli buffer. The remaining final supernatant corresponding to the cytosolic fraction was lysed by addition of 4X Laemmli buffer.
Statistical Analysis
Densitometric analyses were performed using ImageJ software. Results shown in the graphs were analyzed by the paired Student's t‐test or the Wilcoxon matched ranks t‐test depending on the normality of sample distribution. Data are presented as means ± SEM. Statistical significance was set at P < 0.05.
RESULTS
mRNA and Activated Levels of Cathepsin B Are Increased in Adenomas and in Colorectal Tumors of All Stages
Cathepsin B expression was analyzed at both the mRNA and protein levels in a series of human paired specimens at various tumor stages. As shown in Figure 1A, increased transcript levels of cathepsin B were observed in colorectal tumors, regardless of tumor stage, including in adenomas. Of note, increased cathepsin B expression was more prominent in tumors exhibiting APC mutations. By contrast, there did not appear to be a significant difference relative to KRAS mutations (Figure 1B). To establish whether these increased mRNA levels could be correlated with increased cathepsin B protein levels and more importantly with increased activity, expression of the active processed forms of the protease (25 and 30 kDa) was analyzed by Western blot. Both pro‐cathepsin B and active cathepsin B were also increased in colorectal tumors compared to normal tissues (Figure 1C and D). These data hence suggest that increased transcription contributes to a greater expression of active cathepsin B in CRC.
Figure 1.
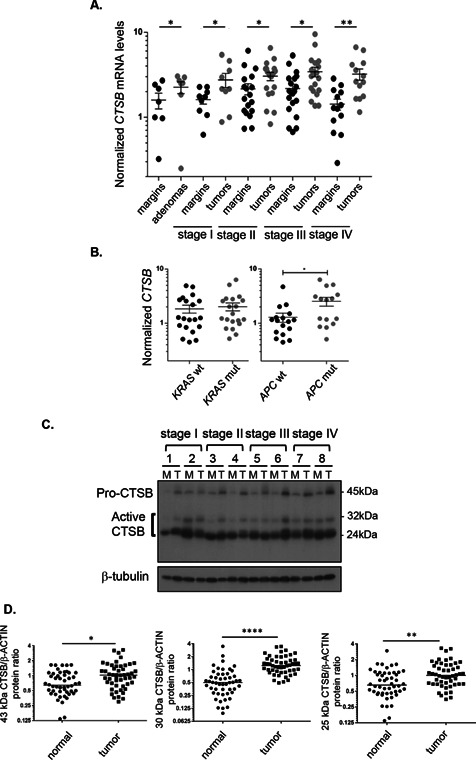
Cathepsin B expression is increased in adenomas and in colorectal cancers of all stages. (A) Relative CTSB mRNA levels were determined by Q‐PCR in human advanced adenomas and adenocarcinomas compared to the paired adjacent healthy tissue. Data presented are the means ± SEM of 7 to 20 tissue samples per stage. Significantly different at *P < 0.05 (Student's t‐test); **P < 0.005 (Student's t‐test). (B) Correlation of induction levels of CTSB transcripts with the presence of KRAS or APC mutations. The induction of CTSB transcripts was analyzed in 15–20 tumors in terms of KRAS and APC mutation status. Data presented are the means ± SEM. Significantly different at *P < 0.05 (Student's t‐test). (C) Representative immunoblot analysis of cathepsin B protein (CTSB) performed on protein extracts from eight paired resection margins and tumor specimens. Tubulin expression is shown as a control of protein loading. (D)Western blot analysis of cathepsin B protein expression in a series of 50 paired specimens (resection margins and primary tumors). Protein levels of cathepsin B pro‐form and active forms were normalized to the intensity of β‐actin staining and to a reference sample, resulting in a dimensionless value. Amounts of normalized cathepsin B proteins in tumor tissues relative to their matched normal samples were analyzed by paired Student's t‐test. Significantly different at *P ≤ 0.05, **P ≤ 0.005, and ****P ≤ 0.0005.
Extracellular Cathepsin B Contributes to Invasiveness of Human CRC Cells but is Dispensable for Their Growth in Soft Agar
Cathepsin B protein levels were next examined in lysates obtained from various human CRC cell lines. As shown in Figure 2A, the proactive and catalytically active processed forms of cathepsin B were detected at various levels in CRC cell lines. Selected cathepsin B presence was also confirmed in conditioned culture medium of CRC cells, again at various levels (Figure 2A, lower panel). However, while the pro‐form of cathepsin B was readily observed in conditioned culture medium of all CRC cells, the catalytically‐active processed forms of cathepsin B were not detected in Western blot analyses. Additionally, using a fluorescence‐based enzymatic assay, no cathepsin B enzyme activity was detected in conditioned medium. Since the pro‐protease form might be activated under acidic pH conditions (peri‐ or extracellular) and by extracellular components of the extracellular matrix, the impact of extracellular inhibition of cathepsin B activation on CRC cell invasion was verified using Biocoat Matrigel chambers. HT‐29, DLD1, and SW480 CRC cell lines secreting different levels of pro‐cathepsin B (Figure 2A) were tested. Experiments were performed using the highly selective and non‐permeant inhibitor Ca074 to reduce extracellular cathepsin B activity. At 10 μM, Ca074 produced a >99% inhibition of recombinant cathepsin B levels while barely reducing intracellular cathepsin B, that is, 5–8%, even upon 12 h exposure to the inhibitor (data not shown). Of note, treatment with 10 μM Ca074 significantly inhibited Matrigel invasion by approximately 45–60% in HT29, DLD1, and SW480 CRC cell lines (Figure 2B). By contrast, treatment with Ca074 had no significant effect on their capacity to form colonies in soft agarose (Figure 2C).
Figure 2.
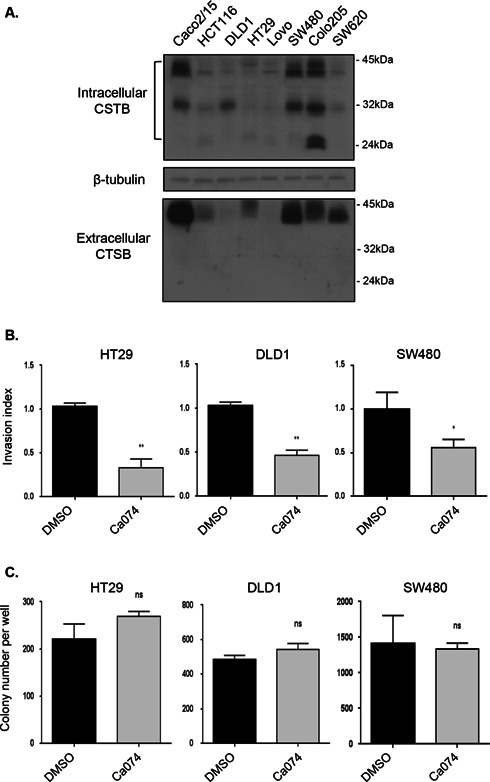
Extracellular cathepsin B contributes to invasiveness of human CRC cells but is dispensable for their growth in soft agar. (A) CRC cell lines were serum‐deprived during 24 h and then lysed in Laemmli buffer and analyzed by Western blotting with specific antibodies against cathepsin B (CTSB) and β‐tubulin. Four milliliter of culture medium from each cell line was concentrated and also analyzed by Western blotting for the expression of extracellular cathepsin B. (B) HT29, DLD1, and SW480 seeded in Matrigel‐coated Transwells were treated with DMSO or Ca074 (10 μM) during 48 h. Thereafter, cells were fixed and stained with DAPI solution for assessment of their capacity to invade Matrigel. The experiments were performed in triplicate and the number of control cells treated with DMSO which had migrated was set at 1. Significantly different at *P ≤ 0.05 and **P ≤ 0.005 (Student's t‐test). (C) HT29, DLD1, and SW480 were cultured in soft agarose in the presence or absence of 10 μM Ca074 for 2–3 wk prior to MTT staining. The number of colonies was determined using ImageJ software. Results are the means ± SEM of at least three independent experiments. ns: not significant (Student's t‐test).
Cathepsin B Silencing in Human CRC Cells Inhibits Growth in Soft Agar and Invasion Capacity
Recombinant lentiviruses encoding anti‐cathepsin B short hairpin RNA (shRNA) were developed in order to stably suppress cathepsin B expression in CRC cells. As shown in Figure 3A, intracellular cathepsin B mRNA and protein levels were decreased in HT29 and DLD1 cells in comparison to a control shRNA which had no effect. Reduction of cathepsin B expression modestly slowed the proliferation rate of HT29 and DLD1 populations in 2D cell culture (Figure 3B). Conversely, cathepsin B silencing significantly reduced the ability of HT29 and DLD1 cells to form colonies in soft agarose (Figure 3C). This indicates that intracellular cathepsin B controls anchorage‐independent growth of CRC cells given the absence of Ca074 effect (Figure 2C). Moreover, cathepsin B silencing also reduced the number of invading HT29 and DLD1 cells to a similar extent as Ca074 treatment (Figure 3D vs. Figure 2B).
Figure 3.
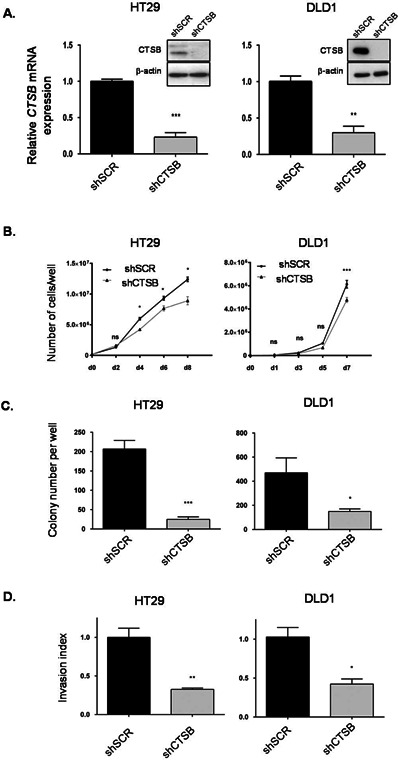
Cathepsin B silencing in human CRC cells inhibits growth in soft agar and invasion capacity. (A) HT29 and DLD1 cells were stably infected with lentiviruses encoding for a control shRNA (scrambled sequence, shSCR) or encoding cathepsin B‐specific shRNAs (shCTSB). Stable cell populations were thereafter lysed and RNA isolated to determine CTSB or PBGD gene expression by Q‐PCR. The relative level of CTSB was normalized to the corresponding PBGD mRNA level. Cell populations were also lysed and protein lysates were analyzed by Western blotting for cathepsin B and β‐actin expression. (B) HT29 and DLD1 cells expressing either shSCR or shCTSB were seeded in a 6‐well plate at 2 × 105 (HT‐29) and 1.2 × 105 (DLD1) cells per well. Cells were harvested and counted after different times. (C) HT29 and DLD1 cells were cultured in soft agarose before MTT staining. The number of colonies was determined using ImageJ software. Results are the means ± SEM of at least three independent experiments. (D) HT29 and DLD1 cell populations were seeded in Matrigel‐coated Transwells and cultured during 48 h. Thereafter, cells were fixed and stained with DAPI solution for assessment of their capacity to invade Matrigel. The experiments were performed in triplicate and the number of control cells (shSCR) which had migrated was set at 1. For all experiments described above: *, significantly different from shSCR cells at P < 0.05 (Student's t‐test). **P < 0.005; ***P < 0.001.
Cathepsin B Silencing in Human CRC Cells Inhibits Tumorigenicity and Metastasis in Immunodeficient Mice
Suppression of cathepsin B expression was found to significantly attenuate the metastatic potential of CRC cells in vivo in experimental metastasis assays. Indeed, immunodeficient mice injected with control CRC cells into the tail vein showed extensive lung metastasis within 28 d, whereas cells expressing shRNA against cathepsin B exhibited reduced lung colonization (Figure 4A). Cathepsin B silencing also altered the capacity of CRC cells to form tumors in mice as assessed by subcutaneous xenograft assays. HT29 cells induced palpable tumors with a short latency period of 9 d after their injection while downregulation of cathepsin B expression in these cells severely impaired their capacity to grow as tumors (Figure 4B).
Figure 4.
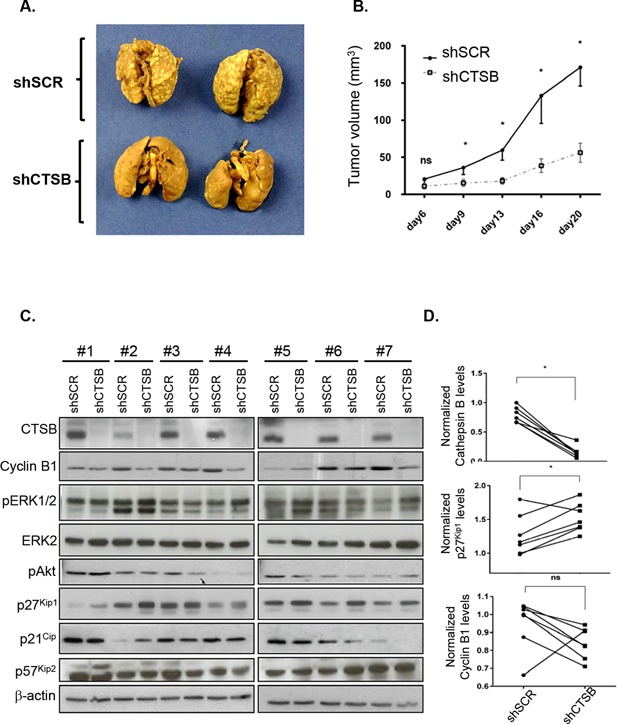
Cathepsin B silencing in human CRC cells inhibits tumorigenicity and metastasis in immunodeficient mice. (A) Representative digital images of mouse lungs 28 d after tail vein injection of 106 HT29 cells expressing either shScrambled (shSCR) or shCathepsin B (shCTSB). (B) HT29 cells expressing either shSCR or shCTSB were injected subcutaneously in immunodeficient mice and the growth of tumors (mm3) over time was measured. The results represent the mean tumor volume obtained from two independent experiments in which at least six mice were injected for each cell line. *, significantly different from shCTSB tumors at P < 0.05 (Student's t‐test). (C) Equal amounts of whole cell lysates from tumors were analyzed by Western blotting for the expression of cathepsin B, cyclin B1, phosphorylated ERK1/2 (pERK1/2), total ERK2, phosphorylated Akt (pAkt), p27Kip1, p21, p57Kip2, and β‐actin. (D) Densitometric analysis of cathepsin B, p27Kip1, and cyclin B1 was determined in each tumor (cells expressing shSCR and cells expressing shCTSB were injected in different flanks of the same mouse) using ImageJ software. *, significantly different from shSCR tumors at P < 0.05 (Wilcoxon matched‐pairs signed‐ranks t‐test).
Expression of factors reported to be involved in colorectal tumor growth was next verified in tumor lysates. As shown in Figure 4C, the expression and activated phosphorylated levels of ERK1/2 MAP Kinases and Akt in tumor lysates were not significantly altered in tumors expressing or not cathepsin B. However, higher levels of the cell cycle inhibitor p27Kip1 were observed in cathepsin B‐deficient tumors while p21Cip and p57Kip2 levels were not significantly affected. Of note, this increase in p27Kip1 expression observed in cathepsin B‐deficient tumors was associated with significant lower cyclin B1 expression in most tumors (Figure 4C and D), suggesting reduced proliferative activity. These data thus suggest that cathepsin B silencing in CRC cells severely reduces their capacity to form tumors in vivo likely through the increased expression of p27Kip1.
Cathepsin B Cleaves the Cell Cycle Inhibitor p27Kip1
In order to verify whether p27Kip1 is in fact a substrate for cathepsin B, both proteins were first overexpressed in 293 T cells and cells subsequently lysed 2 d later for Western blot analysis of their respective expression. As shown in Figure 5A, forced expression of cathepsin B in 293 T cells dose‐dependently reduced p27Kip1 protein levels. Next, to determine whether p27Kip1 could be degraded by cathepsin B in vitro, lysates from 293 T cells overexpressing HA‐tagged p27Kip1 were incubated with purified cathepsin B and analyzed by Western blot. Figure 5B and C shows that cathepsin B degraded p27Kip1 in a time‐dependent manner as visualized by the accumulation of three lower molecular mass species (26, 20, and 12 kDa) in addition to the full‐length p27Kip1 protein (see arrows versus arrowhead).
Figure 5.
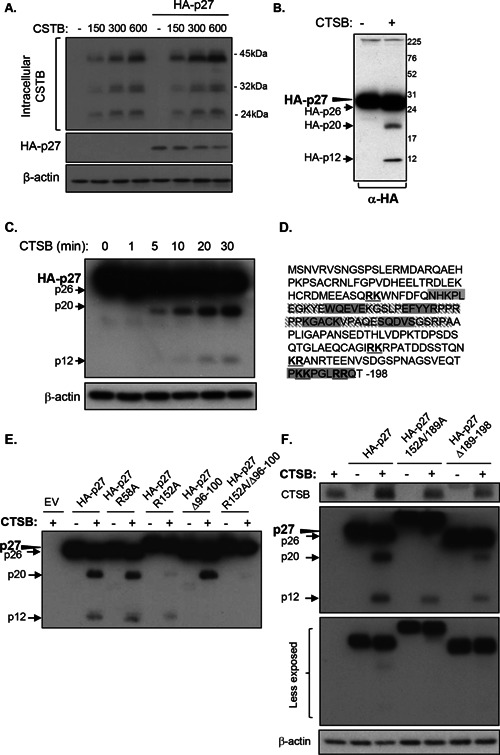
Cathepsin B exhibits endo‐ and exopeptidase activity against p27Kip1. The pcDNA3 vectors without or with the HA‐tagged wild‐type p27Kip1 cDNA were transiently co‐transfected with 0, 150, 300, or 600 ng of the cDNA encoding cathepsin B in 293T cells. After 48 h, lysates were analyzed by Western blotting for the expression of HA‐27Kip1, cathepsin B, and β‐actin. (B and C) The cDNA encoding HA‐tagged p27Kip1 was transfected transiently in 293T cells. After 48 h, cells were lysed and 15 μg of protein lysate was incubated during 30 min (B) or during 1, 5, 10, 20, and 30 min (C) with purified human cathepsin B (300 ng) at 37°C (pH 6.0). Proteins were then solubilized in Laemmli buffer and analyzed by Western blotting for the expression of HA‐27Kip1 and β‐actin. (D) Amino acid sequence of human p27Kip1; putative cleavage sites of cathepsin B are indicated in bold and underlined. The sequence of amino acids deleted in each p27Kip1 deletion mutant generated is highlighted in grey. (E and F) The cDNA encoding HA‐tagged p27Kip1 and indicated mutants were transfected transiently in 293T cells. After 48 h, cells were lysed and 7.5 μg of protein lysates were incubated during 30 min with purified human cathepsin B (300 ng) at 37°C (pH 6.0). Proteins were then solubilized in Laemmli buffer and analyzed by Western blotting for the expression of HA‐27Kip1, cathepsin B, and β‐actin.
Cathepsin B is capable of endopeptidase, peptidyl‐dipeptidase, and carboxydipeptidase activities 18, 19, 20. Cathepsin B also possesses a basic amino acid in the catalytic subsite in position S2 enabling the protease to preferentially split its substrates after Arg–Arg or Lys–Arg or Arg–Lys sequences. At least five of these sequences can be found within the human p27Kip1 sequence (Figure 5D). Therefore, the first amino acid of these doublets was mutated into alanine to test whether it would affect the degradation by cathepsin B. Mutation of arginine 58 (Figure 5E) and lysine 189 (Figure 5F) did not alter the cleavage profile of p27Kip1 by cathepsin B. Mutation of lysine 165 and arginine 194 also had no altering effect (not shown). On the other hand, mutation of arginine 152 into alanine markedly reduced the detection of the 20‐kDa fragment (Figure 5E).
In order to identify other cleavage sites, deletion mutants of p27Kip1 were generated by removal of different sequences of five to ten amino acids (Figure 5D). All of these mutants were expressed in 293 T cells and lysates subsequently exposed to cathepsin B cleavage (data not shown). The lower fragment was not observed with the p27 Δ96–100 mutant following cathepsin B cleavage, suggesting the presence of a cleavage site within this sequence (Figure 5E). This particular sequence contains a glycine, and cathepsin B endopeptidase specificity has been reported to be also guided by the presence of a glycine in position P3′ of the scissile bound 21. Of note, the 12‐ and 20‐kDa fragments were not observed when arginine 152 was mutated in alanine in the Δ96–100 mutant (Figure 5E). Moreover, with the p27Δ189–198 mutant, both the 12‐ and 20‐kDa fragments were still detected (Figure 5F), although the modest shift‐down of 1–2 kDa normally produced following cathepsin B cleavage of full‐length p27Kip1 was not observed (Figure 5F, lower less‐exposed gel). Single point mutation of each amino acid in the 189–198 sequence did not prevent the generation of the 26‐kDa fragment following cathepsin B treatment (Figure 5F for residue 189 as an example). This suggests that this 26‐kDa fragment probably results from the exopeptidase activity of cathepsin B.
The protein stability of wild‐type p27Kip1 was then compared to that of the p27Kip1 R152A/Δ189–198 mutant, which is more resistant to cathepsin B cleavage. 293T cells were transiently transfected with either wild‐type p27Kip1 or p27Kip1 mutant and subsequently treated with cycloheximide to inhibit protein neosynthesis. Thereafter, cells were lysed at different time intervals in order to analyze protein expression levels of p27Kip1 forms. As shown in Figure 6A, following cycloheximide treatment, protein levels of the p27Kip1 mutant decreased much more slowly than that of wild‐type protein. Specifically, 10 h after cycloheximide addition, expression of p27Kip1 protein was clearly decreased while expression of the p27Kip1 mutant remained at control (time 0) levels. Of note, forced expression of cathepsin B in 293 T cells dose‐dependently reduced the wild‐type form of p27Kip1 protein levels while expression of p27Kip1 R152A/Δ189–198 mutant was only very slightly affected (Figure 6B).
Figure 6.
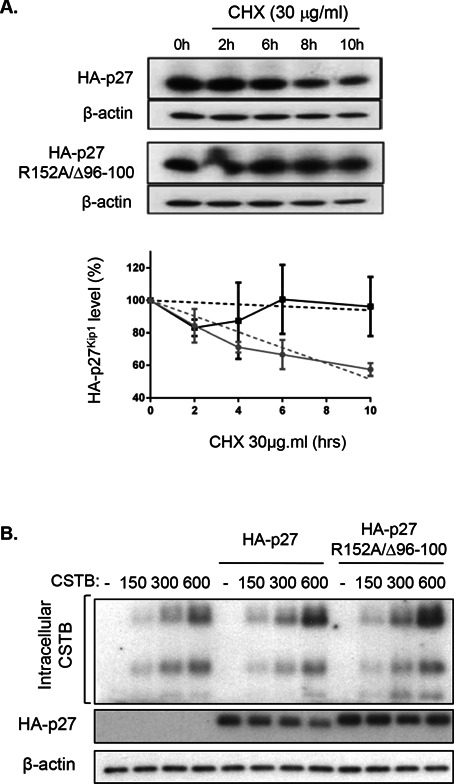
Cathepsin B reduces p27Kip1 stability. (A) The cDNA encoding HA‐tagged wild‐type p27Kip1 and R152A/△96–100 mutant were transiently transfected in 293T cells. Twenty‐four hours following transfection, cells were treated with cycloheximide (CHX, 30 μg/mL) and harvested after 0, 2, 6, 8, or 10 h. Cells were processed and lysed at the same time. Expression of HA‐27Kip1 proteins and β‐actin was analyzed by Western blotting. Representative Western blot analysis is shown in upper panel. The graph illustrates the densitometric analysis of data from three independent experiments. HA‐p27Kip1 expression at 0 h of cycloheximide was set at 100%. Relative HA‐p27Kip1 protein levels were calculated using β‐actin as reference. (B) The pcDNA3 vectors without or with the cDNA encoding HA‐tagged wild‐type p27Kip1 or HA‐tagged R152A/△96–100 mutant were transiently co‐transfected with 0, 150, 300, or 600 ng of the cDNA encoding cathepsin B in 293T cells. After 48 h, protein lysates were analyzed by Western blotting for the expression of HA‐27Kip1, cathepsin B, and β‐actin.
Colocalization of Endogenous p27Kip1 With Cathepsin B Into Lysosomes
To confirm that p27Kip1 also interacts with cathepsin B in CRC cells, their respective putative colocalization was examined by immunofluorescence confocal microscopy. As shown in Figure 7A, the anti‐cathepsin B antibody confirmed the colocalization of cathepsin B (in green) with the lysosomal acidotropic probe LysoTracker (in red). As expected, most of p27Kip1 staining (in green) was observed in the cell nucleus (Figure 7B). However, certain areas of colocalization were observed between endogenous p27Kip1 (in green) and cathepsin B (in red) (Figure 7B, asterisks). Moreover, Western blot analyses revealed the presence of p27Kip1 protein in lysosome‐enriched fractions obtained from differential centrifugation of Caco‐2/15 and SW480 cell lysates (Figure 7C and D). These lysosomal fractions were enriched in lysosome‐associated membrane protein 1 (LAMP1) and exhibited very low or undetectable levels of the nuclear lamin B protein. Of note, p27Kip1 protein was mostly detected in the cytosolic fraction and not in the nuclear fraction contrary to its expected localization. Significant levels of cyclin E and Cdk2 were also found in the cytosolic fraction (Figure 7C). Since p27Kip1 along with cyclins and mitogen‐activated protein kinases are proteins whose functions are spatially and temporally regulated and, therefore, are actively transported between the nucleus and cytoplasm, one could speculate that during the fractionation procedure, these proteins easily exit from the nucleus. On the other hand, cathepsin B silencing in CRC cells specifically enhanced p27Kip1 levels in lysosome‐enriched fractions (Figure 7D).
Figure 7.
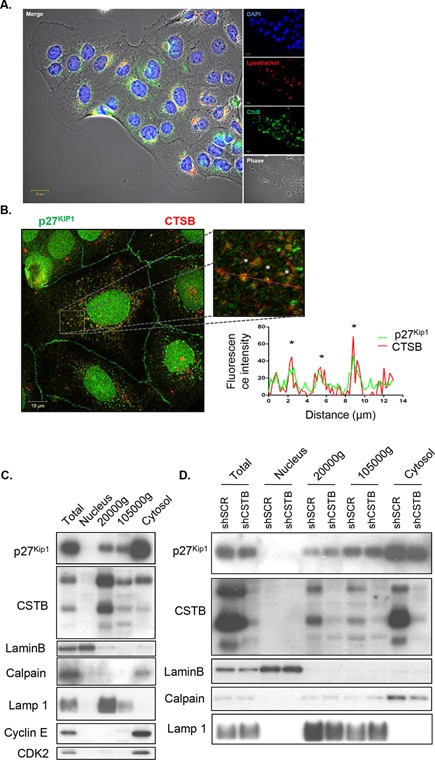
Colocalization of p27 with cathepsin B in lysosomes. (A) Representative confocal microscopy images of Caco‐2/15 CRC cells showing cathepsin B (in green), LysoTracker staining (in red), and DAPI (in blue). (B) Representative confocal microscopy images of Caco‐2/15 CRC cells showing cellular distribution of endogenous p27Kip1 (in green) and cathepsin B (in red) in a double immunofluorescence experiment. Boxed region in the low‐magnification image is enlarged on the right. The graph represents fluorescence profiles along the dashed white line showing areas of colocalization (white asterisks). (C) Caco‐2/15 cell lysates were fractionated by differential centrifugation (see Material and Methods), and equal amounts of proteins from each fraction were analyzed by Western blotting for the expression of p27Kip1 and cathepsin B. Expression of lysosomal (LAMP1), nuclear (lamin B), cytosolic (calpain 2), and nuclear/cytoplasm shuttling markers (cyclin E and CDK2) was analyzed to assess purity of subcellular fractions. (D) SW480 cells stably expressing either shSCR or shCTSB were harvested and lysates were fractionated by differential centrifugation (see Material and Methods). Equal amounts of proteins from each fraction were analyzed by Western blotting for the expression of p27Kip1 and cathepsin B. Expression of lysosomal (LAMP1), nuclear (lamin B), and cytosolic (calpain 2) markers was analyzed to assess purity of subcellular fractions.
DISCUSSION
The most extensive literature to date regarding cathepsin B highlights a key role of this protease in the invasiveness and metastasis of various carcinoma cells 3, 8, 10, 11, 12. The present findings demonstrate that cathepsin B has not only a role in facilitating CRC invasion and metastasis, but also in mediating early premalignant processes. Results herein show that cathepsin B promotes anchorage‐independent CRC cell growth, which translates in vivo to enhanced tumor growth. In addition, cathepsin B was identified as a new protease capable of proteolytic cleavage of the cell cycle inhibitor p27Kip1. This is especially relevant since the loss of p27Kip1 expression has been strongly associated with aggressive tumor behavior and poor clinical outcome in CRC 22, 23.
While many studies have been conducted on cathepsin B expression in CRC 8, 13, 14, 15, 24, 25, 26, the present study specifically analyzed the expression of this protease at both the mRNA and active protein levels as well as its putative association with mutations in APC or KRAS. Results revealed increased cathepsin B mRNA and protein levels in colorectal tumors, irrespective of tumor stage. These data are reminiscent of the immunohistochemistry data reported by Chan et al. 13 showing that cathepsin B protein was expressed in the vast majority of colon cancers analyzed (558 tumors), which was also independent of tumor stage. The present data also revealed that increased transcription of cathepsin B was associated with the presence of mutations in APC but not in KRAS, thus emphasizing the fact that cathepsin B gene expression is already deregulated in early stages of colorectal carcinoma. Indeed, most CRCs acquire loss‐of‐function mutations in both copies of the APC gene, resulting in inefficient breakdown of intracellular β‐catenin and enhanced nuclear signaling 27. Given the importance of the Wnt/APC/β‐catenin pathway in human tumorigenesis initiation, the present data showing an association between cathepsin B expression and APC mutations are particularly noteworthy. Further studies will be of interest to determine whether this association is correlative or causative. Additional evidence stemming from other studies with APCMin/+ mice nonetheless suggests the contribution of this protease in tumorigenesis with mutated APC. These mice are heterozygous for a germ‐line mutation in the mouse homologue of the human APC gene and develop multiple adenomas in the intestine 28. Cathepsin B was also found to be consistently overexpressed in these adenomatous polyps both at the mRNA and protein levels 12. In addition, genetic ablation of cathepsin B resulted in suppression of tumor infiltrating pro‐inflammatory cells and in attenuation of polyposis in these mice 29. In a proteomic screening study, cathepsin B was also recently identified as one of the six upregulated proteins in the tumors of APCΔ580 mice 9. Such observations again suggest that cathepsin B may be implicated early in the process of tumorigenesis. These results hence support a role for cathepsin B in APC‐mutated tumors and lend credence to the possibility of cathepsin B being a therapeutic target for these cancers.
In agreement with other studies that have shown that secretion of cathepsin B is increased in the extracellular environment of several types of cancer cells 7, 11, 30, 31, results obtained herein in CRC cells in culture confirmed the presence of the pro‐form of cathepsin B in conditioned culture medium of all CRC cells. However, the catalytically active processed forms of cathepsin B were not detected by Western blot analysis. In keeping with this finding, no cathepsin B enzyme activity was detected in the conditioned medium. This indicates that CRC cells massively secrete pro‐cathepsin B but not the active forms of cathepsin B and is consistent with previous studies showing that overexpression of cathepsins is frequently accompanied by the secretion of pro‐cathepsins (reviewed in 32). With regard to CRC, it has been reported that cathepsin B moves from the apical region of the cells to the basal plasma membranes, in parallel with malignant progression to late adenomas and early carcinomas. Prior studies using HCT116 human colorectal carcinoma cells have shown that trafficking of cathepsin B to caveolae and its subsequent secretion were regulated by active KRAS 7. Outside of the cells, cathepsin B was moreover ideally positioned to degrade basal membrane components 33, 34 or to activate uPA and MMPs 7, 35, 36, hence promoting cancer cell invasion and metastasis. Since pro‐cathepsin B is catalytically inactive, the contribution of secreted cathepsin B to extracellular matrix degradation can, therefore, be questioned. Nevertheless, several studies have reported that extracellular components of the extracellular matrix can facilitate the processing of cathepsins 19, 37, 38. Furthermore, under acidic pH conditions, pro‐cathepsins are effectively processed into catalytically mature proteases either autocatalytically 39, 40 or by other proteases. Accordingly, inhibition of extracellular cathepsin B activity in the present study by the non‐permeant inhibitor Ca074 markedly attenuated CRC cell invasion through Matrigel suggesting that secreted pro‐cathepsin B was in fact activated under such conditions. Cathepsin B silencing in these same CRC cells also reduced the number of invading cells to a similar extent as that observed with Ca074 in vitro, in addition to inhibiting their metastatic properties in vivo. In keeping with these results, increased serum levels of cathepsin B have been found in patients with CRC 41. Taken together, these results further support the notion that extracellular cathepsin B may play an important function in CRC invasion and metastasis. Of note, intraperitoneal administration of Ca074 was reported to reduce bone metastasis in a model of breast cancer 42, demonstrating the therapeutic potential of targeting extracellular cathepsin B activity. This metastasis suppression by Ca074 was maintained in a late treatment setting, pointing to a role for this protease in metastatic outgrowth.
An additional finding of this study is the requirement of cathepsin B for anchorage‐independent growth and tumorigenesis. Cathepsin B silencing in CRC cells inhibited their capacity to grow in soft agar and to form tumors in mice. One could speculate that intracellular cathepsin B possibly mediates these functions since Ca074 did not alter growth in soft agar. However, in vivo approaches targeting extracellular active cathepsin B are nonetheless needed to clearly confirm such activity. Furthermore, investigation of the molecular mechanisms underlying the inhibitory effect of cathepsin B silencing on tumor growth revealed reduced levels of cyclin B1 and increased levels of p27Kip1 protein in cathepsin B‐deficient tumors. Cyclin B1 is the major controlling cyclin in the G2 phase of the cell cycle and its expression may reflect the proliferative state of the tumor 43. p27Kip1 is a cell cycle regulator which negatively inhibits cyclin‐CDK complexes and acts as a key protein in the decision between proliferation and cell cycle exit. As cells progress toward S‐phase, transcriptional regulation, translational control, and ubiquitin/proteasome‐mediated degradation lead to a decline in p27Kip1 (reviewed in 44, 45). Moreover, increased proteasome‐dependent degradation of p27Kip1 is particularly observed in aggressive colorectal carcinomas and other cancers 23, 46, 47. In keeping with this observation, the absence of p27Kip1 protein expression is a powerful negative prognostic marker in colorectal carcinomas 22, 23, 47. For instance, homozygous p27Kip1 mutation was consistently found to increase the number of intestinal polyps in Apc mutant mice by five‐ to sixfold 48. Herein, cathepsin B was also identified as a novel protease potentially involved in the regulation of p27Kip1 expression. Results show that, in vitro, this protease was able to specifically cleave the p27Kip1 protein. The observation that cathepsin B and p27Kip1 partially colocalized into the lysosomes suggests that some p27Kip1 cleavage occurred in lysosomes. Supporting this notion, mutations of cathepsin B cleavage sites increased p27Kip1 protein stability. Accordingly, endolysosomal degradation of p27Kip1 was also previously demonstrated in Hela and NIH3T3 cells, contributing to the progression of their cell cycle 49. This particular pathway of p27 degradation appears to be mediated by its interaction with the endosomal protein SNX6, a member of the sorting nexin family of vesicular trafficking regulator 49. However, the molecular mechanisms regulating this SNX6‐dependent p27Kip1 proteolytic pathway remain unknown. The present cell models cultured on a plastic substratum (2D culture) did not allow to consistently detect significant modulation of total p27Kip1 protein expression following cathepsin B silencing (data not shown). In addition, Western blot analyses of p27Kip1 expression demonstrated that this cell cycle inhibitory protein was expressed at various levels in CRC cells without any correlation with intracellular cathepsin B expression (Supplementary Figure S1). This suggests that in cultured CRC cells and as previously reported in other cancer cell lines (reviewed in 46), p27Kip1 protein expression may be regulated by multiple oncogenic pathways. By contrast, a decrease in p27Kip1 expression was observed after forced overexpression of cathepsin B in 293 T cells, although p27Kip1 fragments could not be detected in cell lysates. One could speculate that once generated, these fragments are rapidly degraded in the lysosomes. However, a marked increase in p27Kip1 protein levels was found in cathepsin B‐deficient colorectal tumors derived from mice suggesting that this lysosomal proteolytic pathway is likely to occur in the context of tumorigenesis or 3D CRC cell culture.
In conclusion, the present data demonstrate that cathepsin B plays a significant role in colorectal tumor development, invasion, and metastasis. Results suggest that intracellular cathepsin B activity controls premalignant processes such as anchorage‐independent growth and tumor formation while extracellular cathepsin B regulates invasive properties of CRC cells. p27Kip1 is furthermore identified as a new molecular target of cathepsin B in CRC cells. Since p27Kip1 acts as a tumor suppressor, the impairment of its function may increase the tumoral properties of colorectal cells, as shown previously in APCMin/+ mice 48, 50. As such, cathepsin B may, therefore, represent a valuable target for colorectal tumor therapy.
Supporting information
Additional supporting information may be found in the online version of this article at the publisher's web‐site.
Supporting Information Figure S1: Expression of cathepsin B and p27Kip1 in CRC cell lines.
Exponentially growth CRC cell lines were lysed in Laemmli buffer and analyzed by Western blotting with specific antibodies against cathepsin B (CTSB), p27Kip1 and β‐tubulin.
Supporting Information Table S1: Clinicopathological parameters of patients in the series of CRC samples analyzed in Figure 1A.
ACKNOWLEDGMENTS
The authors thank Anne Vézina for technical assistance, Claude Asselin for critical reading of the manuscript, and Pierre Pothier for editing of the manuscript. N.R is a recipient of a Canadian Research Chair in Colorectal Cancer and Inflammatory Signalling. J.C. and F.B. are scholars from the Fonds de la Recherche en Santé du Québec. N.R., J.C., and F.B. are members of the FRQS‐funded “Centre de Recherche du CHUS.”
REFERENCES
- 1. Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell 1990; 61:759–767. [DOI] [PubMed] [Google Scholar]
- 2. DeClerck YA, Mercurio AM, Stack MS, et al. Proteases, extracellular matrix, and cancer: A workshop of the path B study section. Am J Pathol 2004; 16:1131–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gondi CS, Rao JS. Cathepsin B as a cancer target. Expert Opin Ther Targets 2013; 17:281–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yan S, Sloane BF. Molecular regulation of human cathepsin B: Implication in pathologies. Biol Chem 2003; 384:845–854. [DOI] [PubMed] [Google Scholar]
- 5. Hazen LG, Bleeker FE, Lauritzen B, et al. Comparative localization of cathepsin B protein and activity in colorectal cancer. J Histochem Cytochem 2000; 48:1421–1430. [DOI] [PubMed] [Google Scholar]
- 6. Campo E, Munoz J, Miquel R, et al. Cathepsin B expression in colorectal carcinomas correlates with tumor progression and shortened patient survival. Am J Pathol 1994; 145:301–309. [PMC free article] [PubMed] [Google Scholar]
- 7. Cavallo‐Medved D, Dosescu J, Linebaugh BE, Sameni M, Rudy D, Sloane BF. Mutant K‐ras regulates cathepsin B localization on the surface of human colorectal carcinoma cells. Neoplasia 2003; 5:507–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Khan A, Krishna M, Baker SP, Banner BF. Cathepsin B and tumor‐associated laminin expression in the progression of colorectal adenoma to carcinoma. Mod Pathol 1998; 11:704–708. [PubMed] [Google Scholar]
- 9. Hung KE, Faca V, Song K, et al. Comprehensive proteome analysis of an Apc mouse model uncovers proteins associated with intestinal tumorigenesis. Cancer Prev Res (Phila) 2009; 2:224–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tu C, Ortega‐Cava CF, Chen G, et al. Lysosomal cathepsin B participates in the podosome‐mediated extracellular matrix degradation and invasion via secreted lysosomes in v‐Src fibroblasts. Cancer Res 2008; 68:9147–9156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Victor BC, Anbalagan A, Mohamed MM, Sloane BF, Cavallo‐Medved D. Inhibition of cathepsin B activity attenuates extracellular matrix degradation and inflammatory breast cancer invasion. Breast Cancer Res 2011; 13:R115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Marten K, Bremer C, Khazaie K, et al. Detection of dysplastic intestinal adenomas using enzyme‐sensing molecular beacons in mice. Gastroenterology 2002; 122:406–414. [DOI] [PubMed] [Google Scholar]
- 13. Chan AT, Baba Y, Shima K, et al. Cathepsin B expression and survival in colon cancer: Implications for molecular detection of neoplasia. Cancer Epidemiol Biomarkers Prev 2010; 19:2777–2785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Troy AM, Sheahan K, Mulcahy HE, Duffy MJ, Hyland JM, O'Donoghue DP. Expression of cathepsin B and L antigen and activity is associated with early colorectal cancer progression. Eur J Cancer 2004; 40:1610–1616. [DOI] [PubMed] [Google Scholar]
- 15. Hirai K, Yokoyama M, Asano G, Tanaka S. Expression of cathepsin B and cystatin C in human colorectal cancer. Hum Pathol 1999; 30:680–686. [DOI] [PubMed] [Google Scholar]
- 16. Boudreau F, Lussier CR, Mongrain S, et al. Loss of cathepsin L activity promotes claudin‐1 overexpression and intestinal neoplasia. FASEB J 2007; 21:3853–3865. [DOI] [PubMed] [Google Scholar]
- 17. Silverman RH. Viral encounters with 2′,5′‐oligoadenylate synthetase and RNase L during the interferon antiviral response. J Virol 2007; 81:12720–12729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Aronson NN, Jr , Barrett AJ. The specificity of cathepsin B. Hydrolysis of glucagon at the C‐terminus by a peptidyldipeptidase mechanism. Biochem J 1978; 171:759–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bond JS, Barrett AJ. Degradation of fructose‐1,6‐bisphosphate aldolase by cathepsin B. Biochem J 1980; 189:17–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rowan AD, Feng R, Konishi Y, Mort JS. Demonstration by electrospray mass spectrometry that the peptidyldipeptidase activity of cathepsin B is capable of rat cathepsin B C‐terminal processing. Biochem J 1993; 294:923–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Biniossek ML, Nagler DK, Becker‐Pauly C, Schilling O. Proteomic identification of protease cleavage sites characterizes prime and non‐prime specificity of cysteine cathepsins B, L, and S. J Proteome Res 2011; 10:5363–5373. [DOI] [PubMed] [Google Scholar]
- 22. Hershko DD, Shapira M. Prognostic role of p27Kip1 deregulation in colorectal cancer. Cancer 2006; 107:668–675. [DOI] [PubMed] [Google Scholar]
- 23. Loda M, Cukor B, Tam SW, et al. Increased proteasome‐dependent degradation of the cyclin‐dependent kinase inhibitor p27 in aggressive colorectal carcinomas. Nat Med 1997; 3:231–234. [DOI] [PubMed] [Google Scholar]
- 24. Adenis A, Huet G, Zerimech F, Hecquet B, Balduyck M, Peyrat JP. Cathepsin B, L, and D activities in colorectal carcinomas: Relationship with clinico‐pathological parameters. Cancer Lett 1995; 96: 267–275. [DOI] [PubMed] [Google Scholar]
- 25. Talieri M, Papadopoulou S, Scorilas A, et al. Cathepsin D expression in the progression of colorectal adenoma to carcinoma. Cancer Lett 2004; 205:97–106. [DOI] [PubMed] [Google Scholar]
- 26. Shuja S, Sheahan K, Murnane MJ. Cysteine endopeptidase activity levels in normal human tissues, colorectal adenomas and carcinomas. Int J Cancer 1991; 49:341–346. [DOI] [PubMed] [Google Scholar]
- 27. White BD, Chien AJ, Dawson DW. Dysregulation of Wnt/beta‐catenin signaling in gastrointestinal cancers. Gastroenterology 2012; 142:219–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Moser AR, Luongo C, Gould KA, McNeley MK, Shoemaker AR, Dove WF. ApcMin: A mouse model for intestinal and mammary tumorigenesis. Eur J Cancer 1995; 31A:1061–1064. [DOI] [PubMed] [Google Scholar]
- 29. Gounaris E, Tung CH, Restaino C, et al. Live imaging of cysteine‐cathepsin activity reveals dynamics of focal inflammation, angiogenesis, and polyp growth. PLoS ONE 2008; 3:e2916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tedelind S, Jordans S, Resemann H, et al. Cathepsin B trafficking in thyroid carcinoma cells. Thyroid Res 2011; 4:S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Roshy S, Sloane BF, Moin K. Pericellular cathepsin B and malignant progression. Cancer Metastasis Rev 2003; 22:271–286. [DOI] [PubMed] [Google Scholar]
- 32. Bromme D, Wilson SR. Role of cysteine cathepsin in extracellular proteolysis In: Park WC, Mecham RP, editors. Extracellular matrix degradation. Berlin Heidelberg: Springer‐Verlag; 2011. pp. 23–51. [Google Scholar]
- 33. Buck MR, Karustis DG, Day NA, Honn KV, Sloane BF. Degradation of extracellular‐matrix proteins by human cathepsin B from normal and tumour tissues. Biochem J 1992; 282:273–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Premzl A, Zavasnik‐Bergant V, Turk V, Kos J. Intracellular and extracellular cathepsin B facilitate invasion of MCF‐10A neoT cells through reconstituted extracellular matrix in vitro. Exp Cell Res 2003; 283:206–214. [DOI] [PubMed] [Google Scholar]
- 35. Kobayashi H, Ohi H, Sugimura M, Shinohara H, Fujii T, Terao T. Inhibition of in vitro ovarian cancer cell invasion by modulation of urokinase‐type plasminogen activator and cathepsin B. Cancer Res 1992; 52:3610–3614. [PubMed] [Google Scholar]
- 36. Artym VV, Kindzelskii AL, Chen WT, Petty HR. Molecular proximity of seprase and the urokinase‐type plasminogen activator receptor on malignant melanoma cell membranes: Dependence on beta1 integrins and the cytoskeleton. Carcinogenesis 2002; 23:1593–1601. [DOI] [PubMed] [Google Scholar]
- 37. Mason RW, Massey SD. Surface activation of pro‐cathepsin L. Biochem Biophys Res Commun. 1992; 189:1659–1666. [DOI] [PubMed] [Google Scholar]
- 38. Reddy VY, Zhang QY, Weiss SJ. Pericellular mobilization of the tissue‐destructive cysteine proteinases, cathepsins B, L, and S, by human monocyte‐derived macrophages. Proc Natl Acad Sci U S A 1995; 92:3849–3853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pungercar JR, Caglic D, Sajid M, et al. Autocatalytic processing of procathepsin B is triggered by proenzyme activity. FEBS J 2009; 276:660–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Vasiljeva O, Dolinar M, Pungercar JR, Turk V, Turk B. Recombinant human procathepsin S is capable of autocatalytic processing at neutral pH in the presence of glycosaminoglycans. FEBS Lett 2005; 579:1285–1290. [DOI] [PubMed] [Google Scholar]
- 41. Herszenyi L, Plebani M, Carraro P, et al. Proteases in gastrointestinal neoplastic diseases. Clin Chim Acta 2000; 291:171–187. [DOI] [PubMed] [Google Scholar]
- 42. Withana NP, Blum G, Sameni M, et al. Cathepsin B inhibition limits bone metastasis in breast cancer. Cancer Res 2012; 72:1199–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Koliadi A, Nilsson C, Holmqvist M, et al. Cyclin B is an immunohistochemical proliferation marker which can predict for breast cancer death in low‐risk node negative breast cancer. Acta Oncol 2010; 49:816–820. [DOI] [PubMed] [Google Scholar]
- 44. Sherr CJ, Roberts JM. CDK inhibitors: Positive and negative regulators of G1‐phase progression. Genes Dev 1999; 13:1501–1512. [DOI] [PubMed] [Google Scholar]
- 45. Lu Z, Hunter T. Ubiquitylation and proteasomal degradation of the p21(Cip1), p27(Kip1) and p57(Kip2) CDK inhibitors. Cell Cycle 2010; 9:2342–2352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lee J, Kim SS. The function of p27 KIP1 during tumor development. Exp Mol Med 2009; 41:765–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Fujita T, Liu W, Doihara H, Wan Y. Regulation of Skp2‐p27 axis by the Cdh1/anaphase‐promoting complex pathway in colorectal tumorigenesis. Am J Pathol 2008; 173:217–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Philipp‐Staheli J, Kim KH, Payne SR, et al. Pathway‐specific tumor suppression. Reduction of p27 accelerates gastrointestinal tumorigenesis in Apc mutant mice, but not in Smad3 mutant mice. Cancer Cell 2002; 1:355–368. [DOI] [PubMed] [Google Scholar]
- 49. Fuster JJ, Gonzalez JM, Edo MD, et al. Tumor suppressor p27(Kip1) undergoes endolysosomal degradation through its interaction with sorting nexin 6. FASEB J 2010; 24:2998–3009. [DOI] [PubMed] [Google Scholar]
- 50. Aoki K, Kakizaki F, Sakashita H, Manabe T, Aoki M, TAketo MM. Suppression of colonic polyposis by homeoprotein CDX2 through its nontranscriptional function that stabilizes p27Kip1. Cancer Res 2011; 71:593–602. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional supporting information may be found in the online version of this article at the publisher's web‐site.
Supporting Information Figure S1: Expression of cathepsin B and p27Kip1 in CRC cell lines.
Exponentially growth CRC cell lines were lysed in Laemmli buffer and analyzed by Western blotting with specific antibodies against cathepsin B (CTSB), p27Kip1 and β‐tubulin.
Supporting Information Table S1: Clinicopathological parameters of patients in the series of CRC samples analyzed in Figure 1A.