

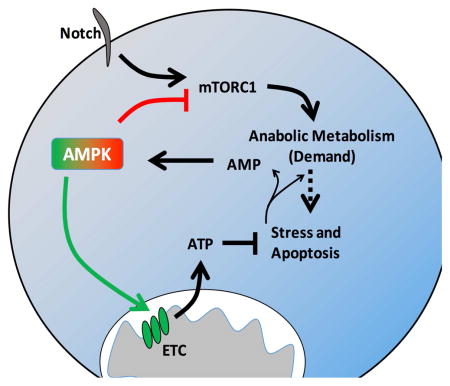
Summary
T cell acute lymphoblastic leukemia (T-ALL) is an aggressive malignancy associated with Notch pathway mutations. While both normal activated and leukemic T cells can utilize aerobic glycolysis to support proliferation, it is unclear to what extent these cell populations are metabolically similar and if differences reveal T-ALL vulnerabilities. Here we show that aerobic glycolysis is surprisingly less active in T-ALL cells than proliferating normal T cells and T-ALL cells are metabolically distinct. Oncogenic Notch promoted glycolysis but also induced metabolic stress that activated 5′ AMP activated kinase (AMPK). Unlike stimulated T cells, AMPK actively restrained aerobic glycolysis in T-ALL cells through inhibition of mTORC1 while promoting oxidative metabolism and mitochondrial Complex I activity. Importantly, AMPK-deficiency or inhibition of Complex I led to T-ALL cell death and reduced disease burden. Thus AMPK simultaneously inhibits anabolic growth signaling and is essential to promote mitochondrial pathways that mitigate metabolic stress and apoptosis in T-ALL.
Graphical Abstract
Introduction
While resting cells typically rely on mitochondrial oxidative phosphorylation to meet bio-energetic needs, cancer cells often utilize a metabolic program known as aerobic glycolysis (Cantor and Sabatini, 2012; Hanahan and Weinberg, 2011). Aerobic glycolysis is characterized by increased glucose import and flux through glycolysis and subsequent production of lactate even under normoxic conditions (Warburg et al., 1927). Tumor cells are thought to utilize aerobic glycolysis to allow diversion of glycolytic intermediates to biosynthetic pathways to generate lipids, nucleotides, and amino acids necessary for cell growth and division (Vander Heiden et al., 2009).
T cell acute lymphoblastic leukemia (T-ALL) is a rapidly proliferating malignancy that, while generally well treated (Pui et al., 2008), has a poor prognosis upon relapse or with advanced age at onset (Bhojwani and Pui, 2013; Oudot et al., 2008). T-ALL is frequently associated with Notch signaling pathway mutations and greater than 60% of human patients exhibit activating mutations in the Notch pathway (Weng et al., 2004). Although Notch can promote glycolytic metabolism in T-ALL cell lines (Palomero et al., 2007) and developing T cells (Ciofani and Zuniga-Pflucker, 2005), recent work has suggested that Notch signaling also drives mitochondrial oxidative metabolism in the context of macrophage polarization (Xu et al., 2015) and in T-ALL cell lines (Palomero et al., 2006). Oncogenic Notch can promote PI3K pathway (Palomero et al., 2007) and c-Myc signaling (Palmer et al., 2015; Palomero et al., 2006) that promotes glutamine oxidation (Herranz et al., 2015). Stimulated normal T cells also activate the PI3K and c-Myc pathways and utilize aerobic glycolysis to rapidly proliferate and carry out immunological functions (Gerriets et al., 2015; Macintyre et al., 2014; Wang et al., 2011). It is unclear, however, to what extent metabolic programs of activated or transformed T cells were similar and if differences may reveal T-ALL vulnerabilities.
In contrast to PI3K, 5′ AMP-activated kinase (AMPK) can inhibit mTORC1 signaling (Gwinn et al., 2008; Inoki et al., 2003). AMPK is activated by the tumor suppressor LKB1(Shaw et al., 2004) and can have growth suppressive functions in cancer settings (Faubert et al., 2013). Further, pharmacological activation of AMPK can slow the growth of some tumors (Hirsch et al., 2009) and AMPK may act to inhibit tumor growth in T-ALL (Mavrakis et al., 2010). Conversely, multiple oncogenic signals, including oncogenic Ras and Myc, can generate metabolic stress (Liu et al., 2012; Moiseeva et al., 2009), and AMPK may promote cancer cell survival under such conditions. Indeed, LKB1 loss sensitizes to metabolic stress(Shackelford et al., 2013) and AMPK may be important to mitigate metabolic stress in myeloid leukemia initiating cells(Saito et al., 2015) and activated T cells in vivo (Blagih et al., 2015).
Here we compared the metabolic programs of primary T-ALL and normal proliferative T cells. As anticipated, primary human T-ALL samples used and required aerobic glycolysis. However, T-ALL glucose metabolism was surprisingly restrained compared to the glycolytic metabolism of normal proliferating T cells and T-ALL and proliferating T cells had different global metabolomes. Consistent with chronic metabolic stress, AMPK was activated and suppressed mTORC1 signaling and glycolysis while supporting mitochondrial metabolism that we found essential for T-ALL cell survival in vivo. Thus, T-ALL cells utilize a distinct metabolic program in which Notch-induced metabolic stress leads to AMPK activation that plays dual roles to inhibit anabolic metabolism while also promoting T-ALL cell survival through positive regulation of mitochondrial metabolism.
Results
Primary T-ALL cells exhibit increased glycolysis that is necessary for cell survival and disease progression
The metabolic profile of primary T-ALL cells is poorly understood. Therefore, we examined the expression levels of the glycolytic proteins Glut1, the primary glucose transporter in T cells (Macintyre et al., 2014), and Hexokinase 2 (HK2), a hexokinase isoform commonly associated with aerobic glycolysis (Gershon et al., 2013; Wolf et al., 2011), in primary human patient peripheral blood samples of T-ALL. Both Glut1 (Figure 1A, S1A) and HK2 (Figure 1B, S1B) were significantly elevated in T-ALL cells compared to naïve T cells from healthy human blood donors. Interestingly, the expression of HK1 (Figure 1C, S1C) was significantly lower in T-ALL cells than in naïve T cells. Human T-ALL cell lines also showed a similar pattern of expression of Glut1, HK1, and HK2 (Figure 1D).
Figure 1. Primary T-ALL cell usage of aerobic glycolysis is essential for survival and disease progression.

A–C. Primary human T-ALL samples and naïve human CD4 T cells were compared for expression of (A) Glut1, (B) HK2 and (C) HK1 by intracellular flow cytometry. D. Human T-ALL cell lines HPB-ALL, SIL-ALL, and DND41 were compared to naïve human CD4 T cells for Glut1, HK2 and HK1 expression by immunoblot. E. Primary human T-ALL and naïve human CD4 T cell glucose uptake was measured. F. OCR and ECAR of primary murine T-ALL and naïve murine T cells were measured. G. Primary human T-ALL samples were treated with vehicle or 10 mM 2DG and the percentage of viable cells that were T-ALL cells (CD4+CD8+) or CD4+ T cells was measured by propidium iodide exclusion flow cytometry. H–I. Primary T-ALL was generated on a Glut1 inducible deletion background and transplanted into sublethally irradiated recipient mice. Mice were treated with vehicle (n=4) or tamoxifen (n=5) for 4 consecutive days starting 4 days after T-ALL transplant and survival was monitored (H) or treated with vehicle (n=4) or tamoxifen (n=5) 10 days after transplant, sacrificed 2 days after treatment completion to (I) measure T-ALL burden. Data are representative of 6 individual human T-ALL samples and 6 independent naïve T cell samples (A–C) see also Supplemental Figure 1A–1C, two independent experiments (D–F), three independent experiments with individual human T-ALL samples (G) see also Supplement Figure 1D–1E or two independent experiments with at least n=4 mice per group (H, I). Data are shown as the mean and standard deviation (* p < 0.05).
We next sought to determine if T-ALL cells used aerobic glycolysis relative to normal resting T cell counterparts. Relative to resting T cells primary human TALL cells had increased rates of glucose uptake (Figure 1E). We then compared the metabolic activity of primary T-ALL cells with resting T cells using a well-characterized primary murine model of Notch driven T-ALL in which retroviral expression of oncogenic intracellular Notch (ICN1) in hematopoietic progenitor cells (HPCs) drives T-ALL initiation and maintenance (Chiang et al., 2008). Primary murine Notch driven T-ALL exhibited increased oxygen consumption rate (OCR) and lactate production as measured by extracellular acidification rate (ECAR) using extracellular flux analysis (Figure 1F) compared to naïve murine T cells. Primary human T-ALL also exhibited increased ECAR relative to naïve human T cells (Figure S1D).
Although T-ALL cells utilized aerobic glycolysis, the dependence of these cells on this metabolic program was unclear. Therefore, we treated primary human T-ALL samples with 2-deoxyglucose (2DG) to inhibit glycolysis. Because each sample was comprised of a mixed population of tumor cells (CD4+CD8+) and non-malignant T cells (CD4+ or CD8+), the effects of 2DG on transformed and non-malignant T cells were compared within the same sample. While the percentage of cancer cells in the total live cell population declined over time with 2DG treatment, the fraction of non-malignant CD4+ T cells remained unchanged or increased (individual patient samples shown in Figures 1G, S1E, S1F). Isolated resting CD4+ T cells from healthy independent donors also did not show reduced viability upon 2DG treatment (Figure S1G).
To directly test the dependence of T-ALL cells on aerobic glycolysis in vivo, Glut1 was conditionally deleted in T-ALL. We generated T-ALL from a Glut1 floxed, inducible Cre background (Glut1fl/fl; Rosa26CreERT2) in which Cre is activated by treatment with tamoxifen to delete Glut1. Primary T-ALL cells were transplanted into secondary recipient mice, which were treated with tamoxifen or vehicle and monitored for survival. Tamoxifen-treated mice survived significantly longer than vehicle-treated mice (Figure 1H), supporting a dependence of T-ALL cells on Glut1. We also examined T-ALL cells with acute deletion of Glut1. In these studies secondary recipients of Glut1fl/fl;Rosa26CreERT2 background primary T-ALL were treated with tamoxifen starting 10 days after transplant, then analyzed after an additional 5 days. This protocol showed a significant reduction in Glut1 expression and the number of T-ALL cells in the spleens of treated mice (Figure 1I and S2A).
To further support a role for glucose metabolism, HK2 was conditionally deleted in T-ALL cells (Figure S2B). This increased animal survival (Figure S2C) and reduced T-ALL burden (Figure S2D). HK2 can promote generation of nucleotides through the pentose phosphate pathway (PPP) (Patra et al., 2013). T-ALL cells isolated five days after HK2 deletion had decreased PPP flux (Figure S2E) and reduced nucleotide and nucleoside levels (Figure S2F, Supplemental Table 1). We also observed an increase in non-phosphorylated glucose and a reduction in fructose 1,6-bisphosphate consistent with reduced glycolytic flux (Figure S2G). Importantly, Cre activation itself had no effect on mouse survival (Figure S3A), tumor burden (Figure S3B–S3E), or PPP flux (Figure S3F).
Glycolysis is selectively restrained in T-ALL
As T-ALL cells exhibited increased glycolytic activity relative to naïve T cells and required this pathway, we sought to compare T-ALL metabolism to that of normally proliferating T cells. Primary TALL was compared to naïve ex vivo murine T cells and T cells that were stimulated for 24 or 48 hours with plate bound anti-CD3 and anti-CD28 using non-targeted mass spectrometry metabolomics analysis. Clustering and principle component analysis (PCA) showed that the metabolomic profile of T-ALL cells is distinct from that of naïve ex vivo T cells as well as 24 and 48 hr activated T cells (Figure 2A, 2B, Supplemental Table 2). Pathway analysis of metabolomics data showed that T-ALL cells had increased concentrations of metabolites associated with the TCA cycle, glutathione metabolism, nucleotide synthesis and fatty acid oxidation when compared to naïve T cells (Supplemental Table 3). Compared to 48 hr stimulated T cells, T-ALL cells instead had increased concentrations of metabolites associated with the oxidation of amino and fatty acids and lower levels of metabolites associated with glycolytic metabolism, amino acid metabolism, nucleotide production, and mitochondrial activity (Supplemental Table 4). In direct metabolic measurements primary murine T-ALL cells were found to have consistent elevation of glucose uptake (Figure 2C), glycolytic flux through enolase (Figure 2D) and PPP flux (Figure 2E) compared to naïve T cells. However, these pathways were significantly lower in T-ALL than activated proliferating T cells. T-ALL cell mitochondrial content and membrane potential were, in contrast, similar to 48 hr stimulated T cells (Figure 2F, Figure S4A).
Figure 2. Primary T-ALL metabolism is distinct from naïve and activated normal T cells.

A–B. Samples of primary murine T-ALL, naïve, and activated T cells were extracted and analyzed using high-resolution LC-QE-MS and show a (A) heat map with unsupervised hierarchical clustering of the top 250 differentially observed analytes by ANOVA and (B) principle component analysis of each sample. C–F. Murine naïve T cells, T cells that were activated for 24 or 48 hrs, and purified primary T-ALL cells were analyzed for (C) glucose uptake, (D) glycolytic flux, (E) PPP glucose oxidation, (F) mitotracker green and TMRE staining. Data are representative of at least 3 biological replicates per group (AB), or at least 4 independent experiments (C–F); see also Supplemental Figure 4C. Data are shown as the mean and standard deviation (* p < 0.05).
Oncogenic Notch regulates glycolytic and oxidative metabolism
Notch likely has diverse effects on metabolism. To examine oncogenic Notch signaling in T-ALL metabolism, oncogenic Notch (ICN) was retrovirally transduced into primary murine hematopoietic progenitor cells (HPCs), the cell of origin of Notch-driven T-ALL (Chiang et al., 2008). ICN transduced cells were cultured for 4 days on OP9 stromal cells and compared to vector control infected cells. Notch-ICN resulted in increased cell size as measured by FSC in primary HPCs (Figure 3A). Conversely, gamma secretase inhibitor (GSI) inhibition of Notch signaling in Notch-dependent human T-ALL cell lines DND41 and HPB-ALL resulted in decreased FSC (Figure 3B). Notch-ICN also increased cell surface localized Glut1 in primary HPCs as measured in cells expressing Glut1myc (Figure 3C) and showed elevated total Glut1 and HK2 expression (Figure 3D). Conversely, GSI treatment of DND41 and HPB-ALL cells decreased expression of HK2 (Figure 3E). Finally, retroviral expression of Notch-ICN resulted in increased glucose uptake in HPCs (Figure 3F).
Figure 3. Oncogenic Notch signaling regulates glycolytic and mitochondrial metabolism.

A, C–D, F–G, I–L. Lineage negative hematopoietic cells were isolated from the bone marrow of wild type (A, D, F–G, I–L) or Glut1myc expressing (C) mice and retrovirally transduced with ICN1 or vector control. Cell size by forward scatter (FSC) (A) and surface Glut1myc expression (C) were measured by flow cytometry. Total Glut1 and HK2 expression were analyzed by immunoblot (D). Virally transduced cells were isolated to measure (F) glucose uptake, (G) cytochrome C expression by immunoblot, (I) basal and (J) maximal OCR. K–L. Transduced cells were analyzed by flow cytometry for (K) mitotracker green staining, (L) TMRE. B, E, H. Human T-ALL cell lines DND41 and HPB-ALL were treated with DMSO vehicle or 100 nM Compound E for 96 hrs and (B) cell size was measured by FSC. Cell lysates were also analyzed by immunoblot for (E) HK2 and (H) cytochrome C expression. Data are representative of at least three independent experiments. Data are shown as the mean and standard deviation (* p < 0.05).
Oncogenic Notch signaling also modulated mitochondrial metabolism. Notch-ICN transduced HPCs had increased expression of cytochrome C (Figure 3G), while GSI treatment of human T-ALL cell lines resulted in reduced cytochrome C (Figure 3H). Viral transduction of Notch-ICN into primary HPC cells also resulted in increased basal and maximal OCR (Figure 3I, 3J), mitochondrial content (Figure 3K, S5A) and membrane potential (Figure 3L, S5B). Reactive oxygen species (ROS) content as measured by DCFDA staining was not significantly altered (Figure S5C).
Notch signaling can activate both the PI3K/AKT/mTOR and c-Myc signaling pathways. To examine how oncogenic Notch signaling regulates metabolism in T-ALL, we treated Notch transduced HPCs with the PI3K pathway inhibitors rapamycin, LY294002 and PP242, and the bromodomain inhibitor JQ1, previously shown to inhibit c-myc activity (Delmore et al., 2011), and measured effects on metabolism. Each treatment resulted in a significant reduction in cell size (Figure S5D), surface Glut1 (Figure S5E) and HK2 expression (Figure S5F).
Oncogenic Notch signaling in T-ALL results in metabolic stress and AMPK pathway activation
Because the PI3K pathway mediated metabolic changes in Notch-ICN transduced cells, we examined this pathway in T cells and T-ALL. In HPCs, Notch-ICN increased phospho-mTOR, yet the mTORC1 downstream effector phospho-S6 ribosomal protein remained surprisingly low (Figure S5G). Similarly, primary murine T-ALL cells displayed increased phospho-mTOR compared to naïve and 48 hr in vitro stimulated T cells, but levels of phospho-S6 and -4EBP1 in T-ALL cells were far below that of stimulated T cells and pS6K levels were reduced relative to total S6K (Figure 4A).
Figure 4. Oncogenic Notch signaling drives metabolic stress and AMPK pathway activation in T-ALL.

A. Primary murine T-ALL, naïve and activated T cells were analyzed by immunoblot. B. Two independent samples of primary human T-ALL and naïve CD4+ T cells were analyzed by immunoblot. C. Lineage negative hematopoietic cells were isolated from the bone marrow of wild type mice and retrovirally transduced with ICN1 or vector control and analyzed by immunoblot. D. Human T-ALL cell lines DND41 and HPB-ALL were treated with vehicle or 100 nM Compound E for 96 hrs and analyzed by immunoblot. E–G. Murine naïve T cells and purified primary T-ALL cells were and samples were extracted and analyzed using high-resolution LC-QE-MS for metabolomics analysis. Data are representative of at least three independent experiments (A, C–D), two independent human T-ALL samples (B) or at least four independent biological replicates (E–G). Data are shown as the mean and standard deviation (* p < 0.05).
These data suggested AMPK signaling pathway may be activated in TALL to suppress mTORC1 activity and oncogenic signals may generate metabolic stresses that activate the AMPK pathway (Liu et al., 2012). Compared to naïve T cells, phosphorylation of AMPK and the AMPK target, ACC, were indeed increased in both primary murine T-ALL and activated T cells (Figure 4A). However, T-ALL cells showed phosphorylation of RAPTOR at Ser792 (Figure 4A), while activated T cells did not. Phosphorylation of this site by AMPK can negatively regulate glycolysis and mTORC1 (Gwinn et al 2008). Naïve T cells also exhibited high levels of phospho-RAPTOR S792 that correlate with low glycolysis and mTORC1 activity. Phospho-ACC was also elevated in two independent primary human T-ALL (Figure 4B) and Notch-ICN was sufficient to activate AMPK in primary HPCs (Figure 4C). Conversely, inhibition of Notch in human T-ALL cell lines decreased AMPK pathway activity (Figure 4D). Because AMPK is regulated in part by ATP, ADP, and AMP, ATP levels were assessed in T-ALL compared to naïve ex vivo T cells. Primary murine T-ALL cells had reduced levels of ATP compared to naïve T cells (Figure 4E), as well as decreased ratios of ATP/AMP (Figure 4F) and ATP/ADP (Figure 4G), suggesting that T-ALL cells experience chronic ATP-insufficiency that may promote AMPK pathway activation.
AMPK signaling suppresses mTORC1 activity in primary T-ALL, resulting in decreased aerobic glycolysis
As T cells exclusively express the catalytic α1 subunit of AMPK (MacIver et al., 2011), T-ALL was generated on an AMPKα1fl/fl;Rosa26CreERT2 background to directly test the role of AMPK in TALL physiology and survival. Primary T-ALL was transplanted into cohorts of sublethally irradiated recipients that were treated with tamoxifen for 4 days after 10 days engraftment. Consistent with the AMPKα1 isoform as the major AMPKα subunit in T-ALL, tamoxifen treatment resulted in a marked decrease in total AMPKα protein expression levels, along with a reduction in the phosphorylation of AMPK target ACC (Figure 5A). The loss of AMPK in T-ALL also resulted in a reduction of inhibitory phosphorylation of RAPTOR (Figure 5A) and increased mTORC1 activity, with higher levels of phospho-S6 kinase, phospho-S6 ribosomal protein, and phospho-4EBP1 (Figure 5A). Glut1 and HK2 were also increased in AMPKα1 deficient T-ALL cells (Figure 5B) and AMPK deletion in vivo led to an increase in ECAR (Figure 5C), and an increase in the expression of genes in the PPP (Figure 5D, Supplemental Table 5). Importantly, alterations in glycolytic flux were not due to Cre activation (Figure S3G). Metabolomic analysis revealed increased levels of metabolites (Figure 5E–G, Supplemental Table 6) associated with mTORC1-regulated anabolic synthesis of nucleotides (Ben-Sahra et al., 2013; Robitaille et al., 2013). AMPK, therefore, actively restrains mTORC1 signaling and de novo nucleotide synthesis of T-ALL cells in vivo.
Figure 5. AMPK signaling negatively regulates mTORC1 and inhibits aerobic glycolysis in T-ALL.

Primary T-ALL was generated on an AMPKα1 inducible deletion background and transplanted into sublethally irradiated recipient mice. Mice were treated with vehicle or tamoxifen for 4 days starting 10 days after transplant and sacrificed 2 days after treatment completion. A–C. Purified T-ALL cells from vehicle and tamoxifen treated groups were isolated and (A–B) analyzed by immunoblot (C) ECAR was measured. D–G. Isolated T-ALL cells from four separate mice in vehicle and tamoxifen treated groups were examined by rtPCR for select PPP gene expression (D) or were (E–G) extracted and analyzed using high-resolution LC-QE-MS. The normalized abundance of select metabolites is shown. Data are representative of two independent experiments with three independent T-ALL samples per group (A–C) or an experiment with four independent T-ALL samples per group (D–G). Data are shown as the mean and standard deviation. Western blot quantifications are normalized to total protein level where applicable or to actin (* p < 0.05).
AMPK pathway signaling promotes oxidative metabolism through regulation of mitochondrial Complex I
AMPK can promote mitochondrial metabolism to mitigate metabolic stress (Lantier et al., 2014). Consistent with this role, we observed a reduction in the OCR (Figure 6A) and ratio of OCR/ECAR was significantly lowered in AMPKα1 deficient T-ALL cells (Figure 6B), indicating that the loss of AMPK resulted in a shift towards increased glycolysis. No decrease was observed in OCR as a result of Cre activation alone (Figure S3H). OCR coupled to ATP production, defined by oligomycin-sensitive OCR, was also significantly reduced in AMPKα1 deficient T-ALL cells (Figure 6C). Further, metabolomics analysis showed an increase in the ratio of NAD+/NADH in AMPKα1 deficient T-ALL cells (Figure 6D) and we observed an increase in ROS levels (Figure 6E), suggesting AMPK was essential to maintain a sufficient reductive mitochondrial capacity. Analysis of mitochondrial gene expression revealed that AMPK selectively regulated the expression of electron transport Complex I (Figure 6F, S6A, Supplemental Table 7), although we observed only modest reduction of NDUFS1 protein, a component of Complex I, in AMPKα1 deficient T-ALL cells (Figure S6B). Importantly, direct measurement of Complex I function revealed reduced activity in AMPKα1 deficient T-ALL cells (Figure 6G, S6C).
Figure 6. AMPK promotes mitochondrial oxidative metabolism in T-ALL through the regulation of Complex I.

Primary T-ALL was generated on an AMPKα1 inducible deletion background and transplanted into sublethally irradiated recipient mice. A–D. Mice were treated with vehicle or tamoxifen for 4 days starting 10 days after transplant and sacrificed 2 days after treatment completion. Purified T-ALL cells from vehicle and tamoxifen treated groups were isolated and (A) OCR, (B) OCR/ECAR ratio and (C) ATP coupled OCR, defined as oligomycin sensitive OCR, were measured or (D) cells were analyzed using high-resolution LC-QE-MS. The normalized abundance of select metabolites is shown. E–G. T-ALL cells were isolated from vehicle and tamoxifen treated mice for (E) Flow cytometry analysis of DCFDA and (F) rtPCR measurement of select mitochondrial energy pathway gene expression (G) analysis of Complex I enzyme activity. Unsupervised hierarchical clustering of gene expression is shown for panels F. Data represent an experiment with four independent T-ALL samples per group (A–D, F–G), or two independent experiments with n = 8 per group (E). Data are shown as the mean and standard deviation (* p < 0.05).
Loss of AMPK signaling or direct pharmacological inhibition of Complex I results in reduced T-ALL cell survival and slowed disease progression
We next tested the effects of AMPK deficiency in T-ALL on disease progression. Despite increased mTORC1 activity, acute deletion of AMPKα1 resulted in reduced numbers and percentages of splenic, lymph node, and bone marrow T-ALL cells (Figure 7A–C). Reductions were associated with increased T-ALL cell staining for Annexin V that indicate early apoptosis and reduced percentages of viable cells (Figure 7D). Cell proliferation, as measured by Ki67 staining, was not significantly altered (Figure S7A). T-ALL cell number and percentage were also decreased in the bone marrow following AMPKα1 deletion at a later time post tamoxifen treatment (Figure S7B), with similar increases in cell death (Figure S7C). Importantly, lower T-ALL numbers correlated with increased animal survival (Figure 7E). Consistent with this role for AMPK to support T-ALL, analysis of two published data sets (Homminga et al., 2011; Sanghvi et al., 2014) indicated no loss of expression of AMPKα1 in human T-ALL compared with control cells (Figure S7D). Therefore, despite actively suppressing T-ALL anabolic metabolism, AMPK is essential for T-ALL cell survival and disease progression.
Figure 7. AMPK and mitochondrial Complex I activity are necessary to maintain T-ALL cell viability and disease progression.

A–E. Primary T-ALL was generated on an AMPKα1 inducible deletion background and transplanted into sublethally irradiated recipient mice. (A–D). Mice were treated with vehicle or tamoxifen for 4 days starting 10 days after transplant and sacrificed 2 days after treatment completion. T-ALL cell burden and percentage present in the (A) spleen, (B) inguinal lymph nodes, and (C) bone marrow were determined by flow cytometry. D. Annexin V and propidium iodide staining was measured by flow cytometry. (E) Mice were treated with vehicle or tamoxifen for 4 days beginning 4 days after transplant and given daily 4-hydroxytamoxifen (5 mg/kg body weight) for 5 days beginning 10 days after transplant to maintain gene deletion and overall survival was measured. F–I. Primary murine T-ALL was generated on a wild type background. T-ALL was isolated and, (F) along with naïve T cells and T cells that were activated in vitro for 48 hrs, were treated with 100 nM rotenone or DMSO vehicle or (G) 100 μM phenformin or PBS vehicle and cell survival was measured over time by propidium iodide exclusion flow cytometry. Viability normalized to vehicle treatment is shown. Isolated T-ALL was treated for 45 minutes with 100 μM phenformin or PBS vehicle and (H) OCR and (I) ECAR were measured. J–L. Primary murine T-ALL was transplanted into sublethally irradiated recipient mice. Mice were dosed daily with vehicle (PBS) or phenformin (100 mg/kg body weight) for 10 consecutive days starting two days after T-ALL transplant. Mice were rested for 2 days, sacrificed and (J) DCFDA staining, (K) spleen cellularity and (L) T-ALL cell burden in the spleen were analyzed for each group. Data are representative of three independent experiments with at least five mice per group (A), an experiment with seven mice per group (B–D), an experiment with ten mice per group (E), three independent experiments (F–I) or two independent experiments with at least five mice per group (J–L). Data are shown as the mean and standard deviation (* p < 0.05).
Because AMPK regulated mitochondrial Complex I, the effects of direct pharmacological inhibition of mitochondrial electron transport on T-ALL were tested. Primary murine T-ALL cells were treated along with naïve ex vivo T cells and 48 hr stimulated T cells with low doses of the Complex I inhibitor rotenone (100 nM, Figure 7F). Interestingly, while the viability of both naïve and in vitro stimulated T cells was not significantly altered by low dose rotenone treatment, primary T-ALL cells were rapidly killed. Similar to rotenone, the clinically relevant Complex I inhibitor phenformin rapidly killed primary T-ALL cells while sparing both naïve and in vitro stimulated T cells (Figure 7G). Phenformin also led to a reduction in the mitochondrial oxygen consumption rate (Figure 7H), coupled with an increase in ECAR (Figure 7I) resulting in a reduction in the OCR/ECAR ratio (Figure S7E).
We next tested the pharmacological impact of phenformin on T-ALL in vivo. Wild type primary T-ALL was transplanted into cohorts of secondary recipient mice and dosed daily with phenformin or vehicle for 10 consecutive days. Mice were rested for 2 days then sacrificed for analysis. T-ALL cells from phenformin treated mice had elevated levels of cytoplasmic ROS (Figure 7J), consistent with Complex I inhibition (Li et al., 2003). Importantly, phenformin treatment reduced the number (Figure 7K) and percentage (Figure 7L) of T-ALL cells in the treated mice and resulted in a significant increase in overall animal survival (Figure S7F).
Discussion
The metabolism of cancer cells has been largely defined relative to resting mature tissues. Comparisons of proliferative normal and transformed cells from similar origins may, however, reveal cancer specific vulnerabilities or selective weaknesses. Here we examined the metabolism of T-ALL cells in comparison to resting and proliferative T cells and found that T-ALL cells utilize aerobic glycolysis, but are restrained from reaching the levels of activated T cells. Specifically, Notch induced metabolic stress that led to AMPK activation that both slowed glycolysis and promoted mitochondrial oxidative metabolism to mitigate stress. Exacerbating this stress by AMPK or mitochondrial inhibition may provide a new approach to treat T-ALL.
Antigen stimulated effector T cells utilize aerobic glycolysis during rapid cellular proliferation to mount immune responses. Previous work has shown that, like many cancers (Liu et al., 2014a), inhibition of T cell glycolysis can result in reduced cell viability and loss of immunological function (Gerriets et al., 2015; Macintyre et al., 2014). Because of this, activated T cells have been proposed as a model for cancer metabolism (Macintyre and Rathmell, 2013). Activated T cells, however, have a short lifespan unless able to transition from glycolysis back to oxidative metabolism (Sukumar et al., 2013; van der Windt et al., 2012). While cancer may select for maximal proliferation, cells with prolonged capacity for replication and survival are also favored. T-ALL cells thus appear reminiscent of long-lived T cells that have adapted to balance glycolysis with mitochondrial oxidative capacity.
Several factors may mediate this difference between activated T cells and T-ALL cells. First, our comparison is between acutely activated T cells and chronic oncogenic signals that drive anabolic growth. Indeed mitochondria are hyperpolarized in T cells of Systemic Lupus Erythematosus patients, suggesting that chronic autoimmunity may lead to mitochondrial compensation (Gergely et al., 2002). Likewise, the immune modulatory protein PD-1 can restrain T cell glycolysis (Patsoukis et al., 2015) and lead to functional exhaustion (Zinselmeyer et al., 2013) in chronic infection or cancer. Mechanisms that reprogram chronically stimulated T cells are unclear, but AMPK was recently shown to be required to allow some T cell mediated immunity (Blagih et al., 2015) and may act similarly in T-ALL. Second, unlike T cell activation that induces a coordinated signal to promote T cell proliferation and viability, T-ALL cells are driven by limited oncogenic signals that may lead to imbalanced metabolic alterations. Although Notch can increase mitochondrial metabolism, this imbalance may promote metabolic stress.
Our data show a dual role for AMPK in T-ALL. AMPK has previously been shown to suppress aerobic glycolysis and progression of B cell leukemia through the inhibition of HIF1α (Faubert et al., 2013). Recent work has also indicated AMPK was necessary to maintain AML tumor initiating cell viability under conditions of metabolic stress through promoting the expression of Glut1 (Saito et al., 2015). We show AMPK acts in T-ALL to negatively regulate aerobic glycolysis and Glut1 while also maintaining mitochondrial function to promote cell survival. These data suggest a context specific role for AMPK. Our findings are consistent with the previously observed role for AMPK in the T cell lineage, where it can inhibit glycolysis and mTORC1 signaling, yet promote T cell survival (MacIver et al., 2011) and function in vivo (Blagih et al., 2015). ARK5, a regulator of AMPK pathway signaling, was also shown to be critical to mitigating ATP stress and promote hepatocellular carcinoma (Liu et al., 2012). Our data demonstrate that AMPK itself plays a critical role in murine T-ALL survival. AMPK activation would protect from metabolic stress both by reducing energy demand through inhibition of mTORC1-regulated anabolic growth and by promoting energy generation through increased mitochondrial Complex I and catabolic metabolism. The loss of AMPK in T-ALL resulted in increased mTORC1 signaling and anabolic metabolism while also leading to an increase in apoptosis. Any protective effect of increased PPP activity, therefore, was insufficient to prevent death. It is interesting that while AMPK was active in both activated T and T-ALL cells, RAPTOR was phosphorylated only in T-ALL cells, suggesting additional regulation.
The paradoxical dual role for AMPK was likely due to AMPK-induced support of mitochondrial metabolism. Indeed, AMPK deletion led to increased NAD/NADH ratio and ROS, demonstrating a requirement for AMPK to maintain cellular reductive capacity. This is consistent with previous work showing that AMPK activity was necessary to maintain tumor cell redox balance (Jeon et al., 2012). AMPK deficiency also led to reduced mitochondrial oxidative metabolism that was characterized by altered mitochondrial Complex I. The effects of AMPK deletion on Complex I may potentially be indirect, such as through HIF1α (Faubert et al., 2013) which can negatively regulate mitochondrial metabolism (Tello et al., 2011). AMPK may also regulate activity of Complex I. While loss of LKB1 and AMPK activation sensitized non-small cell lung cancer cells to mitochondrial inhibition (Shackelford et al., 2013), we found mitochondrial metabolism directly limiting in T-ALL. T-ALL appear under basal metabolic stress and inhibition of mitochondrial Complex I was sufficient to result in rapid T-ALL cell death in vitro and in vivo.
Cancer cells must successfully balance the metabolic demands of rapid cellular proliferation with energetic requirements to maintain viability. Thus, interventions that deliberately skew this balance towards pro-growth activities may instead exacerbate stress and result in a loss of tumor cell viability. Similarly, excessive oncogenic signaling may actually result in the loss of tumor cell viability (Chen et al., 2015). Metabolic stress may be a key and potentially limiting feature of oncogenesis. While our findings here are limited to Notch-induced T-ALL, Ras and Myc can also each drive metabolic stress in tumor cells (Liu et al., 2012; Moiseeva et al., 2009). Further, unrestrained usage of aerobic glycolysis itself may reduce cell life span. Indeed, mutations that result in increased lymphocyte glycolysis can result in immunosuppression due to an inability to generate long-lived memory cells (Angulo et al., 2013; Lucas et al., 2014). Moderated aerobic glycolysis along with alternative metabolic pathways may therefore be required for tumor cells to maintain proliferative capacity and viability over the long term. Mitochondrial metabolism appears limiting and may thus offer new therapeutic targets in the treatment of T-ALL.
Experimental procedures
Human samples and cell lines
DND-41, HPB-ALL and ALL-SIL cell lines were obtained from DSMZ (Braunschweig, Germany). Human T-ALL samples were obtained from the Children’s Oncology Group Biobanking Facility (5 samples), the University of North Carolina Cancer Hospital Tissue Procurement Facility (1 sample), or the Singapore Leukemia Tissue Bank (2 samples). Patient samples were chosen sequentially based solely on a diagnosis of T-ALL and availability of tumor samples obtained from peripheral blood prior to treatment. Samples were collected according to an IRB-approved tissue collection protocol at each institution, and were de-identified prior to use.
Mice
Mice were obtained from Jackson Laboratory or have been previously described (Gershon et al., 2013; Macintyre et al., 2014; Michalek et al., 2011). Eight to twelve week old C57BL/6J mice were used throughout. All procedures were performed under protocols approved by the Duke University Medical Center Institutional Animal Care and Use Committee (IACUC).
T-ALL mouse model
T-ALL was generated in mice as previously described (Herranz et al., 2015), with minor modifications. In brief, lineage negative hematopoietic cells were isolated from murine bone marrow using Miltenyi Lineage Cell Depletion Kit (130-090-858). Isolated cells were cultured for 16 hrs in X-VIVO 15 media (Lonza 04-418Q) with 100 ng/mL murine stem cell factor (Peprotech 250-03), 10 ng/mL murine IL3 (Peprotech 213-13), 10 ng/mL human IL6 (Peprotech 200-06) and 50 ng/mL FLT3-ligand (Peprotech 250-31L). Cells were then treated with freshly made MSCV-ICN1-IRES-GFP or MSCV-ICN1-IRES-NGFR or vector control virus along with 8 μg/mL polybrene on retronectin coated plates and centrifuged for 45 min at 1200 rpm. Cells were rested for 2 hr and plated with fresh media with cytokine supplements for 24 hrs. Spinfection protocol was repeated 24 hrs later. 48 hrs after final spinfection, 5 × 104 transduced cells (identified by GFP or NGFR) along with 2 × 105 bone marrow cells for hemogenic support were injected via tail vein into lethally irradiated (9.0 Gy split into two fractionated doses 4 hrs apart) syngeneic mice. Secondary recipient mice were irradiated with 4.5 Gy and received 2 × 105 T-ALL cells via tail vein.
Retroviral packaging
Retroviral constructs were packaged in Plat-E cells. Plat-E cells were maintained in DMEM supplemented with 10% FBS, with 1 μg/mL puromycin and 10 μg/mL blasticidin. To produce virus, Plat-E cells at 70% confluency in a 6 well dish were transfected with construct plasmid along with Lipofectamine 2000 Transfection Reagent (Life Technologies). Media supernatant was removed 24 hrs post transfection, and fresh virus was collected 72 and 96 hrs post transfection.
Drug treatment of mice
Mice were dosed with tamoxifen (Sigma T5648) dissolved in corn oil at a dosage of 75 mg/kg body weight for 4 consecutive days starting 4 days after T-ALL transplantation for survival curve experiments or 10 days after T-ALL transplantation for acute deletion experiments. Mice were dosed with phenformin hydrochloride (Sigma P7045) dissolved in PBS at a dosage of 100 mg/kg body weight.
Human T-ALL cell line culture
DND-41, HPB-ALL and ALL-SIL cells were cultured between 5 × 105 and 2 × 106 cells per mL media in RPMI 1640 media supplemented with 10% FBS, 2 mM glutamine, 10 mM HEPES and 55 μM β-mercaptoethanol.
T Cell Isolation, Stimulation, and Culture
Murine or human total or CD4 T cells were isolated by magnetic bead negative and cultured in RPMI 1640 media supplemented with 10% FBS, 2 mM glutamine, 10 mM HEPES and 55 μM β-mercaptoethanol. T cells were stimulated on plates with 5 μg/mL anti-CD3ε and anti-CD28 antibodies (Pharmingen) for 24 or 48 hrs as indicated. Naïve T cells were cultured with 10 ng/mL IL-7. Activated T cells were washed off of stimulation plates and cultured with 5 ng/mL IL-2 for assays.
Primary T-ALL cell culture
Murine or human T-ALL cells were cultured in RPMI 1640 media supplemented with 10% FBS, 2 mM glutamine, 10 mM HEPES and 55 μM β-mercaptoethanol. IL-2 (10 ng/mL) and IL-7 (10 ng/mL).
Metabolomics
Nontargeted metabolomic analyses were performed as described using LC Q Exactive Mass Spectrometer (LC-QE-MS) (Thermo Scientific) (Liu et al., 2014b). Metaboanalyst was used to range-scale data and provide PCA and KEGG pathway analysis of metabolites significantly changed (1.5-fold difference, P<0.05)(www.metaboanalyst.ca/).
PCR Arrays
RNA was isolated from cells using RNeasy Plus Mini kit (Qiagen) following the manufacturers’ instructions. 1 μg total RNA was subjected to single-strand cDNA synthesis using the RT2 first strand kit (Qiagen). The cDNA was used in the Glucose Metabolism and Mitochondrial Energy Metabolism RT2 Profiler PCR arrays according to the manufacturer’s instructions and assayed on a CFX384 (Biorad). Data were analyzed using the RT2 Profiler program supplied by Qiagen and normalized to the geometric mean of the housekeeping genes beta-actin, beta-glucuronidase and heat shock protein 90kDa alpha.
Immunoblotting
Immunoblotting was performed as described previously (Jacobs et al., 2008). Primary antibodies were followed by mouse- or rabbit-conjugated horseradish peroxidase (HRP). HRP-conjugated antibodies (anti-mouse or anti-rabbit IgG HRP conjugate, Promega) were detected by enhanced chemiluminescence detection (Thermofisher). This included the following antibodies: ACC (3662, Cell Signaling), phospho-ACC (3661, Cell Signaling), pan-AMPKα (2532, Cell Signaling), phospho-AMPKα (2535, Cell Signaling), 4EBP1 (9644, Cell Signaling), phospho-4EBP1 (2855, Cell Signaling), c-myc (9402, Cell Signaling), phospho-mTOR (5536, Cell Signaling), Activated Notch (ab8925, Abcam), RAPTOR (2280, Cell Signaling), phospho-RAPTOR (2083, Cell Signaling), S6 (2217, Cell Signaling), phospho-p70 S6K (9204, Cell Signaling), p70 S6K (2708, Cell Signaling), phosphor-TSC2 (5584, Cell Signaling), TSC2 (3612, Cell Signaling). Alternatively, primary antibodies were followed by fluorescently labeled anti-mouse or rabbit antibodies (LiCor) and imaged using the Odyssey infrared imaging system (LiCor). This included the following antibodies: Glut1 (ab652, Abcam), hexokinase 2 (2867, Cell Signaling), hexokinase 1 (ab104835, Abcam), cytochrome C (556433, BD Biosciences), β-actin (A5441, Sigma), phospho-S6 (4858, Cell Signaling). Western blots were quantified using ImageJ software.
Metabolic assays
Glycolysis and glucose uptake assays using 3H-glucose or 3H-2-deoxyglucose have been described previously (Gerriets et al., 2015; Macintyre et al., 2014; Wang et al., 2011). Pentose phosphate pathway (PPP) flux and glucose oxidation were determined by the rate of 14CO2 release from 1-14C-glucose as described (Wang et al., 2011). All values were normalized to cell number. OCR and ECAR were measured with an XF24 extracellular flux analyzer (Seahorse Bioscience) as described (Caro-Maldonado et al., 2014). Suspension cells were attached to culture plates using Cell-Tak (BD Bioscience). OCR and ECAR were measured in unbuffered DMEM (Sigma-Aldrich) supplemented with 10mM D-glucose (Sigma-Aldrich) and 10mM L-glutamine, as indicated. OCR and ECAR values were normalized to cell number. For certain experiments, OCR was measured over time following injection of 1 μM oligomycin.
Flow cytometry
The following antibodies were used: anti-human CD4+ VioBlue (Miltenyi), anti-human CD8+ APC (Miltenyi), anti-human CD271 (LNGFR)-APC (Miltenyi) and goat anti-rabbit PE (eBioscience). Cell death was measured by exclusion of 1 μg/ml propidium iodide. Annexin V staining was performed following manufacturer instructions (BD 556547). Intracellular staining for GLUT1, HK2 and HK1 and measurement of surface Glut1 by myc epitope tag (Millipore 05-724) was performed as previously described (Michalek et al., 2011). DCFDA, Mitotracker Green and TMRE staining were measured by manufacturer protocol (Life Technologies). Data were acquired on a MacsQuant Analyzer (Miltenyi Biotec) and analyzed using FlowJo (TreeStar software).
Complex I Activity Assay
Complex I activity assay was performed following manufacturer instructions (ab109721, Abcam). In brief, isolated T-ALL cells were lysed and protein was quantified. 125 μg of protein lysate was loaded per well of immunocapture plate. Biological samples were analyzed in duplicate technical samples. Lysate was incubated on plate for 3 hr, then plate was washed twice and NADH substrate solution was added. OD450 was recorded for each well for 30 minutes and Complex I activity was calculated as the increase in OD450 within each well.
Statistical Analysis
Statistical analyses were performed with Prism software (GraphPad). Data were analyzed using a 2-tailed Student’s t test, and P < 0.05 was considered significant. Survival curve data was analyzed using Log Rank (Mantel-Cox) test. Statistically significant results are indicated (* p < 0.05) and n.s. indicates select non-significant data.
Supplementary Material
HIGHLIGHTS.
Primary T-ALL cells adopt aerobic glycolysis for cell survival and disease progression.
T-ALL glucose metabolism is far lower than the capacity of activated T cells.
Primary T-ALL cells are ATP depleted and exhibit chronic metabolic stress.
AMPK both inhibits anabolic metabolism and is essential for T-ALL cell survival.
Acknowledgments
We thank the Children’s Oncology Group for providing human T-ALL samples and Warren Pear for providing ICN1 constructs. This work was supported by NIH grants R01CA123350 (JCR), R03AI106835 (JCR), R21CA194829 (JCR), NIH R00CA168997 (JWL), R01CA198482 (WKR), F31CA183529 (RJK), K24CA172355 (WKR), and Leukemia and Lymphoma Society (JCR), Duke Cancer Institute Pilot Grant (JCR), Lineberger Cancer Center (K.L.R.) and the German Research Foundation (Deutsche Forschungsgemeinschaft; P.J.S.). The Cancer Science Institute of Singapore supports the Leukemia Tissue Bank.
Footnotes
Author Contributions
J.C.R and R.J.K. designed the study and wrote the manuscript; R.J.K., C.E.B., A.G.N., S.C., V.A.G., A.N.M., P.G.H., P.J.S. and M.O.W. performed the experiments; K.L.R. and A.E.J.Y. provided human T-ALL samples; and P.J.S., A.A.C., W.K.R., T.L., M.O.W., E.D.A., T.R.G., J.W.L. gave technical support and conceptual advice.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Angulo I, Vadas O, Garcon F, Banham-Hall E, Plagnol V, Leahy TR, Baxendale H, Coulter T, Curtis J, Wu C, et al. Phosphoinositide 3-kinase delta gene mutation predisposes to respiratory infection and airway damage. Science. 2013;342:866–871. doi: 10.1126/science.1243292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Sahra I, Howell JJ, Asara JM, Manning BD. Stimulation of de novo pyrimidine synthesis by growth signaling through mTOR and S6K1. Science. 2013;339:1323–1328. doi: 10.1126/science.1228792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhojwani D, Pui CH. Relapsed childhood acute lymphoblastic leukaemia. The Lancet Oncology. 2013;14:e205–217. doi: 10.1016/S1470-2045(12)70580-6. [DOI] [PubMed] [Google Scholar]
- Blagih J, Coulombe F, Vincent EE, Dupuy F, Galicia-Vazquez G, Yurchenko E, Raissi TC, van der Windt GJ, Viollet B, Pearce EL, et al. The Energy Sensor AMPK Regulates T Cell Metabolic Adaptation and Effector Responses In Vivo. Immunity. 2015;42:41–54. doi: 10.1016/j.immuni.2014.12.030. [DOI] [PubMed] [Google Scholar]
- Cantor JR, Sabatini DM. Cancer cell metabolism: one hallmark, many faces. Cancer discovery. 2012;2:881–898. doi: 10.1158/2159-8290.CD-12-0345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caro-Maldonado A, Wang R, Nichols AG, Kuraoka M, Milasta S, Sun LD, Gavin AL, Abel ED, Kelsoe G, Green DR, et al. Metabolic reprogramming is required for antibody production that is suppressed in anergic but exaggerated in chronically BAFF-exposed B cells. J Immunol. 2014;192:3626–3636. doi: 10.4049/jimmunol.1302062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Shojaee S, Buchner M, Geng H, Lee JW, Klemm L, Titz B, Graeber TG, Park E, Tan YX, et al. Signalling thresholds and negative B-cell selection in acute lymphoblastic leukaemia. Nature. 2015;521:357–361. doi: 10.1038/nature14231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang MY, Xu L, Shestova O, Histen G, L’Heureux S, Romany C, Childs ME, Gimotty PA, Aster JC, Pear WS. Leukemia-associated NOTCH1 alleles are weak tumor initiators but accelerate K-ras-initiated leukemia. J Clin Invest. 2008;118:3181–3194. doi: 10.1172/JCI35090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciofani M, Zuniga-Pflucker JC. Notch promotes survival of pre-T cells at the beta-selection checkpoint by regulating cellular metabolism. Nat Immunol. 2005;6:881–888. doi: 10.1038/ni1234. [DOI] [PubMed] [Google Scholar]
- Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, Kastritis E, Gilpatrick T, Paranal RM, Qi J, et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell. 2011;146:904–917. doi: 10.1016/j.cell.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faubert B, Boily G, Izreig S, Griss T, Samborska B, Dong Z, Dupuy F, Chambers C, Fuerth BJ, Viollet B, et al. AMPK Is a Negative Regulator of the Warburg Effect and Suppresses Tumor Growth In Vivo. Cell Metab. 2013;17:113–124. doi: 10.1016/j.cmet.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gergely P, Jr, Grossman C, Niland B, Puskas F, Neupane H, Allam F, Banki K, Phillips PE, Perl A. Mitochondrial hyperpolarization and ATP depletion in patients with systemic lupus erythematosus. Arthritis and rheumatism. 2002;46:175–190. doi: 10.1002/1529-0131(200201)46:1<175::AID-ART10015>3.0.CO;2-H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerriets VA, Kishton RJ, Nichols AG, Macintyre AN, Inoue M, Ilkayeva O, Winter PS, Liu X, Priyadharshini B, Slawinska ME, et al. Metabolic programming and PDHK1 control CD4+ T cell subsets and inflammation. J Clin Invest. 2015;125:194–207. doi: 10.1172/JCI76012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gershon TR, Crowther AJ, Tikunov A, Garcia I, Annis R, Yuan H, Miller CR, Macdonald J, Olson J, Deshmukh M. Hexokinase-2-mediated aerobic glycolysis is integral to cerebellar neurogenesis and pathogenesis of medulloblastoma. Cancer & metabolism. 2013;1:2. doi: 10.1186/2049-3002-1-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, Turk BE, Shaw RJ. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. 2008;30:214–226. doi: 10.1016/j.molcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Herranz D, Ambesi-Impiombato A, Sudderth J, Sanchez-Martin M, Belver L, Tosello V, Xu L, Wendorff AA, Castillo M, Haydu JE, et al. Metabolic reprogramming induces resistance to anti-NOTCH1 therapies in T cell acute lymphoblastic leukemia. Nature medicine. 2015;21:1182–1189. doi: 10.1038/nm.3955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch HA, Iliopoulos D, Tsichlis PN, Struhl K. Metformin selectively targets cancer stem cells, and acts together with chemotherapy to block tumor growth and prolong remission. Cancer research. 2009;69:7507–7511. doi: 10.1158/0008-5472.CAN-09-2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homminga I, Pieters R, Langerak AW, de Rooi JJ, Stubbs A, Verstegen M, Vuerhard M, Buijs-Gladdines J, Kooi C, Klous P, et al. Integrated transcript and genome analyses reveal NKX2-1 and MEF2C as potential oncogenes in T cell acute lymphoblastic leukemia. Cancer cell. 2011;19:484–497. doi: 10.1016/j.ccr.2011.02.008. [DOI] [PubMed] [Google Scholar]
- Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003;115:577–590. doi: 10.1016/s0092-8674(03)00929-2. [DOI] [PubMed] [Google Scholar]
- Jeon SM, Chandel NS, Hay N. AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress. Nature. 2012;485:661–665. doi: 10.1038/nature11066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lantier L, Fentz J, Mounier R, Leclerc J, Treebak JT, Pehmoller C, Sanz N, Sakakibara I, Saint-Amand E, Rimbaud S, et al. AMPK controls exercise endurance, mitochondrial oxidative capacity, and skeletal muscle integrity. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2014;28:3211–3224. doi: 10.1096/fj.14-250449. [DOI] [PubMed] [Google Scholar]
- Li N, Ragheb K, Lawler G, Sturgis J, Rajwa B, Melendez JA, Robinson JP. Mitochondrial complex I inhibitor rotenone induces apoptosis through enhancing mitochondrial reactive oxygen species production. The Journal of biological chemistry. 2003;278:8516–8525. doi: 10.1074/jbc.M210432200. [DOI] [PubMed] [Google Scholar]
- Liu L, Ulbrich J, Muller J, Wustefeld T, Aeberhard L, Kress TR, Muthalagu N, Rycak L, Rudalska R, Moll R, et al. Deregulated MYC expression induces dependence upon AMPK-related kinase 5. Nature. 2012;483:608–612. doi: 10.1038/nature10927. [DOI] [PubMed] [Google Scholar]
- Liu T, Kishton RJ, Macintyre AN, Gerriets VA, Xiang H, Liu X, Abel ED, Rizzieri D, Locasale JW, Rathmell JC. Glucose transporter 1-mediated glucose uptake is limiting for B-cell acute lymphoblastic leukemia anabolic metabolism and resistance to apoptosis. Cell death & disease. 2014a;5:e1470. doi: 10.1038/cddis.2014.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Ser Z, Locasale JW. Development and quantitative evaluation of a high-resolution metabolomics technology. Analytical chemistry. 2014b;86:2175–2184. doi: 10.1021/ac403845u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas CL, Kuehn HS, Zhao F, Niemela JE, Deenick EK, Palendira U, Avery DT, Moens L, Cannons JL, Biancalana M, et al. Dominant-activating germline mutations in the gene encoding the PI(3)K catalytic subunit p110delta result in T cell senescence and human immunodeficiency. Nat Immunol. 2014;15:88–97. doi: 10.1038/ni.2771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macintyre AN, Gerriets VA, Nichols AG, Michalek RD, Rudolph MC, Deoliveira D, Anderson SM, Abel ED, Chen BJ, Hale LP, et al. The Glucose Transporter Glut1 is Selectively Essential for CD4 T Cell Activation and Effector Function. Cell Metab. 2014;20:61–72. doi: 10.1016/j.cmet.2014.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macintyre AN, Rathmell JC. Activated lymphocytes as a metabolic model for carcinogenesis. Cancer & metabolism. 2013;1:5. doi: 10.1186/2049-3002-1-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacIver NJ, Blagih J, Saucillo DC, Tonelli L, Griss T, Rathmell JC, Jones RG. The liver kinase B1 is a central regulator of T cell development, activation, and metabolism. J Immunol. 2011;187:4187–4198. doi: 10.4049/jimmunol.1100367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mavrakis KJ, Wolfe AL, Oricchio E, Palomero T, de Keersmaecker K, McJunkin K, Zuber J, James T, Khan AA, Leslie CS, et al. Genome-wide RNA-mediated interference screen identifies miR-19 targets in Notch-induced T-cell acute lymphoblastic leukaemia. Nat Cell Biol. 2010;12:372–379. doi: 10.1038/ncb2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalek RD, Gerriets VA, Jacobs SR, Macintyre AN, MacIver NJ, Mason EF, Sullivan SA, Nichols AG, Rathmell JC. Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J Immunol. 2011;186:3299–3303. doi: 10.4049/jimmunol.1003613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moiseeva O, Bourdeau V, Roux A, Deschenes-Simard X, Ferbeyre G. Mitochondrial dysfunction contributes to oncogene-induced senescence. Molecular and cellular biology. 2009;29:4495–4507. doi: 10.1128/MCB.01868-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oudot C, Auclerc MF, Levy V, Porcher R, Piguet C, Perel Y, Gandemer V, Debre M, Vermylen C, Pautard B, et al. Prognostic factors for leukemic induction failure in children with acute lymphoblastic leukemia and outcome after salvage therapy: the FRALLE 93 study. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2008;26:1496–1503. doi: 10.1200/JCO.2007.12.2820. [DOI] [PubMed] [Google Scholar]
- Palmer CS, Ostrowski M, Balderson B, Christian N, Crowe SM. Glucose metabolism regulates T cell activation, differentiation, and functions. Frontiers in immunology. 2015;6:1. doi: 10.3389/fimmu.2015.00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palomero T, Lim WK, Odom DT, Sulis ML, Real PJ, Margolin A, Barnes KC, O’Neil J, Neuberg D, Weng AP, et al. NOTCH1 directly regulates c-MYC and activates a feed-forward-loop transcriptional network promoting leukemic cell growth. Proc Natl Acad Sci U S A. 2006;103:18261–18266. doi: 10.1073/pnas.0606108103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palomero T, Sulis ML, Cortina M, Real PJ, Barnes K, Ciofani M, Caparros E, Buteau J, Brown K, Perkins SL, et al. Mutational loss of PTEN induces resistance to NOTCH1 inhibition in T-cell leukemia. Nat Med. 2007;13:1203–1210. doi: 10.1038/nm1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patra KC, Wang Q, Bhaskar PT, Miller L, Wang Z, Wheaton W, Chandel N, Laakso M, Muller WJ, Allen EL, et al. Hexokinase 2 is required for tumor initiation and maintenance and its systemic deletion is therapeutic in mouse models of cancer. Cancer cell. 2013;24:213–228. doi: 10.1016/j.ccr.2013.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patsoukis N, Bardhan K, Chatterjee P, Sari D, Liu B, Bell LN, Karoly ED, Freeman GJ, Petkova V, Seth P, et al. PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nature communications. 2015;6:6692. doi: 10.1038/ncomms7692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pui CH, Robison LL, Look AT. Acute lymphoblastic leukaemia. Lancet. 2008;371:1030–1043. doi: 10.1016/S0140-6736(08)60457-2. [DOI] [PubMed] [Google Scholar]
- Robitaille AM, Christen S, Shimobayashi M, Cornu M, Fava LL, Moes S, Prescianotto-Baschong C, Sauer U, Jenoe P, Hall MN. Quantitative phosphoproteomics reveal mTORC1 activates de novo pyrimidine synthesis. Science. 2013;339:1320–1323. doi: 10.1126/science.1228771. [DOI] [PubMed] [Google Scholar]
- Saito Y, Chapple RH, Lin A, Kitano A, Nakada D. AMPK Protects Leukemia-Initiating Cells in Myeloid Leukemias from Metabolic Stress in the Bone Marrow. Cell stem cell. 2015;17:585–596. doi: 10.1016/j.stem.2015.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanghvi VR, Mavrakis KJ, Van der Meulen J, Boice M, Wolfe AL, Carty M, Mohan P, Rondou P, Socci ND, Benoit Y, et al. Characterization of a set of tumor suppressor microRNAs in T cell acute lymphoblastic leukemia. Science signaling. 2014;7:ra111. doi: 10.1126/scisignal.2005500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shackelford DB, Abt E, Gerken L, Vasquez DS, Seki A, Leblanc M, Wei L, Fishbein MC, Czernin J, Mischel PS, et al. LKB1 inactivation dictates therapeutic response of non-small cell lung cancer to the metabolism drug phenformin. Cancer cell. 2013;23:143–158. doi: 10.1016/j.ccr.2012.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw RJ, Kosmatka M, Bardeesy N, Hurley RL, Witters LA, DePinho RA, Cantley LC. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:3329–3335. doi: 10.1073/pnas.0308061100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sukumar M, Liu J, Ji Y, Subramanian M, Crompton JG, Yu Z, Roychoudhuri R, Palmer DC, Muranski P, Karoly ED, et al. Inhibiting glycolytic metabolism enhances CD8+ T cell memory and antitumor function. J Clin Invest. 2013;123:4479–4488. doi: 10.1172/JCI69589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tello D, Balsa E, Acosta-Iborra B, Fuertes-Yebra E, Elorza A, Ordonez A, Corral-Escariz M, Soro I, Lopez-Bernardo E, Perales-Clemente E, et al. Induction of the mitochondrial NDUFA4L2 protein by HIF-1alpha decreases oxygen consumption by inhibiting Complex I activity. Cell metabolism. 2011;14:768–779. doi: 10.1016/j.cmet.2011.10.008. [DOI] [PubMed] [Google Scholar]
- van der Windt GJ, Everts B, Chang CH, Curtis JD, Freitas TC, Amiel E, Pearce EJ, Pearce EL. Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity. 2012;36:68–78. doi: 10.1016/j.immuni.2011.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, McCormick LL, Fitzgerald P, Chi H, Munger J, et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity. 2011;35:871–882. doi: 10.1016/j.immuni.2011.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warburg O, Wind F, Negelein E. The Metabolism of Tumors in the Body. The Journal of general physiology. 1927;8:519–530. doi: 10.1085/jgp.8.6.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng AP, Ferrando AA, Lee W, Morris JPt, Silverman LB, Sanchez-Irizarry C, Blacklow SC, Look AT, Aster JC. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science. 2004;306:269–271. doi: 10.1126/science.1102160. [DOI] [PubMed] [Google Scholar]
- Wolf A, Agnihotri S, Micallef J, Mukherjee J, Sabha N, Cairns R, Hawkins C, Guha A. Hexokinase 2 is a key mediator of aerobic glycolysis and promotes tumor growth in human glioblastoma multiforme. The Journal of experimental medicine. 2011;208:313–326. doi: 10.1084/jem.20101470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Chi F, Guo T, Punj V, Lee WN, French SW, Tsukamoto H. NOTCH reprograms mitochondrial metabolism for proinflammatory macrophage activation. The Journal of clinical investigation. 2015;125:1579–1590. doi: 10.1172/JCI76468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinselmeyer BH, Heydari S, Sacristan C, Nayak D, Cammer M, Herz J, Cheng X, Davis SJ, Dustin ML, McGavern DB. PD-1 promotes immune exhaustion by inducing antiviral T cell motility paralysis. The Journal of experimental medicine. 2013;210:757–774. doi: 10.1084/jem.20121416. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.