

Abstract
Property tuning by fluorination is very effective for a number of purposes, and currently increasingly investigated for aliphatic compounds. An important application is lipophilicity (log P) modulation. However, the determination of log P is cumbersome for non‐UV‐active compounds. A new variation of the shake‐flask log P determination method is presented, enabling the measurement of log P for fluorinated compounds with or without UV activity regardless of whether they are hydrophilic or lipophilic. No calibration curves or measurements of compound masses/aliquot volumes are required. With this method, the influence of fluorination on the lipophilicity of fluorinated aliphatic alcohols was determined, and the log P values of fluorinated carbohydrates were measured. Interesting trends and changes, for example, for the dependence on relative stereochemistry, are reported.
Keywords: fluorinated carbohydrates, fluorine, fluoroalcohols, lipophilicity, NMR spectroscopy
Potency optimization is a natural focus in drug development. However, potency gain achieved at the expense of physicochemical and pharmacokinetic properties compromises drug efficacy and safety, increasing attrition rates.1 In this regard, the inappropriate use of lipophilicity (log P) to increase potency, which is referred to as “molecular obesity”, has been identified as a key problem.2 The ability to maintain low lipophilicity levels while increasing molecular weight is regarded as one of the keys to a successful drug‐discovery program.3 New efficiency metrics have been introduced for gauging the lipophilicity contribution to potency.1a, 4, 5
Hence, given that late‐stage drug attrition is a very costly and major contemporary problem, new insights for controlling lipophilicity (which is related to important properties such as bioavailability)6 are of great interest. An attractive strategy is substituting substructures for alternatives with lower lipophilicity. Examples include the replacement of gem‐dimethyl groups by oxetanes,7 or using 1,3,4‐oxadiazoles instead of their 1,2,4‐isomers.8
Lipophilicity is also influenced by fluorination.9, 10 Whereas it is still frequently stated that fluorination increases lipophilicity, this is typically limited to aromatic substrates.9, 11 For aliphatic compounds, C—H→C—F, or even CH3→CF3 exchange, can lead to a log P decrease.9, 10a As fluorine is introduced in lead optimization for the optimization/introduction of an ever‐increasing list of properties, it is important to understand how the compound lipophilicity will be influenced when fluorination is applied for this purpose, or indeed how it can be directly used to modulate lipophilicity.
For aromatic substrates, the influence of fluorination on log P has been studied in detail. In contrast, few systematic studies are available for aliphatic compounds.12 In a seminal contribution,12b Carreira, Müller, and co‐workers showed that two competing effects affect lipophilicity, namely changes in polarity (polar C—F bond) and in the hydrophobicity of the surface (non‐polarizable fluorine atoms). They also showed for a series of trifluoromethylated aromatic compounds, that this balance depends on the absolute log P value of the compound: For highly lipophilic compounds, changes in polarity tend to dominate (log P tends to decrease upon F introduction), whereas for polar compounds, changes in the hydrophobic surface area dominate (log P tends to increase). Later on, Müller introduced a straightforward bond vector analysis as a qualitative method for assessing the polarity of partially fluorinated alkyl and alkoxy groups, and its repercussions on compound lipophilicity.13
The presence of functional groups close to fluorination sites further affects log P, often in unpredictable ways, which are yet to be fully explored. A clear barrier for progress in this field is the actual log P determination process. Whereas many methods are available, these are cumbersome (requiring calibration curves). HPLC‐based methods14 are less suitable when accurate log P values are required and subject to limitations.15 Above all, quantification typically rests on UV spectroscopy, which hampers the measurement of non‐UV‐active solutes (even commercially). NMR‐based methods12a, 16 are cumbersome or not easily amenable for use in typical multiuser NMR facilities. The calculation of log P is also possible (clog P), but accuracy is strongly dependent on the training sets used, stereochemistry cannot be taken into account, and errors of multiple log P units are no exception.17
Herein, we present a practical and experimentally straightforward method for the accurate determination of the lipophilicity (±0.01 log P) of fluorinated compounds that is based on 19F NMR spectroscopy. The method is illustrated by determining the log P values of fluorinated alkanols, as representatives of a biologically relevant group, and we present the first log P values of fluorohydrin diastereomers as well as of mono‐ and polyfluorinated carbohydrates. The remarkable power of even single fluorination to modulate log P in both directions is highlighted.
The principle of the method (Figure 1) is based on the use of an internal (fluorinated) reference (ref): A mixture of ref and an unknown compound (X) is partitioned between (non‐deuterated) octanol and H2O. An aliquot of each phase is transferred to an NMR tube, and its 19F NMR spectrum is taken. By reprocessing each FID, the integration ratio between the peaks of X and ref can be determined. These ratios are defined as ρ aq and ρ oct, and correspond to the ratio of the respective concentrations. If the peaks represent a different number (n) of fluorine substituents, then a correction factor is applied [Eq. (1)]. The ratio of the ρ values is equal to the ratio of the respective P values [Eq. (2)]. Rewriting this as Eq. (3) then results in Eq. (4), which shows that the log P value of the unknown compound can be obtained by adding the logarithm of the ratio of the measured ρ values to the known log P value of the reference compound.
Figure 1.

Principle of the log P determination method. See the Supporting Information for a detailed procedure.
There are a number of practical advantages associated with this method. Because of the compensation effect inherent to the determination of a ratio of a ratio, systematic errors are eliminated, and no quantitative measurement is required for an absolute amount of material, phase volumes (respecting solubilities), and NMR aliquot volumes. Small impurities are tolerated given only the compound signals are integrated.
Accurate quantitative integration is crucial to the method. Further to standard data processing (window function (LB), zero filling, phasing, and baseline correction), a good signal to noise ratio (S/N) is essential, which is significantly enhanced for fluorine signals by proton decoupling. The use of inverse gated decoupling minimizes any potential interference from nuclear Overhauser effects (nOes; see the Supporting Information, Table S1).18 Narrowing the spectral width (SW) increases digital resolution and the S/N ratio, but we experimentally found that these settings did not influence the determined log P value (Table S2). The frequency offset point (O1P) is ideally set equidistant between the fluorine signals to ensure that both are equally excited (important when applying a reduced SW). Indeed, the ρ values significantly changed upon varying the O1P value (Table S3), but virtually no effect was seen on the log P value owing to the aforementioned compensation effect. Accurate integration also rests on complete nuclear relaxation, requiring a sufficiently long pulse delay time (D1). It was determined that the ρ values did depend on the D1 value, with a non‐negligible effect on the log P values (±0.04 units, Table S4). This is due to the solvent dependence of the fluorine spin–lattice relaxation times (T 1), invalidating the compensation effect for this parameter. Hence, the T 1 values of a number of compounds were determined prior to log P measurement (Table S5), revealing that the T 1 values are generally higher in water than in octanol, and tend to be higher for monofluorinated compounds (up to 8 s) than for the corresponding di‐ and trifluorinated compounds (up to 4.5 s). Given that T 1 determinations are time‐consuming, very conservative D1 settings (ideally 5×T 1) of 30 s (octanol) and 60 s (water) were chosen. Finally, the S/N ratio is also positively influenced by an increased number of scans (NS). However, reducing SW and increasing NS significantly increases experiment time, and for typical experiments, SW=300 ppm and NS=64 were used.
Because of the compensation effect, the identity of the reference compound is less important. However, when a reduced SW is used, the window must encompass both chemical shifts. Furthermore, to achieve an optimal integration ratio between 0.1 and 10, the relative compound quantities should be adjusted, and a reference compound with an appropriate relative log P value (guided by clog P of the unknown) should be selected. The method rests on the assumption that the intermolecular interactions between X and ref are negligible or at least do not influence the log P value, which was confirmed experimentally (Table S6). In any case, this compares favorably to most high‐throughput shake‐flask log P determinations, where compounds are typically added to octanol/water as a solution in DMSO.19
The method was validated by the determination of known log P values. The complete set of results is given in the Supporting Information (Tables S7–S12), and shows that the values are generally very similar to reported data (Table S12) despite the fact that a number of different log P values can often be found in the literature. Furthermore, the internal consistency of the method was confirmed by a control experiment (Tables S9–S11). Lipophilicity measurements were typically carried out in triplicate, and excellent reproducibility was observed (generally <0.005 log P units). Deviations occurring by different reprocessings of the same FID were very minimal (Table S8). We then embarked on determining the log P values of a range of known and novel fluorinated alkanols (Figure 2). The color coding visualizes how for the library investigated, for a given scaffold, monofluorination lowers the log P value—in contrast to aromatic monofluorination—which then increases upon further fluorination, with few exceptions.
Figure 2.
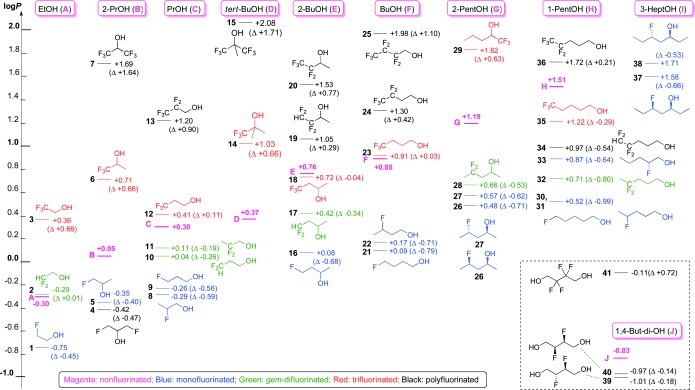
Lipophilicity map of fluorinated alcohols. See the Supporting Information, Figure S1–S10 for detailed maps and further comments.
The increase in log P upon alcohol β‐trifluorination (ca. 0.65 units; compounds 3 and 6), as well as the steady reduction in this increase—even reverting into a log P decrease—as the trifluorinated carbon atom is positioned further away from the alcohol group upon chain elongation (12, 23, 35) was known,12a and could be explained by the increasing importance of the CF3 polarity upon increasing the lipophilicity of the parent alcohol (see the Carreira/Müller observation).12b However, our determination that a shift of approximately 0.65 log P is maintained for the much more lipophilic 29, a novel compound, shows that the actual β‐trifluorination effect is independent of the absolute log P value. Double β‐trifluorination, as in 7 and 15, causes a significant log P increase. Pentafluorinated substrates (13, 20, 24, and 36) show a further increase in lipophilicity. Interestingly, the ΔΔlog P value for the corresponding trifluorinated derivatives is strongly dependent on whether the pentafluoroethyl group is in the α‐position of the alcohol group or not (12/13, 18/20 vs. 23/24, 35/36).
In all cases except 2 (Δ=+0.01), the introduction of a gem‐difluoro group (Figure S4) leads to a decrease in log P, which can be substantial (32: Δ=−0.80) regardless of whether it occurred at a terminal or internal alkane position. Interestingly, β‐difluorination (as in 11) leads to a decrease in log P, which is markedly different from the increase caused by β‐trifluorination as discussed above.
Monofluorination, as expected, always leads to a decrease in the log P value regardless of the position; this change can be very substantial: For the 1‐pentanol series, a decrease of 0.99 log P units was observed when the 4‐ or the 5‐position was fluorinated (30 and 31). For the same parent compound, fluorination in the 2‐position (33) results in a much smaller log P decrease. For the parent compounds F and C, there is a small log P difference between 3‐ versus 4‐ and 2‐ versus 3‐fluorination despite the fact that in the latter case (8), the fluorine is in the β‐position of the alcohol. We also determined the log P values of diastereomeric fluorohydrins. The syn diastereomer 37 is less lipophilic than anti diastereomer 38, likely because the (more polar) conformation with aligned C—O/C—F dipoles will be more stabilized in water than the equivalent conformer of the anti isomer (Figure S6). Interestingly, a second monofluorination at a different position only leads to a minor further decrease (5→4). The vicinally difluorinated diols 39 and 40 only have a slightly reduced log P value compared to J, with a minimal difference between the diastereomers.
The decrease in log P observed upon replacing CH with CF is not seen when the carbon atom already had a fluorine substituent: for the compounds investigated, the gem‐difluorinated substrates were always more lipophilic than the corresponding monofluorinated substrate(s). Equally, replacing a CHF2 group with a CF3 group leads to an increase in log P, which is substantial with an α‐hydroxy group (2→3). The same effect was seen when going from CF2CF2H to CF2CF3 (19→20; 34→36), but it is more pronounced the greater the distance to the hydroxy group. The much lower lipophilicity of the tetrafluorinated compound 34 compared to trifluorinated 35 (ΔΔlog P=0.25) is interesting, and reveals a higher polarity of the CF2CF2H group compared to CH2CF3 despite it having one more fluorine substituent, which is likely due to a conformation effect (see Figure S9). The identification of such space‐filling groups is of great interest in medicinal chemistry.
The influence of fluorination on rigid cyclohexanols was also investigated (Figure 3). For K, equatorial or axial fluorination at the 2‐position gave very similar effects on log P (42, 43; Δlog P=−0.47±0.01). In contrast, axial monofluorination of cyclohexanol L in the 2‐ or 3‐position led to very different changes of log P: for vicinal trans fluorohydrin 44, an increase in log P was observed. This represents, to the best of our knowledge, the first example of an increase in log P induced by monofluorination of an aliphatic alcohol. Interestingly, the clog P values of 44 are vastly underestimated (Table S14). Conversely, 1,3‐diaxial fluorohydrin 45 showed a significant decrease in log P, exceeding that observed for acyclic 1,3‐fluorohydrins. The aligned C—O and C—F dipoles in 45 will be responsible for the significant decrease in log P. In contrast, the C—O/C—F dipoles are opposed in 44, which leads to a log P increase. The unavoidable antiperiplanar C—F/ C—OH orientation in α‐trifluoromethylated alcohols will similarly contribute to the strong increase in their log P values (see above).
Figure 3.
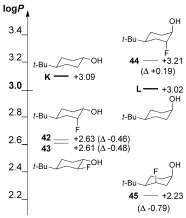
Lipophilicities of conformationally rigid cyclohexanols.
Fluorinated carbohydrates find application as mechanism‐based inhibitors,20 for the stabilization of glycosidic bonds,21 and have historically also been used as probes for sugar epitope mapping.22 Briefly, a decreased binding affinity upon monodeoxyfluorination indicates a hydrogen bond (HB) donor function for the replaced alcohol, whereas a similar binding affinity suggests that it functions as an HB acceptor. However, it is difficult to take the altered lipophilicity of fluorinated carbohydrates into account in binding data interpretations, in part because there are no quantitative data regarding the influence of deoxyfluorination on carbohydrate lipophilicity. Given the difficult quantification of carbohydrate concentrations, as well as the fact that the clog P values for diastereomeric sugars are identical, these substrates were deemed an ideal test case to demonstrate the usefulness of our log P determination method, taking into account that the fluorine signals of both anomers, and possibly the furanose forms, need to be integrated. The results (Figure 4) show that the influence of monodeoxyfluorination on lipophilicity is already significant, and clearly depends upon position and sugar stereochemistry (46–50). However, the log P difference between 49 and 50, which only differ in the stereochemistry at the fluorine‐substituted 2‐position, is only 0.1 log P units. The lipophilicity of sugars in which two hydroxy groups have been removed further increases by another log P unit: 2,3‐Dideoxy‐2,3‐difluoroglucose 52 is about 2.1 log P units more lipophilic than Glc. Interestingly, 2,3‐dideoxymonofluorinated 51 is much more hydrophilic. The change from 51 to 52 represents CH—H for CH—F replacement, and this is only the second example here with a log P increase for such a change. A further increase by one log P unit is achieved by trideoxyfluorination as in 2,3,4‐trideoxy‐2,3,4‐trifluoroglucose 55.23 Interestingly, this 103 fold increase in the P value can also be achieved by mere dideoxygenation if the individual CHOH groups are both replaced by CF2 groups (as in 53, 54). For these sugar derivatives, the relative configuration of the remaining CHOH group also strongly impacts on the lipophilicity.
Figure 4.
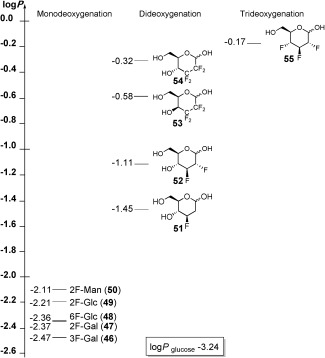
Lipophilicities of fluorinated carbohydrates.
In conclusion, we have presented a new and facile log P determination method, which is suitable for fluorinated compounds, whether they are UV‐active or not, and applicable to lipophilic and hydrophilic compounds with a log P range of up to ±3 and for different solvents besides octanol. The lipophilicities of a range of acyclic and conformationally rigid fluorohydrins were reported. We showed that monofluorination can lead to very large decrease in log P, that relative stereochemistry has a measurable effect on log P, which is especially pronounced for rigid, cyclic fluorohydrins, and that the Δlog P value upon introducing a certain motif does not always depend on the absolute log P value. We have also quantified the lipophilicity changes upon sugar deoxyfluorination, showing the significant dependence on position, stereochemistry, number of fluorination sites, and fluorination motif. Given the contemporary emphasis on developing three‐dimensional C(sp3)‐containing scaffolds,24 the need for facile analysis of non‐UV‐active compounds will increase considerably. Hence, the convenience of this method, which does not require specialist/cumbersome operations such as solvent suppression or concentration determinations, brings log P measurements of fluorinated compounds within reach of scientists with access to NMR spectrometers, which should rapidly and significantly boost knowledge in this important field, and easily allow for experimental verification of log P predictions. Further method development is in progress in our laboratory.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
miscellaneous_information
Acknowledgements
We thank the EPSRC (fellowship EP/K016938/1 and core capability EP/K039466/1), Dextra (CASE studentship), and the European Community (INTERREG IVa Channel Programme, AI‐Chem, project 4494/4196) for funding. We thank Prof. David O’Hagan for a gift of 2,3,4‐trideoxy‐2,3,4‐trifluoroglucose (55), and Dextra Laboratories for a donation of monodeoxofluorinated sugars.
References
- 1.
- 1a. Tarcsay A., Keseru G. M., J. Med. Chem. 2013, 56, 1789–1795; [DOI] [PubMed] [Google Scholar]
- 1b. Gleeson M. P., Hersey A., Montanari D., Overington J., Nat. Rev. Drug Discovery 2011, 10, 197–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.
- 2a. Arnott J. A., Planey S. L., Expert Opin. Drug Discovery 2012, 7, 863–875; [DOI] [PubMed] [Google Scholar]
- 2b. Hann M. M., MedChemComm 2011, 2, 349–355; [Google Scholar]
- 2c. Leeson P. D., Springthorpe B., Nat. Rev. Drug Discovery 2007, 6, 881–890. [DOI] [PubMed] [Google Scholar]
- 3. Perola E., J. Med. Chem. 2010, 53, 2986–2997. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Shultz M. D., Bioorg. Med. Chem. Lett. 2013, 23, 5992–6000; [DOI] [PubMed] [Google Scholar]
- 4b. Freeman‐Cook K. D., Hoffman R. L., Johnson T. W., Future Med. Chem. 2013, 5, 113–115. [DOI] [PubMed] [Google Scholar]
- 5. Tarcsay A., Nyiri K., Keseru G. M., J. Med. Chem. 2012, 55, 1252–1260. [DOI] [PubMed] [Google Scholar]
- 6. Yoshida F., Topliss J. G., J. Med. Chem. 2000, 43, 2575–2585. [DOI] [PubMed] [Google Scholar]
- 7. Wuitschik G., Carreira E. M., Wagner B., Fischer H., Parrilla I., Schuler F., Rogers‐Evans M., Mueller K., J. Med. Chem. 2010, 53, 3227–3246. [DOI] [PubMed] [Google Scholar]
- 8. Boström J., Hogner A., Llinas A., Wellner E., Plowright A. T., J. Med. Chem. 2012, 55, 1817–1830. [DOI] [PubMed] [Google Scholar]
- 9. Smart B. E., J. Fluorine Chem. 2001, 109, 3–11. [Google Scholar]
- 10.
- 10a. Böhm H. J., Banner D., Bendels S., Kansy M., Kuhn B., Müller K., Obst‐Sander U., Stahl M., ChemBioChem 2004, 5, 637–643; [DOI] [PubMed] [Google Scholar]
- 10b. Purser S., Moore P. R., Swallow S., Gouverneur V., Chem. Soc. Rev. 2008, 37, 320–330; [DOI] [PubMed] [Google Scholar]
- 10c. O’Hagan D., Chem. Soc. Rev. 2008, 37, 308–319. [DOI] [PubMed] [Google Scholar]
- 11. Mueller K., Faeh C., Diederich F., Science 2007, 317, 1881–1886. [DOI] [PubMed] [Google Scholar]
- 12.For examples, see:
- 12a. Muller N., J. Pharm. Sci. 1986, 75, 987–991; [DOI] [PubMed] [Google Scholar]
- 12b. Huchet Q. A., Kuhn B., Wagner B., Fischer H., Kansy M., Zimmerli D., Carreira E. M., Müller K., J. Fluorine Chem. 2013, 152, 119–128; [Google Scholar]
- 12c. Samsonov S. A., Salwiczek M., Anders G., Koksch B., Pisabarro M. T., J. Phys. Chem. B 2009, 113, 16400–16408; [DOI] [PubMed] [Google Scholar]
- 12d. Orliac A., Routier J., Charvillon F. B., Sauer W. H., Bombrun A., Kulkarni S. S., Pardo D. G., Cossy J., Chem. Eur. J. 2014, 20, 3813–3824; [DOI] [PubMed] [Google Scholar]
- 12e. Chernykh A. V., Radchenko D. S., Chernykh A. V., Kondratov I. S., Tolmachova N. A., Datchenko O. P., Kurkunov M. A., Zozulya S. X., Kheylik Y. P., Bartels K., Daniliuc C. G., Haufe G., Eur. J. Org. Chem. 2015, 29, 6466–6471. [Google Scholar]
- 13. Müller K., Chimia 2014, 68, 356–362. [DOI] [PubMed] [Google Scholar]
- 14. Young R. J., Green D. V. S., Luscombe C. N., Hill A. P., Drug Discovery Today 2011, 16, 822–830. [DOI] [PubMed] [Google Scholar]
- 15. Caron G., Vallaro M., Ermondi G., MedChemComm 2013, 4, 1376–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mo H., Balko K. M., Colby D. A., Bioorg. Med. Chem. Lett. 2010, 20, 6712–6715. [DOI] [PubMed] [Google Scholar]
- 17. Waring M. J., Expert Opin. Drug Discovery 2010, 5, 235–248. [DOI] [PubMed] [Google Scholar]
- 18. Claridge T., High‐Resolution NMR Techniques in Organic Chemistry, Pergamon, New York, 1999. [Google Scholar]
- 19. Shalaeva M., Caron G., Abramov Y. A., O’Connell T. N., Plummer M. S., Yalamanchi G., Farley K. A., Goetz G. H., Philippe L., Shapiro M. J., J. Med. Chem. 2013, 56, 4870–4879, and references therein. [DOI] [PubMed] [Google Scholar]
- 20. Kim J.‐H., Resende R., Wennekes T., Chen H.‐M., Bance N., Buchini S., Watts A. G., Pilling P., Streltsov V. A., Petric M., Liggins R., Barrett S., McKimm‐Breschkin J. L., Niikura M., Withers S. G., Science 2013, 340, 71–75. [DOI] [PubMed] [Google Scholar]
- 21.
- 21a. Lee S. S., Greig I. R., Vocadlo D. J., McCarter J. D., Patrick B. O., Withers S. G., J. Am. Chem. Soc. 2011, 133, 15826–15829; [DOI] [PubMed] [Google Scholar]
- 21b. Namchuk M. N., McCarter J. D., Becalski A., Andrews T., Withers S. G., J. Am. Chem. Soc. 2000, 122, 1270–1277. [Google Scholar]
- 22.
- 22a. Glaudemans C. P. J., Chem. Rev. 1991, 91, 25–33; [Google Scholar]
- 22b. Lemieux R. U., Chem. Soc. Rev. 1989, 18, 347–374. [Google Scholar]
- 23.
- 23a. Bresciani S., Lebl T., Slawin A. M. Z., O’Hagan D., Chem. Commun. 2010, 46, 5434–5436; [DOI] [PubMed] [Google Scholar]
- 23b. Corr M. J., O’Hagan D., J. Fluorine Chem. 2013, 155, 72–77. [Google Scholar]
- 24. Lovering F., Bikker J., Humblet C., J. Med. Chem. 2009, 52, 6752–6756. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
miscellaneous_information