

Abstract
A redox‐neutral palladium(II)‐catalyzed conversion of aryl, heteroaryl, and alkenyl boronic acids into sulfinate intermediates, and onwards to sulfones and sulfonamides, has been realized. A simple Pd(OAc)2 catalyst, in combination with the sulfur dioxide surrogate 1,4‐diazabicyclo[2.2.2]octane bis(sulfur dioxide) (DABSO), is sufficient to achieve rapid and high‐yielding conversion of the boronic acids into the corresponding sulfinates. Addition of C‐ or N‐based electrophiles then allows conversion into sulfones and sulfonamides, respectively, in a one‐pot, two‐step process.
Keywords: electrophiles, oxidation, palladium, sulfur, synthetic methods
The sulfonyl group, which is embedded in sulfones, sulfonamides, sulfonate esters, and sulfinic acids, is a structural motif with numerous uses. These important functional groups contribute significant physiochemical properties,1 as well as varied synthetic utility,2 and a wide range of pharmaceutical and agrochemical applications are known.3, 4
Sulfones and sulfonamides are the most prominent sulfonyl‐containing molecules, and general and simple methods for their construction are in high demand. Classical syntheses which are commonly employed include sulfide oxidation5 and sulfonyl chloride‐amine coupling,6 to access sulfones and sulfonamides, respectively. Limitations of such strategies include the use of odorous thiols and functional‐group‐restricting oxidative conditions in the sulfone synthesis, and harsh acidic treatment of arenes to access sulfonyl chloride precursors by electrophilic aromatic sulfonation. In addition, electrophilic substitution processes impose constraints on the substitution patterns which can be readily prepared.
Alternative routes to a variety of sulfonyl derivatives can be achieved from the direct insertion of sulfur dioxide into suitably functionalized substrates. For example, the SO2‐surrogate DABSO,7 DABCO⋅(SO2)2 (DABCO=1,4‐diazabicyclo[2.2.2]octane), has been combined with preformed organometallic reagents to generate metal sulfinates which can then undergo in situ conversion into sulfones8 and sulfonamides.9–11 A palladium‐catalyzed preparation of ammonium sulfinates has also been reported by our laboratory, and allows (hetero)aryl iodides and DABSO to be efficiently combined using a palladium(0) catalyst and iPrOH as reductant (Scheme 1 a).12 A similar transformation was developed by Shavnya and co‐workers with the formation of sulfinates from aryl halides using K2S2O5 as the sulfur dioxide source and formate as the reductant (Scheme 1 b).13 While these processes serve as effective means of accessing sulfonyl‐containing compounds, they suffer from either the use of specialized and expensive phosphine ligands or high catalyst and ligand loadings, and are also slow reactions (typically 16–18 h). The need for supporting ligands presumably arises from the operation of a Pd0/II cycle.
Scheme 1.
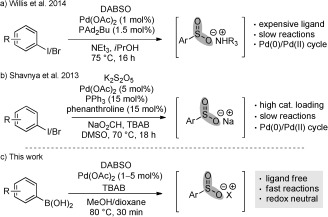
Catalytic sulfnate synthesis. a,b) Examples involving a Pd0/II cycle. c) This work, involving a redox neutral PdII system.
To address these shortcomings we targeted the development of a palladium(II) catalytic system employing boronic acids as substrates in combination with DABSO. Such a process would be redox‐neutral, should allow greater functional‐group tolerance, and should avoid the requirement for specialized, often costly, phosphine ligands. Toste and co‐workers recently disclosed a process for the conversion of aryl boronic acids into sulfinates using K2S2O5 and gold(I) catalysis.14 This elegant study demonstrates the viability of a redox‐neutral sulfinate synthesis, albeit in a gold(I) manifold, but suffers from the use of high catalyst loadings and limited onwards sulfinate reactivity with products obtained in moderate yields. Herein, we outline the successful development of a one‐pot, redox‐neutral palladium(II)‐catalyzed preparation of sulfinate derivatives from boronic acid substrates. A phosphine‐free catalyst allows the rapid conversion of boronic acids into the corresponding sulfinates, and then onwards to a variety of sulfones and sulfonamides (Scheme 1 c).15–17
Initial investigations quickly revealed that the union of p‐tert‐butylphenylboronic acid, DABSO, and tert‐butyl bromoacetate could be achieved by the use of Pd(OAc)2 as the catalyst with Et3N as the base in a 1,4‐dioxane/MeOH solvent mixture. The addition of TBAB resulted in a small increase in yield, thus allowing isolation of the sulfone 1 a in 83 % (Scheme 2 a). A brief assessment of the scope of boronic acids and electrophiles compatible with this one‐pot, one‐step process was carried out (Scheme 2 b). Aryl boronic acids bearing electron‐donating and electron‐withdrawing substituents proved to be effective substrates (1 a–e), and simple heteroaromatic variants were also well tolerated (1 f). Alkenyl boronic acids were found to be lower yielding (1 g). Aromatic BF3K salts were compatible with this system (1 e). In terms of suitable electrophiles, only activated alkyl halides (1 a,h–j) proved to be effective in this process with simple alkyl halides affording none of the desired products (e.g., butyl bromide). A further limitation with this one‐step format, was that the solvent for the sulfinate functionalization was confined to 1,4‐dioxane/MeOH, thus resulting in the incompatibility of more elaborate electrophiles such as epoxides and O‐hydroxylaminesulfonic acid. Such couplings generally require water as a solvent.18
Scheme 2.
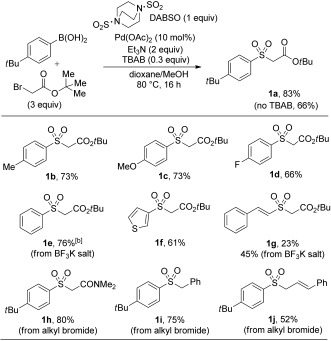
Boronic acid and electrophile scope in the one‐step palladium(II)‐catalyzed sulfone synthesis. Reaction conditions: Boronic acid (0.25 mmol, 1 equiv), Pd(OAc)2 (10 mol %), DABSO (1 equiv), Et3N (2 equiv), MeOH/1,4‐dioxane (1:1) [0.16 m], RX (0.75 mmol). Isolated yields. [a] 100 °C. TBAB=tetra‐n‐butylammonium bromide.
To achieve a more general process we were motivated to pursue the proposed synthesis of sulfinate derivatives with subsequent in situ trapping in a two‐step format. We postulated that the palladium sulfinate generated after Pd—C SO2 insertion was responsible for the observed low reactivity with simple electrophiles, and that a Lewis acid or ligand could aid Pd—O(S) bond cleavage to release an alternative metal sulfinate and turnover the palladium(II) catalyst. While initial attempts were unsuccessful, upon introducing a Brønsted acid in the form of TFA into the reaction [p‐tolylboronic acid with DABSO and Pd(OAc)2, without Et3N], the sulfinic acid 2 b (for structure see Scheme 3) was observed in 75 % yield (HPLC). When performing the same transformation in the absence of both TFA and Et3N, 2 b was formed in 92 % yield, thus translating into a 88 % yield of the sulfone 1 b after base treatment and alkylation (Scheme 3).
Scheme 3.
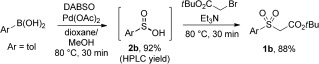
Base‐free, one‐pot, two‐step sulfone synthesis. Pd(OAc)2 (10 mol %).
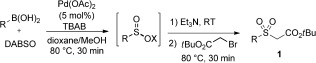
With this key observation that the desired boronic acid sulfination could be achieved by simple treatment with Pd(OAc)2 and DABSO in a short reaction time (30 min), further assessment of the reaction conditions revealed that lower palladium(II) loadings could effect this transformation and that MeOH was integral as a cosolvent (see the Supporting Information for details). On examination of the boronic acid scope of the reaction, using these optimized reaction conditions, it was quickly established that varying the electronics of the aryl boronic acid was detrimental to reactivity. Substrates bearing p‐fluoro and p‐tert‐butyl functionalities resulted in immediate biphenyl and palladium(0) formation. This side‐reaction could be suppressed by employing TBAB as an additive in a low loading (0.25 equiv). A full examination of the reaction scope was then conducted, and by employing triethylamine and tert‐butyl bromoacetate, as the electrophilic trap, a range of sulfones could be accessed (Table 1).19 Substrates bearing electron‐donating and electron‐withdrawing groups, as well as ortho‐, meta‐, and para‐substitution patterns were all well tolerated in this system. Pleasingly, sensitive functional groups such as phenol (1 m), amide (1 n), amine (1 p), and indole (1 u) moieties were compatible with sulfones obtained in good yields.
Table 1.
Scope with respect to the boronic acid in the one‐pot, two‐step palladium(II)‐catalyzed sulfone synthesis.[a]
|
|
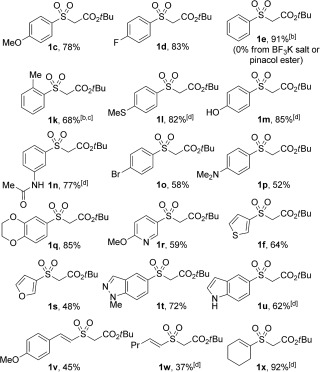
[a] Reaction conditions: 1) Boronic acid (0.25 mmol, 1 equiv), Pd(OAc)2 (5 mol %), DABSO (1 equiv), TBAB (0.25 equiv), 1,4‐dioxane/MeOH (1:1) [0.16 m], 80 °C. 2) Et3N (2 equiv) RT, 1 min then tert‐butyl bromoacetate (3 equiv), 80 °C, 30 min. [b] Without TBAB. [c] Sulfination step for 3 h. [d] Sulfination step for 1 h.
The incorporation of the methylthio‐substituted boronic acid (1 l) promotes the use of this chemistry over traditional sulfide oxidation strategies, where access to such mixed oxidation‐state S products would not be possible. Pleasingly, a variety of heteroaromatic groups were well tolerated in this reaction, with thiophene and furan substrates delivering the corresponding products in acceptable yields (1 f and 1 s), while examples incorporating pyridine (1 r), benzodioxane (1 q), and indazole (1 t) functionalities performed well. Alkenyl boronic acids could be employed as substrates, with styryl and straight‐chain alkenyl variants giving products in modest yields (1 v,w), and the cyclohexenyl boronic acid afforded the corresponding sulfone in high yield (1 x).
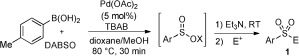
We next looked to expand the scope of sulfonyl‐containing products beyond those accessible with the one‐step procedure (Scheme 2), and were pleased to find that a variety of C‐based electrophiles could be employed (Table 2). Alkylations could be achieved by maintaining 1,4‐dioxane/MeOH as the solvent and simple alkyl iodides were found to perform well (1 y,z). Diaryl sulfones were accessible using either an aryl iodonium salt (1 aa)20 or through SNAr chemistry (1 ab and 1 ac).21 Modification of the reaction conditions for the derivatization of the sulfinic acid derivatives allowed the chemistry to be extended to epoxide ring opening. For example, use of cyclohexene oxide delivered the sulfone 1 ad in good yield. The β‐hydroxy sulfone 1 ae was prepared using this one‐pot process in moderate yield, and is significant as it corresponds to the methyl ester derivative of the anti‐androgen pharmaceutical Casodex.22
Table 2.
Scope with respect to the C electrophile in the one‐pot, two‐step palladium(II)‐catalyzed sulfone synthesis.[a]
|
|
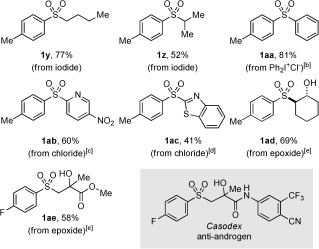
[a] Reaction conditions for sulfinate derivatization step: E+ (3 equiv), 1,4‐dioxane/MeOH, 80 °C, 1 h. [b] Ph2I+Cl− (3 equiv), 1,4‐dioxane/MeOH, 80 °C, 6 h. [c] E+ (2 equiv), DMAc, 100 °C, 1 h. [d] E+ (3 equiv), DMSO, 80 °C, 2 h. [e] Epoxide (5 equiv), H2O, 90 °C, 6 h. DMAc= N,N‐dimethylacetamide, DMSO=dimethylsulfoxide.
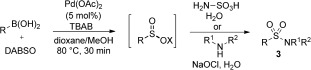
The methodology could be extended to allow the use of N‐electrophiles. Both primary and substituted sulfonamides could be prepared by combining the in situ generated sulfinates with either O‐hydroxylaminesulfonic acid or N‐chloroamines (also generated in situ; Table 3).10a
Table 3.
The use of N electrophiles to access sulfonamides.[a]
|
|
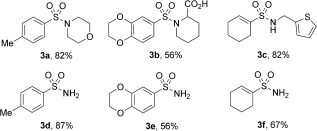
Sulfinate derivatization step: [a] Et3N, RT then H2NSO3H (2 equiv), H2O, RT, 2 h or R1R2NH (5 equiv), NaOCl (3 equiv), H2O, RT, 16 h.
Finally, we were able to demonstrate the application of this one‐pot sulfone synthesis on a preparative scale. Using a 1 mol % catalyst loading and a slightly extended sulfination reaction time, the sulfone 1 b was isolated in 90 % yield on a 3.5 gram scale (Scheme 4). Importantly, this larger scale reaction was performed open to air.
Scheme 4.
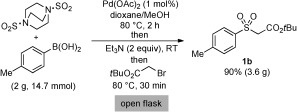
Preparative scale synthesis of the sulfone 1 b.
In summary, we have developed a one‐pot, redox‐neutral and ligand‐free synthesis of sulfinic acid derivatives. By simply treating boronic acid substrates with DABSO and low loadings of Pd(OAc)2, sulfinic acid intermediates were formed and subsequently coupled with a variety of electrophiles in situ, to access a broad range of sulfones and sulfonamides. In addition to this, a one‐step reaction was also developed, thus allowing preparation of sulfones using activated alkyl halides.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
miscellaneous_information
Acknowledgements
This work was supported by the EPSRC and Syngenta.
References
- 1.
- 1a. McGrath N. A., Brichacek M., Njardarson J. T., J. Chem. Educ. 2010, 87, 1348; [Google Scholar]
- 1b. Smith D. A., Jones R. M., Curr. Opin. Drug Discovery Dev. 2008, 11, 72; [PubMed] [Google Scholar]
- 1c. Otzen T., Wempe E. G., Kunz B., Bartels R., Lehwark‐Yvetot G., Hänsel W., Schaper K.‐J., Seydel J. K., J. Med. Chem. 2004, 47, 240; [DOI] [PubMed] [Google Scholar]
- 1d. Kleeman A., Engel J., Kutscher B., Reichert D., Pharmaceutical Substances, Syntheses Patents, Applications, Thieme, Stuttgart, 1999. [Google Scholar]
- 2.
- 2a. Simpkins N. S., Sulphones in Organic Synthesis, Pergamon Press, Oxford, 1993; [Google Scholar]
- 2b. Crowley P. J., Fawcett J., Kariuki B. M., Moralee A. C., Percy J. M., Salafia V., Org. Lett. 2002, 4, 4125. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Xu W.‐M., Han F.‐F., He M., Hu D.‐Y., He J., Yang S., Song B.‐A., J. Agric. Food Chem. 2012, 60, 1036; [DOI] [PubMed] [Google Scholar]
- 3b. Noutoshi Y., Ikeda M., Saito T., Osada H., Shirasu K., Front. Plant Sci. 2012, 3, 245; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3c. Sun Z.‐Y., Botros E., Su A.‐D., Kim Y., Wang E., Baturay N. Z., Kwon C.‐H., J. Med. Chem. 2000, 43, 4160. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Silvestri R., De Martino G., La Regina G., Artico M., Massa S., Vargiu L., Mura M., Loi A. G., Marceddu T., La Colla P., J. Med. Chem. 2003, 46, 2482; [DOI] [PubMed] [Google Scholar]
- 4b. Neamati N., Mazumder A., Zhao H., Sunder S., Burke T. R., Schultz R. J., Pommier Y., Antimicrob. Agents Chemother. 1997, 41, 385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.
- 5a. Kozak J. A., Dake G. R., Angew. Chem. Int. Ed. 2008, 47, 4221; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 4289; [Google Scholar]
- 5b. Jana N. K., Verkade J. G., Org. Lett. 2003, 5, 3787; [DOI] [PubMed] [Google Scholar]
- 5c. Sato K., Hyodo M., Aoki M., Zheng X.‐Q., Noyori R., Tetrahedron 2001, 57, 2469; [Google Scholar]
- 5d. Trost B. M., Curran D. P., Tetrahedron Lett. 1981, 22, 1287. [Google Scholar]
- 6. Hamada T., Yonemitsu O., Synthesis 1986, 852. [Google Scholar]
- 7.
- 7a. Emmett E. J., Willis M. C., Asian J. Org. Chem. 2015, 4, 602; [Google Scholar]
- 7b. Deeming A. S., Emmett E. J., Richards‐Taylor C. S., Willis M. C., Synthesis 2014, 2701. [Google Scholar]
- 8.
- 8a. Chen C. C., Waser J., Org. Lett. 2015, 17, 736; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8b. Deeming A. S., Russell C. J., Hennessy A. J., Willis M. C., Org. Lett. 2014, 16, 150; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8c. Rocke B. N., Bahnck K. B., Herr M., Lavergne S., Mascitti V., Perreault C., Polivkova J., Shavnya A., Org. Lett. 2014, 16, 154; [DOI] [PubMed] [Google Scholar]
- 8d. Richards‐Taylor C. S., Blakemore D. C., Willis M. C., Chem. Sci. 2014, 5, 222; [Google Scholar]
- 8e. Emmett E. J., Hayter B. R., Willis M. C., Angew. Chem. Int. Ed. 2013, 52, 12679; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 12911. [Google Scholar]
- 9.
- 9a. Wang X., Xue L., Wang Z., Org. Lett. 2014, 16, 4056; [DOI] [PubMed] [Google Scholar]
- 9b. Emmett E. J., Richards‐Taylor C. S., Nguyen B., Garcia‐Rubia A., Hayter B. R., Willis M. C., Org. Biomol. Chem. 2012, 10, 4007; [DOI] [PubMed] [Google Scholar]
- 9c. Nguyen B., Emmett E. J., Willis M. C., J. Am. Chem. Soc. 2010, 132, 16372. [DOI] [PubMed] [Google Scholar]
- 10a. Deeming A. S., Russell C. J., Willis M. C., Angew. Chem. Int. Ed. 2015, 54, 1168; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 1184; [Google Scholar]
- 10b. Waldmann C., Schober O., Haufe G., Kopka K., Org. Lett. 2013, 15, 2954; [DOI] [PubMed] [Google Scholar]
- 10c. Woolven H., González‐Rodríguez C., Marco I., Thompson A. L., Willis M. C., Org. Lett. 2011, 13, 4876. [DOI] [PubMed] [Google Scholar]
- 11.For the use of K2S2O5, see:
- 11a. Ye S., Wu J., Chem. Commun. 2012, 48, 10037; For a radical process that exploits DABSO, see: [Google Scholar]
- 11b. Zheng D., An Y., Li Z., Wu J., Angew. Chem. Int. Ed. 2014, 53, 2451; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 2483; [Google Scholar]
- 11c. Zheng D., Li Y., An Y., Wu J., Chem. Commun. 2014, 50, 8886. [DOI] [PubMed] [Google Scholar]
- 12. Emmett E. J., Hayter B. R., Willis M. C., Angew. Chem. Int. Ed. 2014, 53, 10204; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 10368. [Google Scholar]
- 13. Shavnya A., Coffey S. B., Smith A. C., Mascitti V., Org. Lett. 2013, 15, 6226. [DOI] [PubMed] [Google Scholar]
- 14. Johnson M. W., Bagley S. W., Mankad N. P., Bergman R. G., Mascitti V., Toste F. D., Angew. Chem. Int. Ed. 2014, 53, 4404; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 4493. [Google Scholar]
- 15.Wu and co‐workers have reported a palladium(0)‐catalyzed aryl boronic acid based synthesis of aminosulfonamides: Ye S., Wu J., Chem. Commun. 2012, 48, 7753. [Google Scholar]
- 16.Buchwald and co‐workers have reported the palladium(0)‐catalyzed coupling of aryl boronic acids and phenyl chlorosulfate to deliver aryl sulfonyl chlorides: DeBergh J. R., Niljianskul N., Buchwald S. L., J. Am. Chem. Soc. 2013, 135, 10638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.After submission of this manuscript, related chemistry, employing aryl boronic acids as substrates in the palladium‐catalyzed synthesis of sulfones appeared online: Shavnya A., Hesp K. D., Mascitti V., Smith A. C., Angew. Chem. Int. Ed. 2015, 54, 13571; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 13775. [Google Scholar]
- 18. Narayana Murthy S., Madhav B., Prakash Reddy V., Rao K. Rama, Nageswar Y. V. D., Tetrahedron Lett. 2009, 50, 5009. [Google Scholar]
- 19.The sulfinate intermediate is depected as Ar—S(O)OX to allow for the possibility that a boron sulfinate or the sulfinic acid is the relevant intermediate during the reaction.
- 20.
- 20a. Umierski N., Manolikakes G., Org. Lett. 2013, 15, 188; [DOI] [PubMed] [Google Scholar]
- 20b. Umierski N., Manolikakes G., Org. Lett. 2013, 15, 4972. [DOI] [PubMed] [Google Scholar]
- 21.
- 21a. Liang S., Zhang R.‐Y., Xi L.‐Y., Chen S.‐Y., Yu X.‐Q., J. Org. Chem. 2013, 78, 11874; [DOI] [PubMed] [Google Scholar]
- 21b. Maloney K. M., Kuethe J. T., Linn K., Org. Lett. 2011, 13, 102. [DOI] [PubMed] [Google Scholar]
- 22. Schellhammer P. F., Expert Opin. Pharmacother. 2002, 3, 1313. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
miscellaneous_information