

Abstract
Molecular structures of the most prominent chiral non‐racemic hypervalent iodine(III) reagents to date have been elucidated for the first time. The formation of a chirally induced supramolecular scaffold based on a selective hydrogen‐bonding arrangement provides an explanation for the consistently high asymmetric induction with these reagents. As an exploratory example, their scope as chiral catalysts was extended to the enantioselective dioxygenation of alkenes. A series of terminal styrenes are converted into the corresponding vicinal diacetoxylation products under mild conditions and provide the proof of principle for a truly intermolecular asymmetric alkene oxidation under iodine(I/III) catalysis.
Keywords: alkenes, chirality, diacetoxylation, hydrogen bonding, hypervalent iodine
Enantioselective catalysis has been recognized as a core technology toward the supply of chiral molecular entities with defined absolute configuration. Different methods have been devised over past decades and synthetic molecular catalysts based on transition‐metal complexes1 or small organic compounds2 represent the most advanced concepts in the field.3 Within the context of small organic catalysts, chiral hypervalent iodine reagents have enabled the development of significant advances in catalytic oxidation reactions that do not rely on common transition metals.4, 5 Examples include the ester derivative 1 6, 7 and the amide derivative 2 a,8, 9 which has been identified as a particularly successful reagent.
In contrast to the rapidly growing number of successful examples of chiral molecular iodine(I/III) catalysts,5, 6, 10 defined structural information on the accurate enantiocontrol of these compounds is notably missing. A definite understanding of the mode of action of chiral hypervalent iodine reagents can form the basis to enlarge the differentiation of prochiral face recognition from the established intramolecular reaction control to more challenging topics, such as the asymmetric oxidation of prochiral substrates by intermolecular reaction control, and particularly in terms of catalysis. Within this context, no definite mechanistic investigation has so far been reported for the parent iodine(III) reagents 1 and 2 a bearing lactic esters and amides as chiral entities (Figure 1).11, 12 Regarding the important iodine 2 a, structural information could now be obtained for the derivatives 2 b and 2 c, which contain sterically enlarged anilide groups. As unambiguously determined by X‐ray analyses (Figure 2) for both compounds, the amide NH groups engage in hydrogen bonding with the acetoxy groups located at the iodine center.13a These hydrogen bonds have the expected values for NH–O contacts of 2.201 and 2.243 Å, and for N‐H‐O bond angles of 163.3 and 160.0°,13a and generate two nine‐membered rings.14, 15
Figure 1.

Representative chiral iodine reagents 1 and 2 a and successful enantioselective intramolecular reactions with 2 a.
Figure 2.
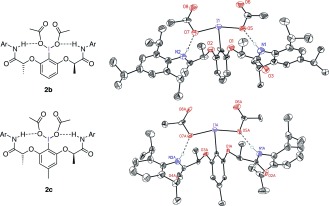
Intramolecular hydrogen‐bonding properties of chiral hypervalent iodine compounds 2 b (Ar=2,4,6‐iPr3C6H2) and 2 c (Ar=2,6‐iPr2C6H3). All hydrogen atoms except the N‐H groups are omitted for clarity.
The main stereochemical consequence of this hydrogen bonding rests in the effective creation of a supramolecular helical chirality around the central iodine atom.13b,c This is best envisioned through the respective top view representations (Figure 3, top). The chiral helicity is of C 2 symmetry and forms with complete diastereoselectivity as demonstrated by 1H NMR spectroscopy. X‐Ray data and circular dichroism (CD) spectroscopy (Figure 3, bottom) confirm that the helical chirality is obtained with identical configuration for both 2 b and 2 c and thus directly originates from the absolute configuration of the chiral lactamide as the stereoinducing group. CD and one‐ and two‐dimensional 1H NMR data14 furthermore confirm that the H‐bonding persists in solution. With the demonstration of the effective hydrogen‐bonding motif, the lateral lactamide chains are recognized as a source of chiral induction for the generation of the supramolecular helical chirality. It is the helical chirality that is expected to exercise efficient enantioselection in asymmetric catalytic oxidation reactions with 2 a. In this context, we studied the requirements to obtain intermolecular enantioselective oxidation reactions based on using the chiral motif 2 as the catalyst. While intramolecular reactivity has been widely explored with reagent 2 a,8 related intermolecular asymmetric transformations have remained without demonstration. In addition, the general scope of compounds 2 as chiral catalysts still remains to be explored, since the Kita intramolecular spirolactonization currently stands out as the single successful example.8a,b,f
Figure 3.
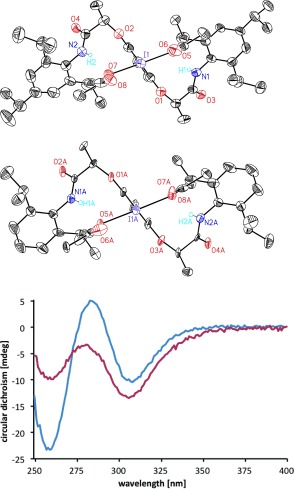
Top: Intramolecular hydrogen‐bonding properties of chiral hypervalent iodine compounds 2 b and 2 c result in helical chiral assemblies (all hydrogen atoms except the N‐H group are omitted for clarity). Bottom: CD spectra for compounds 2 b (red) and 2 c (blue).
In early work on chiral hypervalent iodine reagents Wirth devised an enantioselective dioxygenation reaction under stoichiometric conditions,16 which was later extended using the chiral bislactate derivative 1.7k Herein, we now provide the first catalytic enantioselective version based on the use of hypervalent iodine compounds 2 of the bislactamide motif. The initial optimization process focused on a suitable reoxidant, for which peracetic acid was identified.14 It is noteworthy that this oxidant is superior to the commonly employed mCPBA,10 which promotes epoxidation in the present case. Our current hypothesis is that owing to its lower reactivity, peracetic acid does not promote such a background reaction.14, 17 The iodine(I/III)‐catalyzed diacetoxylation of styrene 4 a was then investigated for several structurally related aryl iodine(I) catalyst precursors 3. First, the established compound 3 a was employed. At a catalyst loading of 20 mol %, a reasonable conversion was obtained and the product 5 a was obtained with 84 % ee (Table 1, entry 1). A similar outcome was obtained for new derivative 3 b as the catalyst precursor to 2 b (83 % ee, entry 2). While an attempt to lower the catalyst loading led to a decreased conversion (entry 3), the introduction of a 4‐methyl group into the central arene core of 2 a greatly enhanced the reactivity (57 % isolated yield of 5 a, 83 % ee, entry 4). Further modification of the aniline substitution led to identification of 3 c as the most active and enantioselective catalyst precursor (86 % ee, entry 5). The yield of isolated product could be enhanced by addition of trifluoromethansulfonic acid (TfOH) as Brønsted acidic co‐catalyst (entry 6), which resulted in a reasonable conversion even at a 5 mol % catalyst loading (entry 7).
Table 1.
Enantioselective catalytic diacetoxylation of styrenes: Optimization. 
| En‐ | Catalyst | Loading | Yield | ee |
|---|---|---|---|---|
| try[a] | [mol %] | [%][a] | [%][b] | |
| 1 |
|
20 | 61 | 84 |
| 2 |
|
20 | 58 | 83 |
| 3 | 10 | 43 | 83 | |
| 4 |
|
10 | 57 | 83 |
| 5 |
|
10 | 55 | 86 |
| 6 | 10[c] | 71 | 85 | |
| 7 | 5[c] | 61 | 83 |

[a] Yield after purification. [b] Determined by HPLC on chiral stationary phase after conversion into the free diol. [c] With additional 5 mol % of TfOH.
New aryl iodide 3 c displays additional advantageous features. In contrast to other catalyst precursors 3, it is fully soluble at the start of the reaction and can be fully recovered afterwards, while compounds 3 a, 3 b, and 3 d suffer degradation over time. In the same context, the isolated active catalyst 2 c has an unprecedentedly high stability in solution and prolonged life‐time in the isolated form.14 It is further noteworthy that the enantioselectivity of the catalytic reaction is identical to that from a stoichiometric reaction with preformed 2 c. This result corroborates again the involvement of hydrogen bonding in the catalytic transformation.14
The optimized conditions proved to be generally applicable, and a total of twenty‐four differently substituted styrenes 4 a–x could be converted into the corresponding dioxygenation products 5 a–x in an enantioselective and completely chemoselective manner (Figure 4). Besides parent styrene 4 a, several para‐substituted styrenes 4 b–l could be cleanly diacetoxylated and the products 5 b–l were obtained with up to 94 % ee. Likewise, meta‐substituted products 5 m–p were obtained with up to 90 % ee, and ortho‐substituents led to diacetoxylation in 86–90 % ee (products 5 q–s). Higher‐substitution pattern was equally tolerated as demonstrated for products 5 t–w. Finally, 3‐vinyl estra‐1,3,5(10)‐trien‐17‐one 4 x was converted into the corresponding diol 5 x under catalyst control and with complete chemoselectivity.14 In view of these results and given the inherent advantage that hypervalent iodine catalysis does not suffer from potential product contamination by the presence of residual transition metal ions, the metal‐free catalysis described provides reactivity that can complement established enantioselective transition‐metal‐based procedures.18 The benign use of peracetic acid as the terminal oxidant adds to the overall attractiveness of the process since no by‐products other than the solvent acetic acid and water are produced.
Figure 4.

Scope of the enantioselective catalytic diacetoxylation of styrenes.
The observed results can be rationalized by the underlying catalytic cycle depicted in Figure 5. At the reaction outset, peracetic acid oxidizes iodine(I) precursor 3 c to the active catalyst 2 c. Its established H‐bonding motif offers a chance to study the enantioselection in the prochiral alkene recognition. As explored in detail by Gade and Kang, triflic acid activation of PhI(OAc)2 via protonolysis is a prerequisite to accelerate diacetoxylation reactions.19 This points to a different role of the two acetate groups in 2 c. One engages in hydrogen bonding for the generation of a cyclic stereochemical arrangement. The second one should be prone to dissociation and thus to formation of the iodine(III) catalyst state A, which is no longer of C 2‐symmetry, but rather C 1‐symmetry. This catalyst state with its free coordination site at iodine(III) engages in the required efficient prochiral face differentiation of the unsaturated hydrocarbon substrate within a preferential coordination C. Subsequent nucleophilic attack of acetate to the exposed re‐face establishes the correct S‐configured benzylic C—O bond formation at intermediate B. Intramolecular nucleophilic addition of the acetate regenerates iodine(I) catalyst state 3 c and provides the Woodward dioxolane intermediate 6,7i, 20 which upon aqueous addition generates the two regioisomeric acetoxy alcohols 7 and 7′ that can be unambiguously detected in the crude reaction product. Treatment of this crude mixture with acetic anhydride furnishes 5 as the uniform product.
Figure 5.
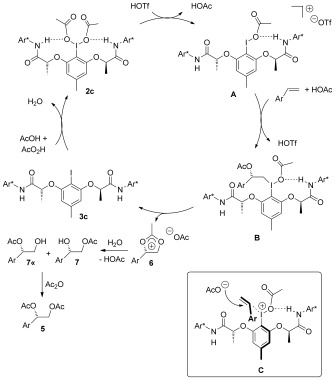
Catalytic cycle.
In summary, we have uncovered hydrogen bonding as the significant structural force in chiral hypervalent iodine reagents 2. Based on this structural insight, we have developed a first method for an iodine(III)‐catalyzed enantioselective vicinal dioxygenation of alkenes under entirely intermolecular reaction control. The reaction proceeds under mild conditions and provides the corresponding oxidation products with up to 94 % ee. The successful intermolecular diacetoxylation reaction using 2 c suggests for the first time that the broad potential of asymmetric hypervalent iodine catalysis can indeed be realized for intermolecular reactions. It can be expected that building on the structure‐determining features of hydrogen bonding as a guiding principle in catalyst development will lead to additional catalytic asymmetric reactions.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
miscellaneous_information
Acknowledgements
Financial support for this project was provided from the Spanish Ministry for Economy and Competitiveness (CTQ2011‐25027 grant to K.M., and Severo Ochoa Excellence Accreditation 2014‐2018 to ICIQ, SEV‐2013‐0319), the Deutsche Forschungsgemeinschaft (WO 1924/1‐1 to T.H.W.) and Cellex‐ICIQ Programme (fellowship to C.M.). We thank E. Escudero‐Adán for the X‐ray structural analyses.
References
- 1.
- 1a. Noyori R., Asymmetric Catalysis In Organic Synthesis, Wiley, Chichester, 1994; [Google Scholar]
- 1b. Catalytic Asymmetric Synthesis (Ed.: I. Ojima), 2nd ed., Wiley‐VCH, Weinheim, 2000; [Google Scholar]
- 1c. Transition Metals for Organic Synthesis (Eds.: M. Beller, C. Bolm), 2nd ed., Wiley‐VCH, Weinheim, 2004; [Google Scholar]
- 1d. Comprehensive Asymmetric Catalysis (Eds.: E. N. Jacobsen, A. Pfaltz, H. Yamamoto), Springer, Berlin, 1999. [Google Scholar]
- 2.
- 2a. Berkessel A., Groeger H., Asymmetric Organocatalysis, Weinheim, Wiley‐VCH, 2005; [Google Scholar]
- 2b.Special issue Organocatalysis, List B., Chem. Rev. 2007, 107, 5413; [Google Scholar]
- 2c. MacMillan D. C., Nature 2008, 455, 304; [DOI] [PubMed] [Google Scholar]
- 2d.For particularly general organocatalytic oxidation reactions: Zhu Y., Wang Q., Cornwall R. G., Shi Y., Chem. Rev. 2014, 114, 8199. [DOI] [PubMed] [Google Scholar]
- 3.For the emerging field of directed evolution:
- 3a. Reetz M. T., J. Am. Chem. Soc. 2013, 135, 12480; [DOI] [PubMed] [Google Scholar]
- 3b. Renata H., Wang Z. J., Arnold F. H., Angew. Chem. Int. Ed. 2015, 54, 3351; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 3408; [Google Scholar]
- 3c. Romero P. A., Arnold F. H., Nat. Rev. Mol. Cell Biol. 2009, 10, 866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.For general reviews on iodine(III) reagents:
- 4a. Zhdankin V. V., Hypervalent Iodine Chemistry Preparation, Structure and Synthetic Applications of Polyvalent Iodine Compounds, Wiley, Chichester, 2014; [Google Scholar]
- 4b.“Hypervalent Iodine Chemistry. Modern Developments in Organic Synthesis”: Topics in Current Chemistry, Vol. 224 (Ed.: T. Wirth), Springer, Berlin, 2003; [Google Scholar]
- 4c. Stang P. J., Zhdankin V. V., Chem. Rev. 1996, 96, 1123; [DOI] [PubMed] [Google Scholar]
- 4d. Zhdankin V. V., Stang P. J., Chem. Rev. 2002, 102, 2523; [DOI] [PubMed] [Google Scholar]
- 4e. Zhdankin V. V., Stang P. J., Chem. Rev. 2008, 108, 5299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.
- 5a. Uyanik M., Ishihara K., J. Synth. Org. Chem. Jpn. 2012, 70, 1116; [Google Scholar]
- 5b. Singh F. V., Wirth T., Chem. Asian J. 2014, 9, 950; [DOI] [PubMed] [Google Scholar]
- 5c. Romero R. M., Wöste T. H., Muñiz K., Chem. Asian J. 2014, 9, 972. [DOI] [PubMed] [Google Scholar]
- 6.Initial reports:
- 6a. Fujita M., Okuno S., Lee H. J., Sugimura T., Okuyama T., Tetrahedron Lett. 2007, 48, 8691; [Google Scholar]
- 6b. Fujita M., Ookubo Y., Sugimura T., Tetrahedron Lett. 2009, 50, 1298. [Google Scholar]
- 7.Stoichiometric and catalytic transformations with 1:
- 7a. Röben C., Souto J. A., González Y., Lishchynskyi A., Muñiz K., Angew. Chem. Int. Ed. 2011, 50, 9478; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 9650; [Google Scholar]
- 7b. Kong W., Feige P., de Haro T., Nevado C., Angew. Chem. Int. Ed. 2013, 52, 2469; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 2529; [Google Scholar]
- 7c. Souto J. A., González Y., Iglesias A., Zian D., Lishchynskyi A., Muñiz K., Chem. Asian J. 2012, 7, 1103; [DOI] [PubMed] [Google Scholar]
- 7d. Fujita M., Yoshida Y., Miyata K., Wakisaka A., Sugimura T., Angew. Chem. Int. Ed. 2010, 49, 7068; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 7222; [Google Scholar]
- 7e. Takesue T., Fujita M., Sugimura T., Akutsu H., Org. Lett. 2014, 16, 4634; [DOI] [PubMed] [Google Scholar]
- 7f. Shimogaki M., Fujita M., Sugimura T., Eur. J. Org. Chem. 2013, 7128; [DOI] [PubMed] [Google Scholar]
- 7g. Fujita M., Mori K., Shimogaki M., Sugimura T., RSC Adv. 2013, 3, 17717; [Google Scholar]
- 7h. Fujita M., Mori K., Shimogaki M., Sugimura T., Org. Lett. 2012, 14, 1294; [DOI] [PubMed] [Google Scholar]
- 7i. Fujita M., Wakita M., Sugimura T., Chem. Commun. 2011, 47, 3983. [DOI] [PubMed] [Google Scholar]
- 8.Stoichiometric and catalytic transformations with 2 a:
- 8a. Uyanik M., Yasui T., Ishihara K., Angew. Chem. Int. Ed. 2010, 49, 2175; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 2221; [Google Scholar]
- 8b. Uyanik M., Yasui T., Ishihara K., Tetrahedron 2010, 66, 5841; [Google Scholar]
- 8c. Farid U., Wirth T., Angew. Chem. Int. Ed. 2012, 51, 3462; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 3518; [Google Scholar]
- 8d. Farid U., Malmedy F., Claveau R., Albers L., Wirth T., Angew. Chem. Int. Ed. 2013, 52, 7018; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 7156; [Google Scholar]
- 8e. Mizar P., Wirth T., Angew. Chem. Int. Ed. 2014, 53, 5993; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 6103; [Google Scholar]
- 8f. Zhang D.‐Y., Xu L., Wu H., Gong L.‐Z., Chem. Eur. J. 2015, 21, 10314; [DOI] [PubMed] [Google Scholar]
- 8g. Alhalib A., Kamouka S., Moran W. J., Org. Lett. 2015, 17, 1453. [DOI] [PubMed] [Google Scholar]
- 9.Stoichiometric and catalytic transformations with related compounds:
- 9a. Rodríguez A., Moran W. J., Synthesis 2012, 1178; [Google Scholar]
- 9b. Uyanik M., Yasui T., Ishihara K., Angew. Chem. Int. Ed. 2013, 52, 9215; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 9385; [Google Scholar]
- 9c. Wu H., He Y.‐P., Xu L., Zhang D.‐Y., Gong L.‐Z., Angew. Chem. Int. Ed. 2014, 53, 3466; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 3534. [Google Scholar]
- 10.For general reviews on iodine(I/III) catalysis in homogeneous oxidation:
- 10a. Richardson R. D., Wirth T., Angew. Chem. Int. Ed. 2006, 45, 4402; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 4510; [Google Scholar]
- 10b. Ochiai M., Miyamoto K., Eur. J. Org. Chem. 2008, 4229; [Google Scholar]
- 10c. Ochiai M., Chem. Rec. 2007, 7, 12; [DOI] [PubMed] [Google Scholar]
- 10d. Dohi T., Kita Y., Chem. Commun. 2009, 2073; [DOI] [PubMed] [Google Scholar]
- 10e. Uyanik M., Ishihara K., Chem. Commun. 2009, 2086. [DOI] [PubMed] [Google Scholar]
- 11.As to the only noteworthy exception, one of us reported on a related hypervalent iodine with chiral amidoalcohol substituents that provide a hydrogen‐bonding scenario: see Ref. [9b].
- 12.For X‐ray structural data on some derivatives of compound 1 see Refs. [7d,i].
- 13a. Desiraju G. R., Angew. Chem. Int. Ed. 2011, 50, 52; [Google Scholar]; Angew. Chem. 2011, 123, 52; [Google Scholar]
- 13b. Lehn J. N., Supramolecular Chemistry. Concepts and Perspectives, Wiley‐VCH, Weinheim, 1995; [Google Scholar]
- 13c. Hydrogen Bonded Supramolecular Structures (Eds.: Z. Li, L.‐Z. Wu), Springer, Berlin, 2015. [Google Scholar]
- 14.See Supporting Information for details.
- 15. CCDC 1404588 (2 b) and 1404589 (2 c) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre.
- 16.
- 16a. Hirt U. H., Spingler B., Wirth T., J. Org. Chem. 1998, 63, 7674; [Google Scholar]
- 16b. Wirth T., Hirt U. H., Tetrahedron: Asymmetry 1997, 8, 23; [Google Scholar]
- 16c. Hirt U. H., Schuster M. F. H., French A. N., Wiest O. G., Wirth T., Eur. J. Org. Chem. 2001, 1569; [Google Scholar]
- 16d. Boye A. C., Meyer D., Ingison C. K., French A. N., Wirth T., Org. Lett. 2003, 5, 2157. [DOI] [PubMed] [Google Scholar]
- 17.At present, we cannot rule out that a minor contribution of background reaction is involved. The high enantioselectivities of products 5 a–s suggest that such a process, if present, does not influence them significantly. For discussions on such a background reaction, see:
- 17a. Kang Y.‐B., Gade L. H., J. Org. Chem. 2012, 77, 1610; [DOI] [PubMed] [Google Scholar]
- 17b. Zhong W., Liu S., Yang J., Meng X., Li Z., Org. Lett. 2012, 14, 3336; [DOI] [PubMed] [Google Scholar]
- 17c.For a discussion on the challenge of overcoming the peracid background reaction in (chiral) iodine catalysis, see: Richardson R. D., Desaize M., Wirth T., Chem. Eur. J. 2007, 13, 6745. [DOI] [PubMed] [Google Scholar]
- 18.
- 18a. Kolb H. C., VanNieuwenhze M. S., Sharpless K. B., Chem. Rev. 1994, 94, 2483; [Google Scholar]
- 18b. Zaitsev A. B., Adolfsson H., Synthesis 2006, 1725. [Google Scholar]
- 19. Kang Y.‐B., Gade L. H., J. Am. Chem. Soc. 2011, 133, 3658. [DOI] [PubMed] [Google Scholar]
- 20. Woodward R. B., F. V. Brutcher, Jr. , J. Am. Chem. Soc. 1958, 80, 209. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
miscellaneous_information