

Abstract
An olefin‐directed palladium‐catalyzed oxidative regio‐ and stereoselective arylation of allenes to afford 1,3,6‐trienes has been established. A number of functionalized allenes, including 2,3‐ and 3,4‐dienoates and 3,4‐dienol derivatives, have been investigated and found to undergo the olefin‐directed allene arylation. The olefin moiety has been proven to be a crucial element for the arylating transformation.
Keywords: allenes, arylation, directing groups, oxidation, palladium
Allenes, a class of compounds with the interesting and special substructure of two cumulative carbon–carbon double bonds,1 have been demonstrated as powerful building blocks for the construction of complicated natural products, as well as pharmacologically active compounds.2, 3 Therefore, much attention has been focused on transition‐metal‐catalyzed cyclizations of functionalized allenes, especially those catalyzed by palladium.4 The PdII‐promoted reactions of allenes with a nucleophilic functionality, mostly an N‐ or O‐containing functional group, would produce cyclic intermediates Int ‐1 or Int ‐1′ (Scheme 1 a). A subsequent cross‐coupling reaction would lead to product A or A′ respectively. However, the utilization of a π‐bond‐containing group as a nucleophile for such cyclizations is highly limited.5 On this basis, we envisioned that enallene (1, FG=vinyl) may undergo annulation under the catalysis of PdII to provide cyclized product B by reaction with an external nucleophile via Int ‐2 (Scheme 1 b).
Scheme 1.
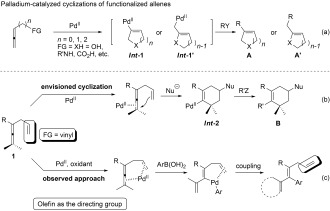
a) Traditional Pd‐catalyzed cyclization of functionalized allenes. b) Envisioned Pd‐catalyzed cyclization of enallenes with an external nucleophile. c) Observed approach of olefin‐directed Pd‐catalyzed oxidative arylation of allenes. FG=functional group. Nu=nucleophilic unit.
Based on this concept, we initially chose a readily accessible 2,3‐dienoate as the standard substrate.6 When allyl‐substituted 2,3‐dienoate 1 a was treated with Pd(OAc)2 (5 mol %), PhB(OH)2 (1.3 equiv), and BQ (1.1 equiv) in the presence of NaOAc (1.2 equiv) in THF at 50 °C for 23 h, the envisioned product 4 a was not observed (Scheme 2). Surprisingly, instead the phenylated triene product (E)‐3 aa was obtained in 74 % yield as a single stereoisomer (Scheme 2, cf. Scheme 1 c). The stereochemistry was determined by NOE measurements. It should be noted that the reaction worked even better without NaOAc, producing (E)‐3 aa in 83 % yield as shown in Scheme 3. The exclusive stereoselectivity for the E isomer in this allene arylation indicates coordination of the olefin group during the reaction.
Scheme 2.
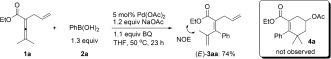
Pd‐catalyzed oxidative phenylation of allene 1 a. BQ=1,4‐benzoquinone.
Scheme 3.
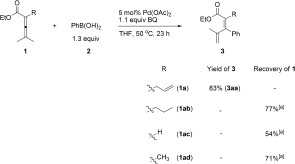
Investigation of different substituents on allenes for the Pd‐catalyzed oxidative allene‐arylation. [a] Yield determined by 1H NMR analysis using anisole as the internal standard.
We next investigated how the olefin group in the substrate influenced the outcome of the reaction (Scheme 3). To demonstrate the necessity of the allyl group, we examined the reactivity of allenes with different substituents: 2,3‐dienoates with a propyl substituent (1 ab), hydrogen (1 ac), or methyl group (1 ad) all failed to undergo the arylating transformation, indicating that the olefin group of 1 a is an indispensable assisting/directing group7, 8 for the allene arylation. Coordination of the C=C bond during the reaction would account for the high stereoselectivity for the E isomer (Scheme 1 c).
With these inspiring results in hand, we set out to optimize the reaction conditions (for details, see the Supporting Information). Solvent screening showed that acetone was the best solvent for this transformation, improving the yield further to 91 % (yield determined by 1H NMR analysis using anisole as the internal standard). Other solvents such as 1,4‐dioxane, 1,2‐dichloroethane, and toluene also gave good yields. Catalyst screening showed that Pd(TFA)2 (TFA=trifluoroacetate) produced the corresponding triene in only 40 % yield, while [Pd(PPh3)2Cl2] and [Pd(CH3CN)2Cl2] failed to promote the transformation. We were pleased to obtain (E)‐3 aa in 90 % yield (87 % yield of isolated product) with a catalyst loading of 1 mol %. Both Pd(OAc)2 and BQ are required for the reaction to occur. Finally, 50 °C was found to be the best temperature for this reaction.
Under the optimal conditions, we next examined the scope of arylboronic acids in the reaction with 2,3‐dienoate 1 a. Arylboronic acids bearing electron‐donating substituents such as 3‐Me, 2‐MeO, 3‐MeO, and 4‐MeO all reacted well and produced the corresponding trienes in good yields (Table 1, entries 2 and 4–6), while the para‐tBu‐substituted arylboronic acid led to a notable decrease in yield probably due to steric effects (Table 1, entry 3). For a series of electron‐deficient arylboronic acids, LiOAc.2 H2O (50 mol %) was required as an additive to ensure an efficient transformation: halogenated arenes proved to be compatible with the reaction conditions (Table 1, entries 7 and 8). Other electron‐withdrawing substituents, such as 3‐NO2, 4‐NO2, 4‐formyl, and 4‐acetyl could be present in the aryl unit, leading to the corresponding trienes in good yields (Table 1, entries 9–12). Finally, it is worth noting that 2‐naphthylboronic acid also works well, affording 3 am in 71 % yield (Table 1, entry 13).
Table 1.
Scope of functionalized arylboronic acids.[a]

| Entry | Ar | t [h] | Yield of 3 [%][b] |
|---|---|---|---|
| 1 | Ph | 21 | 87 (3 aa) |
| 2 | 3‐Me‐C6H4 | 18 | 94 (3 ab) |
| 3 | 4‐tBu‐C6H4 | 16 | 64 (3 ac) |
| 4 | 2‐MeO‐C6H4 | 21 | 76 (3 ad) |
| 5 | 3‐MeO‐C6H4 | 20 | 77 (3 ae) |
| 6 | 4‐MeO‐C6H4 | 16 | 80 (3 af) |
| 7[c] | 3‐Br‐C6H4 | 14 | 82 (3 ag) |
| 8[c] | 4‐F‐C6H4 | 17 | 90 (3 ah) |
| 9[c,d] | 3‐O2N‐C6H4 | 21 | 91 (3 ai) |
| 10[c] | 4‐O2N‐C6H4 | 17 | 80 (3 aj) |
| 11[c] | 4‐formyl‐C6H4 | 15 | 70 (3 ak) |
| 12[c] | 4‐acetyl‐C6H4 | 16 | 85 (3 al) |
| 13[c] | 2‐naphthyl | 17 | 71 (3 am) |
[a] The reaction was conducted at 50 °C in acetone (1 mL) with 1 a (0.2 mmol), arylboronic acid 2 (1.3 equiv), and BQ (1.1 equiv) in the presence of Pd(OAc)2 (1 mol %). [b] Yield of isolated product after column chromatography. [c] LiOAc.2H2O (50 mol %) was added to the reaction mixture. [d] Product 3 ai was obtained in only 41 % yield in the absence of LiOAc.2 H2O.
We further investigated the oxidative allene arylation using different allenes (Scheme 4). The reaction of substrates with phenyl or two methyl substituents on the olefin moiety worked well, producing 3 b and 3 c in 78 and 75 % yield, respectively. Furthermore, cycloalkylidene allenes could also be employed, affording products 3 d, 3 e, and 3 e′ in excellent yields. To demonstrate the broad scope of allenes in our olefin‐directed arylation reaction, we chose more general allene‐containing structures: 3,4‐dienoate 1 g (R3=CH2CO2Et)9 also showed excellent reactivity. It is worth noting that the reaction of 3,4‐dienol 1 h (R3=CH2CH2OH), an allene containing a free OH group, produced triene 3 h instead of proceeding via oxypalladation as shown in Scheme 1a. The corresponding yield is lower probably due to the instability of the starting material and possible reaction with the OH group.10 Surprisingly, benzyl and tosyl groups could be introduced to improve the corresponding yield significantly as shown by the formation of 3 i and 3 j in 93 and 80 % yield, respectively. Finally, it is interesting to note that 54 % yield of 3 k could be still obtained using a trisubstituted allene (R3=H),11 and the stereochemistry was further confirmed by NOE measurements (for details, see the Supporting Information).
Scheme 4.
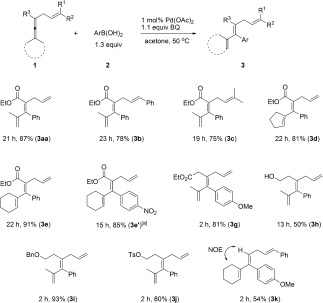
Scope of allenes for the olefin‐directed Pd‐catalyzed oxidative arylation. [a] LiOAc.2H2O (50 mol %) was added to the reaction.
A C=C bond in the chain has been demonstrated as an indispensable directing group for the allene arylation reaction developed, while 2,3‐dienoates lacking this double bond failed to undergo the arylation (see Scheme 3). We subsequently examined two other substrates (1 l and 1 m) with π‐bond‐containing groups. Treatment of benzyl‐substitued 2,3‐dienoate 1 l under the standard conditions of Table 1 failed to give the desired product, and 1 l was recovered in 88 % yield (Scheme 5). Interestingly, an alkynyl‐substituent was found to work as a directing group for the allene arylation. Thus, reaction of the alkynyl‐substituted 2,3‐dienoate 1 m afforded the corresponding dienyne product 3 m in 71 % yield.
Scheme 5.

Directing‐group investigation for the allene arylation.
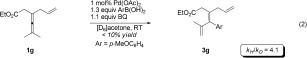
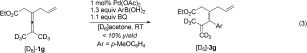
To gain a deeper insight into the reaction mechanism, the deuterium kinetic isotope effect (KIE) was determined from the reaction of a 1:1 mixture of 1 g and [D6]‐1 g at room temperature for 10 min [Eq. (1)]. The product ratio 3 g/[D5]‐3 g (ca. 31 % conv.) measured was 3.3:1, while the ratio of the recovered 1 g and [D6]‐1 g was 1:1.6. From these ratios the KIE was determined to be kH/kD=4.1. Furthermore, parallel kinetic experiments using 1 g and [D6]‐1 g provided an intermolecular KIE (kH/kD, from initial rate) value of 4.1 [Eqs. (2) and (3)].12 These results indicate that the allenylic C—H bond cleavage is the rate‐determining step in the olefin‐directed allene arylation reaction and that the cleavage of the C—H bond has to occur before any irreversible steps.13, (1), (2), (3)
 |
(1) |
 |
(2) |
 |
(3) |
Based on the observed kinetic isotope effects, the stereochemical outcome, and the experiments in Scheme 3, a possible mechanism for the reaction is proposed in Scheme 6. Simultaneous coordination of the allyl C=C bond and the allenic C=C bond of substrate 1 to the PdII center14 would generate chelate Int ‐3, followed by allene attack to afford vinylpalladium intermediate Int ‐4 involving allenylic C—H bond cleavage. Further, transmetalation of Int ‐4 with ArB(OH)2 would produce Int ‐5, which on subsequent reductive elimination would lead to 1,3,6‐triene 3 (path a). However, transmetalation of the PdII species with ArB(OH)2 via In t‐3′ could also occur before allene attack (path b).15 The pathway via Int ‐4 (path a) seems more likely than that via Int ‐3′ (path b) considering the fact that the PdII center is more electrophilic in Int ‐3 than in Int ‐3′. Furthermore, a pathway via Int ‐3′ would not give a large competitive isotope effect [Eqs. (2) and (3)] unless the transmetalation (Int ‐3→Int ‐3′) is reversible, which seems unlikely.16
Scheme 6.
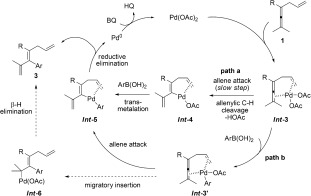
Proposed mechanism for the olefin‐directed Pd‐catalyzed oxidative arylation of allenes. HQ=hydroquinone.
In conclusion, we have developed an efficient olefin‐directed palladium‐catalyzed oxidative regio‐ and stereoselective17 arylation of allenes to afford 1,3,6‐trienes. The reaction showed a broad substrate scope for the arylboronic acids and allene substrates. The catalyst loading could be decreased to as low as 1 mol %, giving products in good to excellent yields. Mechanistic studies indicate that the allenylic C—H bond cleavage is the rate‐limiting step. The olefin unit was proven to be essential to realize the transformation, and this observation has important mechanistic implications for our previously developed oxidative carbocyclizations involving allenes.18 In these PdII‐catalyzed reactions, it has now been confirmed that the allene attack on PdII requires an additional coordination of an olefin or acetylene. Finally, because of the regio‐ and stereoselective formation of multisubstituted trienes, this method will be useful in synthetic and materials chemistry. Further studies on the scope, mechanism, and synthetic application of this reaction are currently under way in our laboratory.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
miscellaneous_information
Financial support from the European Research Council (ERC AdG 247014), The Swedish Research Council, The Berzelii Center EXSELENT, and the Knut and Alice Wallenberg Foundation is gratefully acknowledged. We thank Maoping Pu and Timofei Privalov for fruitful discussions.
References
- 1.
- 1a. Schuster H. F., Coppola G. M., Allenes in Organic Synthesis, Wiley, New York, 1984; [Google Scholar]
- 1b. Patai S., The Chemistry of Ketenes, Allenes, and Related Compounds, Part 1, Wiley, New York, 1980. [Google Scholar]
- 2.
- 2a. Ma S. in Handbook of Organopalladium Chemistry for Organic Synthesis (Eds.: E. Negishi, A. de Meijere), Wiley, New York, 2002, p. 1491; [Google Scholar]
- 2b. Ma S. in Topics in Organometallic Chemistry; (Ed.: J. Tsuji), Springer, Heidelberg, 2005, p. 183–210. [Google Scholar]
- 3.For reviews, see:
- 3a. Zimmer R., Dinesh C. U., Nandanan E., Khan F. A., Chem. Rev. 2000, 100, 3067; [DOI] [PubMed] [Google Scholar]
- 3b. Marshall J. A., Chem. Rev. 2000, 100, 3163; 11749316 [Google Scholar]
- 3c. Lu X., Zhang C., Xu Z., Acc. Chem. Res. 2001, 34, 535; [DOI] [PubMed] [Google Scholar]
- 3d. Bates R. W., Satcharoen V., Chem. Soc. Rev. 2002, 31, 12; [DOI] [PubMed] [Google Scholar]
- 3e. Ye J., Ma S., Org. Chem. Front. 2014, 1, 1210; [Google Scholar]
- 3f. Wei L.‐L., Xiong H., Hsung R. P., Acc. Chem. Res. 2003, 36, 773; [DOI] [PubMed] [Google Scholar]
- 3g. Rivera‐Fuentes P., Diederich F., Angew. Chem. Int. Ed. 2012, 51, 2818; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 2872; [Google Scholar]
- 3h. Ma S., Acc. Chem. Res. 2009, 42, 1679; [DOI] [PubMed] [Google Scholar]
- 3i. Alcaide B., Almendros P., Aragoncillo C., Chem. Soc. Rev. 2010, 39, 783; [DOI] [PubMed] [Google Scholar]
- 3j. Ma S., Aldrichimica Acta 2007, 40, 91; [Google Scholar]
- 3k. Jeganmohan M., Cheng C.‐H., Chem. Commun. 2008, 3101; [DOI] [PubMed] [Google Scholar]
- 3l. Hashmi A. S. K., Angew. Chem. Int. Ed. 2000, 39, 3590; [PubMed] [Google Scholar]; Angew. Chem. 2000, 112, 3737; [Google Scholar]
- 3m. Sydnes L. K., Chem. Rev. 2003, 103, 1133; [DOI] [PubMed] [Google Scholar]
- 3n. Hoffmann‐Röder A., Krause N., Angew. Chem. Int. Ed. 2002, 41, 2933; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2002, 114, 3057; [Google Scholar]
- 3o. Aubert C., Fensterbank L., Garcia P., Malacria M., Simonneau A., Chem. Rev. 2011, 111, 1954; [DOI] [PubMed] [Google Scholar]
- 3p. Krause N., Winter C., Chem. Rev. 2011, 111, 1994; [DOI] [PubMed] [Google Scholar]
- 3q. López F., Mascareñas J. L., Chem. Eur. J. 2011, 17, 418; [DOI] [PubMed] [Google Scholar]
- 3r. Yu S., Ma S., Angew. Chem. Int. Ed. 2012, 51, 3074; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 3128; [Google Scholar]
- 3s. Yu S., Ma S., Chem. Commun. 2011, 47, 5384. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a.For reviews, see: Ma S., Acc. Chem. Res. 2003, 36, 701; [DOI] [PubMed] [Google Scholar]; Ye J., Ma S., Acc. Chem. Res. 2014, 47, 989; [DOI] [PubMed] [Google Scholar]; Ma S., Chem. Rev. 2005, 105, 2829; [DOI] [PubMed] [Google Scholar]
- 4b. Piera J., Närhi K., Bäckvall J.‐E., Angew. Chem. Int. Ed. 2006, 45, 6914; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 7068; [Google Scholar]
- 4c. Piera J., Persson A., Caldentey X., Bäckvall J.‐E., J. Am. Chem. Soc. 2007, 129, 14120; [DOI] [PubMed] [Google Scholar]
- 4d. Persson A. K. Å., Bäckvall J.‐E., Angew. Chem. Int. Ed. 2010, 49, 4624; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 4728; [Google Scholar]
- 4e. Deng Y., Bartholomeyzik T., Bäckvall J.‐E., Angew. Chem. Int. Ed. 2013, 52, 6283; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 6403; [Google Scholar]
- 4f. Pardo‐Rodríguez V., Marco‐Martínez J., Buñuel E., Cárdenas D. J., Org. Lett. 2009, 11, 4548. [DOI] [PubMed] [Google Scholar]
- 5.For selected examples of platinum‐ or gold‐catalyzed cyclizations of allenes with a π‐bond‐containing group as a nucleophilic group, see:
- 5a. Liu Z., Wasmuth A. S., Nelson S. G., J. Am. Chem. Soc. 2006, 128, 10352; [DOI] [PubMed] [Google Scholar]
- 5b. Chen B., Fan W., Chai G., Ma S., Org. Lett. 2012, 14, 3616; [DOI] [PubMed] [Google Scholar]
- 5c. Kong W., Fu C., Ma S., Chem. Commun. 2009, 4572; [DOI] [PubMed] [Google Scholar]
- 5d. Qiu Y., Fu C., Zhang X., Ma S., Chem. Eur. J. 2014, 20, 10314; [DOI] [PubMed] [Google Scholar]
- 5e. Funami H., Kusama H., Iwasawa N., Angew. Chem. Int. Ed. 2007, 46, 909; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 927; [Google Scholar]
- 5f. Lee J. H., Toste F. D., Angew. Chem. Int. Ed. 2007, 46, 912; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 930. [Google Scholar]
- 6. Lang R. W., Hansen H.‐J., Org. Synth. 1984, 62, 202. [Google Scholar]
- 7.For a recent review of transition‐metal‐catalyzed π‐bond‐assisted C—H bond functionalization, see: Gandeepan P., Cheng C.‐H., Chem. Asian J. 2015, 10, 824. [DOI] [PubMed] [Google Scholar]
- 8.For selected examples of π‐bond‐directed C—H bond functionalization, see:
- 8a. Gandeepan P., Cheng C.‐H., J. Am. Chem. Soc. 2012, 134, 5738; [DOI] [PubMed] [Google Scholar]
- 8b. Zhao J., Asao N., Yamamoto Y., Jin T., J. Am. Chem. Soc. 2014, 136, 9540; [DOI] [PubMed] [Google Scholar]
- 8c. Kiyooka S.‐i., Takeshita Y., Tetrahedron Lett. 2005, 46, 4279; [Google Scholar]
- 8d. Tobisu M., Hyodo I., Onoe M., Chatani N., Chem. Commun. 2008, 6013; [DOI] [PubMed] [Google Scholar]
- 8e. Gandeepan P., Cheng C.‐H., Org. Lett. 2013, 15, 2084; [DOI] [PubMed] [Google Scholar]
- 8f. Chernyak N., Gevorgyan V., J. Am. Chem. Soc. 2008, 130, 5636; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8g. Chernyak N., Gevorgyan V., Adv. Synth. Catal. 2009, 351, 1101; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8h. Chernyak N., Gorelsky S. I., Gevorgyan V., Angew. Chem. Int. Ed. 2011, 50, 2342; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 2390; [Google Scholar]
- 8i. Minami Y., Shiraishi Y., Yamada K., Hiyama T., J. Am. Chem. Soc. 2012, 134, 6124; [DOI] [PubMed] [Google Scholar]
- 8j. Minami Y., Yamada K., Hiyama T., Angew. Chem. Int. Ed. 2013, 52, 10611; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 10805; [Google Scholar]
- 8k. Maekawa T., Segawa Y., Itami K., Chem. Sci. 2013, 4, 2369; [Google Scholar]
- 8l. Ferreira E. M., Stoltz B. M., J. Am. Chem. Soc. 2003, 125, 9578. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Henderson M. A., Heathcock C. H., J. Org. Chem. 1988, 53, 4736; [Google Scholar]
- 9b. Ma S., Yu F., Li Y., Gao W., Chem. Eur. J. 2007, 13, 247. [DOI] [PubMed] [Google Scholar]
- 10.3,4‐Dienol 1 h is unstable at room temperature and exposed to air. For details, see the Supporting Information.
- 11.For the CdI2‐mediated coupling reaction of trisubstituted allenes, see: Tang X., Zhu C., Cao T., Kuang J., Lin W., Ni S., Zhang J., Ma S., Nat. Commun. 2013, 4, 2450. [DOI] [PubMed] [Google Scholar]
- 12.For details, see the Supporting Information.
- 13.
- 13a. Simmons E. M., Hartwig J. F., Angew. Chem. Int. Ed. 2012, 51, 3066; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 3120; [Google Scholar]
- 13b. Chen C.‐S., Fujimoto Y., Girdaukas G., Sih C. J., J. Am. Chem. Soc. 1982, 104, 7294. [Google Scholar]
- 14. Hartley F. R., Chem. Rev. 1969, 69, 799. [Google Scholar]
- 15.
- 15a. Suzuki A., Angew. Chem. Int. Ed. 2011, 50, 6722; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 6854; [Google Scholar]
- 15b. Amatore C., Duc G. L., Jutand A., Chem. Eur. J. 2013, 19, 10082. [DOI] [PubMed] [Google Scholar]
- 16.With an irreversible arylpalladation (In t‐3′→Int‐6), a rate‐limiting β‐hydride elimination would not give an isotope effect in the competition experiment. For mechanistic details, see the Supporting Information.
- 17.For a review on control of regio‐ and stereoselectivity in allene chemistry, see: Ma S., Pure Appl. Chem. 2007, 79, 261. [Google Scholar]
- 18.
- 18a. Deng Y., Bartholomeyzik T., Persson A. K. Å., Sun J., Bäckvall J.‐E., Angew. Chem. Int. Ed. 2012, 51, 2703; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 2757; [Google Scholar]
- 18b. Deng Y., Bäckvall J.‐E., Angew. Chem. Int. Ed. 2013, 52, 3217; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 3299; [Google Scholar]
- 18c. Bartholomeyzik T., Mazuela J., Pendrill R., Deng Y., Bäckvall J.‐E., Angew. Chem. Int. Ed. 2014, 53, 8696; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 8840; [Google Scholar]
- 18d. Persson A. K. Å., Jiang T., Johnson M., Bäckvall J.‐E., Angew. Chem. Int. Ed. 2011, 50, 6155; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 6279; [Google Scholar]
- 18e. Jiang T., Persson A. K. Å., Bäckvall J.‐E., Org. Lett. 2011, 13, 5838; [DOI] [PubMed] [Google Scholar]
- 18f. Jiang T., Bartholomeyzik T., Mazuela J., Willersinn J., Bäckvall J.‐E., Angew. Chem. Int. Ed. 2015, 54, 6024; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 6122. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
miscellaneous_information