

Abstract
Hepatocellular carcinoma (HCC) is the most common type of primary liver cancer and the third leading cause of cancer‐related deaths worldwide. Limitations in HCC treatment result due to poor prognosis and resistance against traditional radiotherapy and chemotherapies. The multikinase inhibitor sorafenib is the only FDA approved drug available for advanced HCC patients, and development of second‐line treatment options for patients who cannot tolerate or develop resistance to sorafenib is an urgent medical need. In this study, we established sorafenib‐resistant cells from Huh7 and Mahlavu cell lines by long‐term sorafenib exposure. Sorafenib‐resistant HCC cells acquired spindle‐shape morphology, upregulated mesenchymal markers, and showed significant increase in both migration and invasion abilities compared to their parental counterparts. Moreover, after long‐term sorafenib treatment, HCC cells showed induction of hepatocyte growth factor (HGF) synthesis and secretion along with increased levels of c‐Met kinase and its active phosphorylated form, indicating autocrine activation of HGF/c‐Met signaling. Importantly, the combined treatment of the resistant cells with c‐Met kinase inhibitor SU11274 and HGF neutralizing antibody significantly reversed the increased invasion ability of the cells. The combined treatment also significantly augmented sorafenib‐induced apoptosis, suggesting restoration of sorafenib sensitivity. These results describe, for the first time, compensatory upregulation of HGF synthesis leading to autocrine activation of HGF/c‐Met signaling as a novel cellular strategy in the acquisition of sorafenib resistance. Therefore, we suggest that combinatorial therapeutic strategies with HGF and c‐Met inhibitors comprise promising candidates for overcoming sorafenib resistance.
Keywords: c‐Met, hepatocarcinogenesis, hepatocyte growth factor, liver cancer, sorafenib
Hepatocellular carcinoma is the most common type of primary liver cancer and the third leading cause of cancer‐related deaths worldwide.1, 2 Late diagnosis of HCC patients along with resistance of HCC tumor cells to most conventional anticancer agents present major obstacles for the clinical management of HCC.3, 4
Recent advances in molecular targeted therapies allowed the identification of sorafenib, a multi‐targeted tyrosine kinase inhibitor as the first systemic agent to show survival benefit in patients with advanced HCCs in a double‐blind, randomized, placebo‐controlled trial (SHARP trial).5, 6 Although sorafenib has since become the mainstay therapy recommended for advanced HCC patients, tumor response to sorafenib is poor, with the median overall survival remaining less than 1 year and the majority of patients eventually showing disease progression while on a therapeutic regimen.7, 8 Several studies now indicate that primary and/or acquired resistance in the tumor is a major challenge in sorafenib therapy.9, 10 Both tumor and stromal cells are believed to contribute in the resistance and several molecular pathways such as PI3K/Akt and STAT3 are identified as mediators of this process.11, 12, 13 Although the main molecular mechanisms underlying the acquired resistance to sorafenib are largely unknown, it is becoming clear that developing inhibitors that simultaneously inhibit the pathways involved in sorafenib resistance are needed to achieve broader and more potent antitumor efficacy. For this purpose, several clinical trials are ongoing to test different molecularly targeted agents in combination with sorafenib or as second‐line therapies after sorafenib failure.8, 14
Of particular interest are the inhibitors of c‐Met, a high‐affinity tyrosine kinase receptor for HGF.15, 16, 17 Although HGF/c‐Met signaling is not active in liver during physiologic conditions, many studies have reported that this signaling pathway is required for liver regeneration, hepatocyte survival, and tissue remodeling after acute injury.18, 19, 20 Following c‐Met phosphorylation and activation, multiple downstream effector proteins and cascades such as MAPK/ERK and PI3K/Akt pathways are activated and through these pathways c‐Met can trigger distinct cellular processes including cell proliferation, survival, invasion, epithelial remodeling, and angiogenesis.20, 21, 22
Importantly, the c‐Met pathway is one of the most frequently deregulated pathways in human cancer, and aberrant c‐Met signaling through autocrine, paracrine ligand production, genomic amplification, or mutational activation has been documented in most solid tumors including HCC.23, 24 Increased expression of c‐Met has been observed in more than 80% of HCC patients where it is correlated with poor prognosis and short survival.25, 26, 27 However, controversially, it has also been reported that deficiency of c‐Met increases chemically induced tumor initiation without effecting tumor promotion in liver.28 Importantly, increasing evidence implicates HGF/c‐Met signaling as a common mechanism of resistance to anti‐angiogenic therapies including currently used EGFR and vascular endothelial growth factor receptor inhibitors.24 Recent studies have also indicated a correlation between HGF‐induced EMT and drug resistance.29, 30, 31, 32, 33 However, the activation mechanisms of HGF/c‐Met signaling during long‐term sorafenib treatment have not been clarified in detail and the role of this signaling pathway in sorafenib resistance needs further investigation.
To study acquired resistance to sorafenib, we developed two sorafenib‐resistant HCC cell lines by long‐term exposure to this drug. In the established (Huh7‐soR and MV‐soR) cells, we evaluated the expression and activation status of the HGF/c‐Met pathway, and associated molecular and behavioral changes. Then, we aimed to reveal the mechanism of HGF/c‐Met activation in these cells and investigate whether the restoration of sorafenib sensitivity and the reversal of the invasive phenotype can be achieved by HGF and/or c‐Met inhibition. Our results indicated a functional role for the HGF/c‐Met axis in the acquired sorafenib resistance and showed for the first time the autocrine activation of HGF/c‐Met signaling in sorafenib‐resistant HCC cells.
Materials and Methods
Cell lines and culture conditions
Human HCC cell lines were cultured as previously described.34 To generate sorafenib‐resistant colonies, cells were treated with stepwise increasing concentrations of sorafenib up to their parental cell IC50 doses. Over a period of 3–5 months, sorafenib resistance colonies were generated from Huh7 and Mahlavu parental cells (Huh7‐soR and MV‐soR, respectively).
Reagents
Sorafenib was prepared as a 15 mM stock solution in DMSO (Sigma‐Aldrich, St. Louis, MO, USA). c‐Met inhibitor SU‐11274 (448101) was purchased from Calbiochem (San Diego, CA, USA) and used for selective c‐Met inhibition. Hepatocyte growth factor was purchased from R&D Systems (Minneapolis, MN, USA). Anti‐HGF antibody (ab10678) was purchased from Abcam (Cambridge, MA, USA) and used for neutralization of secreted HGF. Mouse IgG1, kappa monoclonal (MOPC‐21) (ab18443) was used as isotype control.
Cell viability assay
Briefly, parental and soR cells were grown in 96‐well plates (5 × 103 per well) with indicated concentrations of sorafenib and after 72 h cell viability was assessed using MTT reagent (Sigma‐Aldrich) according to the manufacturer's instructions.
Western blot analysis
The cells were lysed using RIPA lysis buffer containing 1 mM Na3VO2, 1 mM NaF, and 1% protease inhibitor cocktail (Roche Diagnostics, Indianapolis, IN, USA), and the lysates were subjected to Western blot analysis as described previously.34 Primary antibodies are described in Doc. S1.
Immunofluorescence studies
For staining of the actin filaments, a 5 mg/mL stock solution of tetramethylrhodamine–phalloidin (Sigma P1951) was prepared in methanol and used according to the manufacturer's instructions.
Immunofluorescence staining for phospho‐Met, c‐Met, and HGF were carried out as described previously.35 Staining was carried out using primary antibodies against phospho‐Met (Y‐1234/1235) (cs‐3129), c‐Met (sc‐161), and HGF (sc‐1387) prepared in blocking solution. Cells were visualized using an Olympus BX50 fluorescence microscope (Olympus, Tokyo, Japan).
Migration and invasion assay
In vitro motility and invasion assays were carried out as described previously.34 Briefly, cells were cultured in DMEM with 5% FBS and treated with either 1.0 μM SU11274, anti‐HGF antibody (1 μg/mL), or both. Cells treated with 0.1% DMSO and mouse IgG were used as controls. The number of migrated and invaded cells was counted in five areas under a bright‐field inverted microscope. Fold inductions were calculated using average numbers of migrated and invaded cells from at least three replicates.
Analysis of gene expression
Total RNA was isolated using the RNeasy mini kit (Qiagen, Valencia, CA, USA) and RNA concentration was detected using NanoDrop (Thermo Fisher Scientific, MA, USA). One microgram of RNA was then converted to cDNA using a RevertAid First Strand cDNA Synthesis kit (Thermo Fisher Scientific, Waltham, MA, USA) with random primers. For real‐time quantitative RT‐PCR, expression levels were determined in triplicate on a Light Cycler instrument (Roche 480), using the SYBR Green PCR Master Mix (Applied Biosystems, Thermo Fisher Scientific, MA, USA). Relative gene expression was normalized to GAPDH and calculated by using the 2−ΔΔCt method. Primer pairs used are given in Doc. S1.
Quantitative PCR for analysis of HGF copy number
Quantitative PCR was done on genomic DNA purified from parental and soR cell lines using the GeneJET Genomic DNA Purification Kit (Thermo Fisher Scientific). Reactions were done in quadruplicate with 20 ng genomic DNA. Data were normalized to RNase P which encodes the RNA moiety for the RNase P enzyme and calculated by using the 2−ΔΔCt method. Primer pairs used are given in Doc. S1.
Enzyme‐linked immunosorbent assay
Hepatocyte growth factor concentration in the supernatants of parental and soR cells was detected by an HGF Human ELISA Kit (KAC2211; Invitrogen, Carlsbad, CA, USA) following the manufacturer's instructions. Briefly, parental and soR cells were seeded into six‐well plates in 0.1% BSA. Following 48 h of cultivation, cultured media were collected and ELISA was carried out.
Apoptosis assay
Cells were grown in DMEM with 10% FBS containing 3 μM sorafenib and treated with either 1 microMolar, anti‐human HGF antibody, or both. After 48 h, cells were collected, resuspended in annexin V binding buffer, and stained using an annexin V–FITC/propidium iodide staining kit (BD Biosciences, San Jose, CA, USA) according to the manufacturer's instructions. Cells were then immediately analyzed using a FACSCalibur flow cytometer (BD Biosciences).
Statistical analysis
Statistical analysis was carried out using GraphPad Prism (GraphPad Software, Inc, California, USA). Statistical methods included anova and Student's t‐test. Differences between groups were considered significant at *P < 0.05, **P < 0.001, and ***P < 0.0001.
Results
Hepatocellular carcinoma cell lines became resistant to long‐term sorafenib treatment and showed upregulation of EMT markers
In our previous studies, we characterized HCC cell lines into two groups as “well‐differentiated” and “poorly differentiated” according to their differentiation status.36, 37 Poorly differentiated HCC cell lines show a mesenchymal phenotype and increased invasion ability and overexpress c‐Met receptor. Well‐differentiated cell lines, which have limited motility and invasion ability, show an epithelial phenotype and lack c‐Met expression.36, 37 For this study, we chose one HCC cell line from each group: (i) the Mahlavu cell line, which shows mesenchymal features and augmented motility and invasion and expresses c‐Met receptor; and (ii) the Huh7 cell line, which shows epithelial features and lacks invasive ability and c‐Met receptor expression. For both cell lines, sorafenib resistance was obtained by exposing cell lines to increasing concentrations of sorafenib over each cell passage. Over 3–5 months, Huh7 and MV cell lines became sorafenib‐resistant (Huh7‐soR and MV‐soR). The viability curves obtained by MTT assay indicated that established cell lines showed significantly decreased sorafenib sensitivity compared to parental cells (Fig. 1a). A visible effect of long‐term sorafenib exposure was the morphological changes that occurred during the development of sorafenib resistance. Under light microscopy, the resistant cells showed a notable spindle‐shaped morphology (Fig. 1b) and, by F‐actin staining, we demonstrated that the altered morphology was accompanied by the elongation of actin stress fibers in resistant cells (Fig. 1c). Several studies reported EMT to be a critical step in tumor invasion and drug resistance.9, 38 Analysis of epithelial and mesenchymal gene expressions in our cell lines indicated that, when compared to the epithelial‐like parental Huh7 cell line, Huh7‐soR cells lost E‐cadherin and CK19 expressions and significantly upregulated the expressions of several mesenchymal markers such as vimentin (>15‐fold), snail (>3.5‐fold), slug (>80‐fold), and zeb2 (>365‐fold) (Fig. 1d), suggesting the acquisition of a mesenchymal phenotype. The MV parental cell line is classified among HCC cell lines with a mesenchymal phenotype and endogenously expresses high basal levels of several mesenchymal markers.31 MV‐soR cells also showed further upregulation of snail (>2.5‐fold) and slug (>4.5‐fold) and suppression of E‐cadherin expression (Fig. 1d).
Figure 1.
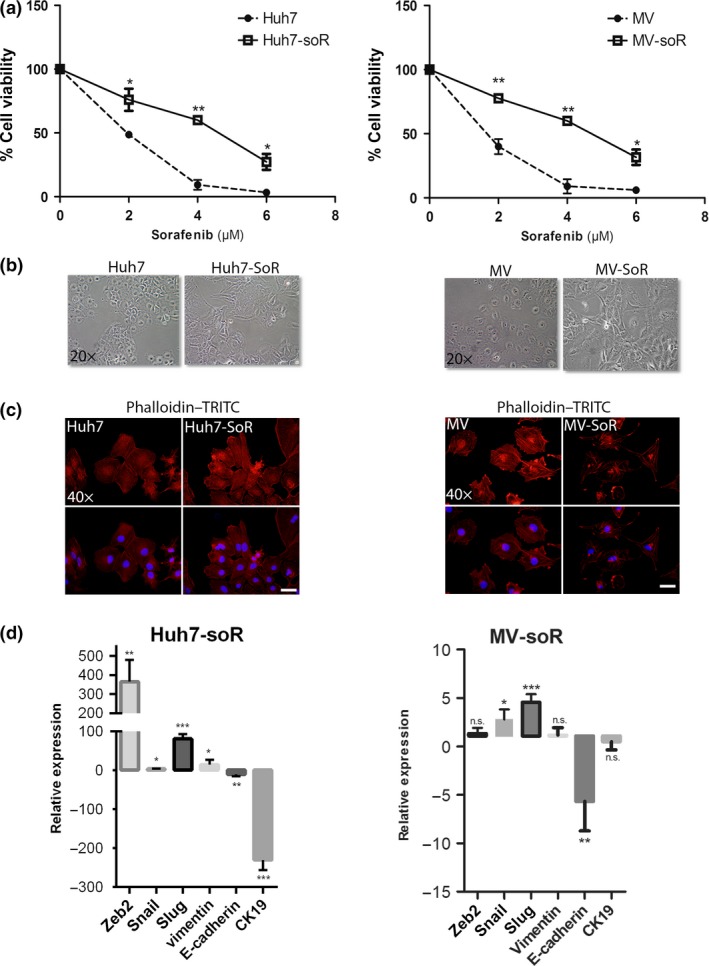
Long‐term sorafenib treatment induces sorafenib‐resistant hepatocellular carcinoma cells with altered morphology and gene expression. (a) Parental (Huh7 and Mahlavu [MV]) and sorafenib‐resistant (Huh7‐soR and MV‐soR) cells were treated with indicated doses of sorafenib. After 48 h, MTT assay was carried out to determine viability curves. (b) Altered spindle‐shaped morphology of resistant cells was visualized under a light microscope. Representative images of parental and soR cells were taken at ×20 magnification. (c) Parental and soR cells were stained with tetramethylrhodamine (TRITC)‐labeled phalloidin (red) to show F‐Actin filament organization and counterstained with DAPI (blue) to show the nuclei. Representative images of cells were captured using a fluorescence microscope (magnification, ×40). (d) Epithelial and mesenchymal marker gene expressions were analyzed by real‐time PCR. Huh7‐soR cells showed downregulation of epithelial markers E‐cadherin and CK‐19, whereas mesenchymal markers vimentin, snail, slug and zeb2 were induced. MV‐soR cells also showed further upregulation of snail and slug expression and downregulation of E‐cadherin (n = 3). *P < 0.05; **P < 0.001. Error bars indicate SD. NS, not significant.
Hepatocyte growth factor synthesis and secretion is upregulated in resistant cells
In several studies in HCC, it had been reported that HGF could induce morphological changes, upregulate EMT markers, and stimulate cell invasion and migration of HCC cells through activation of c‐Met signaling. To assess the role of HGF/c‐Met signaling in the acquisition of sorafenib resistance, we first analyzed HGF synthesis and secretion in the resistant cells. By RT‐PCR, we showed that HGF transcription was induced in Huh7‐soR cells, even though HGF transcription was not at detectable levels in Huh7 parental cells. Similarly, compared to MV parental cells, HGF transcription was upregulated in MV‐soR cells (Fig. 2a). DNA copy number analysis by quantitative PCR indicated that induction of HGF transcription happens without a change in HGF copy number between parental and resistant cells (Fig. S1). We also carried out immunofluorescence staining and showed that the Huh7‐soR and MV‐soR cells expressed higher levels of HGF protein (Fig. 2b). Moreover, when we used ELISA to measure HGF in the culture media, we showed that sorafenib‐resistant cells had increased HGF secretion (Fig. 2c). In order to assess if exogenous introduction of HGF could lead to development of sorafenib resistance, we treated Huh7 and MV cells with 20 ng/mL HGF 1 day before sorafenib treatment. After 48 h, the cell viability was assessed using MTT assay. Our results showed that 20 ng/mL HGF significantly attenuated sorafenib‐induced cell death, indicating that HGF treatment can confer sorafenib resistance in HCC cells (Fig. 2d).
Figure 2.
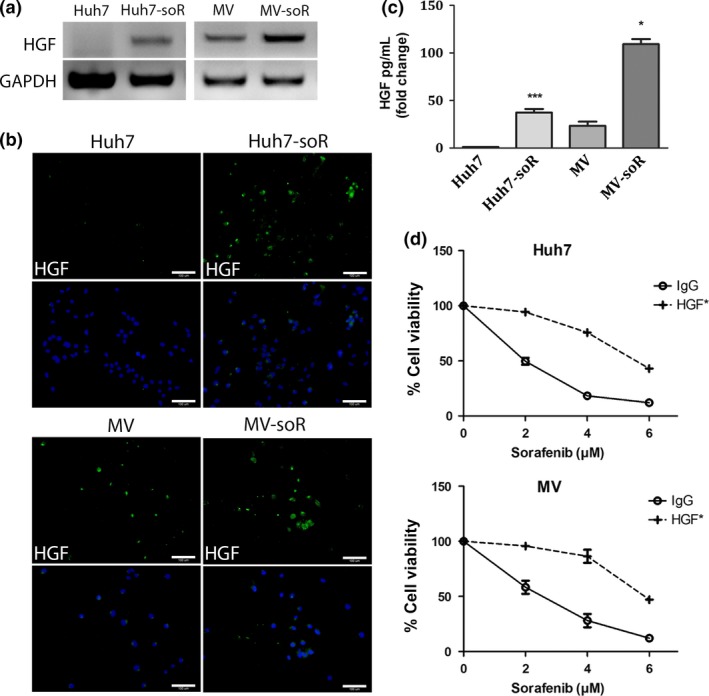
Hepatocyte growth factor (HGF) synthesis and secretion are increased in sorafenib‐resistant (soR) cells. (a) RNA from parental and sorafenib‐resistant Huh7 and Mahlavu (MV) cells were extracted and RT‐PCR was carried out with HGF‐specific primers. (b) Immunofluorescence microscopy was used to detect HGF expression using HGF‐specific antibody (green). DAPI (blue) was used to stain the cell nuclei. Representative images of parental and soR cells were captured under a fluorescence microscopy (magnification, ×20). (c) HGF concentration in the supernatants of parental and soR cells was detected using an HGF Human ELISA Kit following the manufacturer's instructions. Parental and soR cells were seeded into six‐well plates in 0.1% BSA. Following a 48‐h cultivation, cultured media were collected and ELISA was carried out. (d) Huh7 and MV parental cells were treated with 20 ng/mL HGF for 24 h then were treated with indicated doses of sorafenib. After 48 h, MTT assay was carried out to determine viability curves (n = 3). *P < 0.05, ***P < 0.0001. Error bars indicate SD.
c‐Met is activated in sorafenib‐resistant cell lines
We then analyzed whether increased secretion of HGF by resistant cells induces expression and activation of its receptor tyrosine kinase c‐MET. In the parental Huh7 cell line, basal c‐Met expression is at very low levels; whereas in the parental MV cell line, c‐Met is expressed but not constitutively activated.37 Western blot analysis showed that c‐Met levels are increased in Huh7‐soR and MV‐soR cells. Moreover, sorafenib‐resistant cells displayed a dramatic elevation in c‐Met phosphorylation within the activation loop compared with parental cells, suggesting the activation of c‐Met signaling in these cells (Fig. 3a,b). Similarly, immunofluorescence staining using specific antibodies revealed a higher presence of c‐Met‐ and p‐Met‐expressing cells in the resistant cell lines (Fig. 3c,d).
Figure 3.
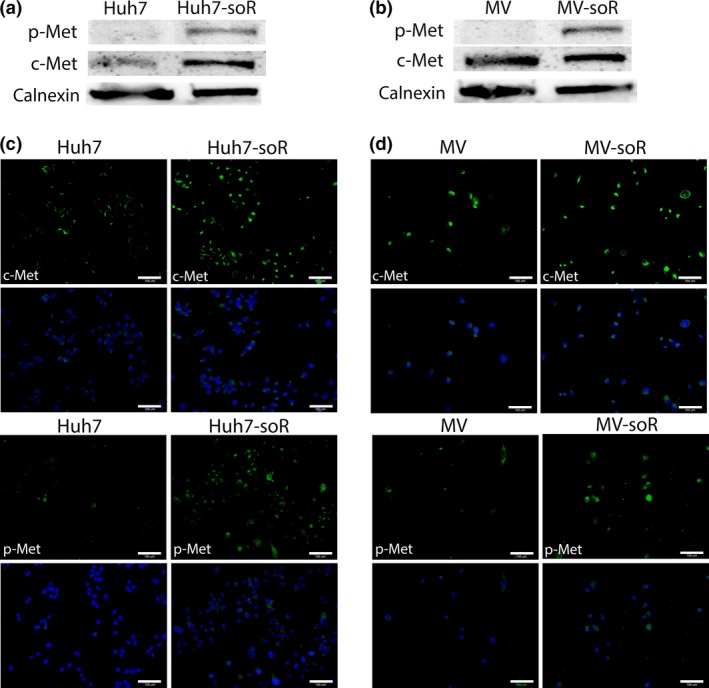
c‐Met is activated in sorafenib‐resistant (soR) cells. Huh7‐soR and Mahlavu (MV)‐soR cells showed activation of c‐Met signaling after long‐term sorafenib treatment. (a, b) Western blot analysis showed an increase in basal levels of c‐Met protein expression and induction of p‐Met (Y1234/1235) in soR cells. Calnexin was used as loading control. (c, d) Huh7‐soR and MV‐soR cells were stained with antibodies specific to c‐Met (green) (upper panels) and phospho‐Met (Y‐1234/1235) (green) (lower panels) to detect Met‐expressing and Met‐active cells. DAPI (blue) was used to stain nuclei. Scale bar = 100 μm.
Activation of c‐Met signaling is reversed by c‐Met inhibitor, SU11274
As expected, parallel to c‐Met activation we detected upregulated phosphorylation of Akt and of ERK1/2 in soR‐HCC cells and increased phosphorylation of β‐catenin at Ser675, which leads to its accumulation in the nucleus and increases its transcriptional activity. To assess the effects of c‐Met inhibition on c‐Met and its downstream signaling pathways, we treated soR‐HCC cells with 1 μM SU11274, a selective small molecule c‐Met inhibitor, for 48 h. Treatment with SU11274 abolished c‐Met phosphorylation in Huh7‐soR and MV‐soR cells, and also was able to effectively abrogate the phosphorylation of the downstream effectors, including Akt and ERK (Fig. 4). Importantly, activation of Akt was previously shown to mediate acquired sorafenib resistance and, in several studies, PI3K/Akt inhibitors were shown to partly restore sorafenib sensitivity.11, 39, 40, 41 Here we show that upstream inhibition of c‐Met can also block activation of Akt in addition to other downstream effectors and thus can serve to target parallel pathways important in the development of resistance.
Figure 4.
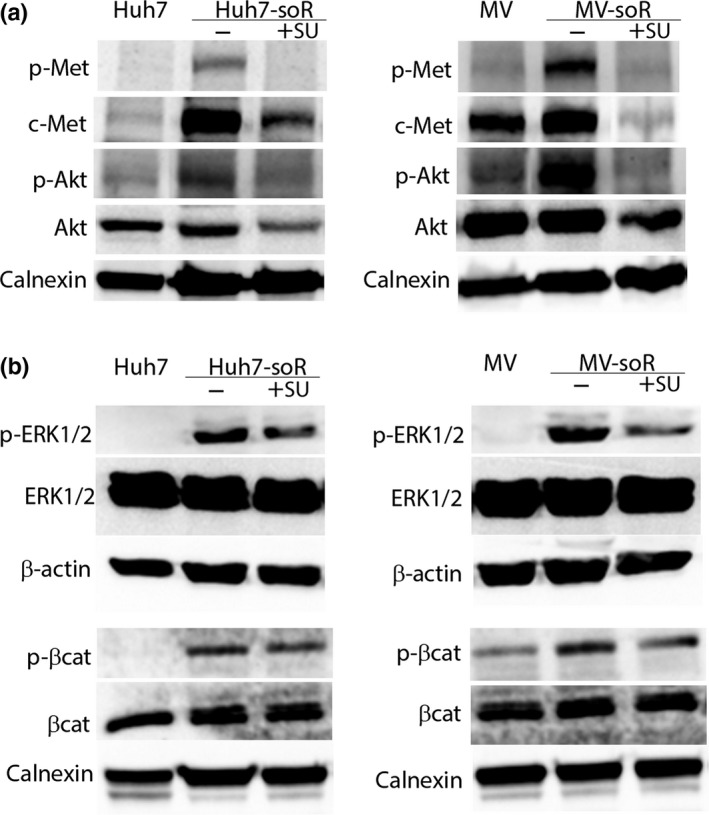
Activation of c‐Met and its downstream pathways in sorafenib‐resistant (soR) cells can be reversed by c‐Met inhibitor, SU11274. Huh7‐soR and Mahlavu (MV)‐soR cells were treated with either 0.1% DMSO or 1.0 μM SU11274 (+SU) for 48 h. To analyze protein expression, whole cell lysates were prepared and immunoblotted for c‐Met and Akt (a) and ERK1/2 and β‐catenin (b), and their phosphorylated forms. Calnexin and β‐actin were used as loading controls.
c‐Met inhibition reversed the increased migration and invasion capability of sorafenib‐resistant cells
As one of the major concerns with anti‐angiogenic inhibitors including sorafenib is the observed increase in aggressive behavior of the tumor cells after therapy, we evaluated the migration and invasion abilities of Huh7‐soR and MV‐soR cells. Huh7‐soR cells showed a 9.6‐fold increase in migration capacity and a 19.8‐fold increase in invasion capacity compared to Huh7 parental cells (Fig. 5a). Similarly, MV‐soR cells showed 8.6‐fold increases in both migration and invasion capacities (Fig. 5b). We then analyzed whether c‐Met inhibition by SU11274 can reverse the augmented invasive behavior of resistant cells. In our studies, c‐Met inhibition resulted in a 27% reduction in migration capacity and a 33.3% reduction in invasion capacity of Huh7‐soR cells. Notably, c‐Met inhibition had a greater effect on MV‐soR cells and caused a 55.8% reduction in migration and a 70.9% reduction in invasion capacity of these cells. In our previous studies, we reported that Egr1 mediates HGF‐induced cell invasion through regulation of MMPs.42 To understand the molecular mechanisms that lead to increased invasive behavior in sorafenib‐resistant HCC cells, we analyzed the expression of Egr1 and MMP9 by Western blotting and showed that resistant cells have increased levels of Egr1 protein and show induction of MMP9 expression. Moreover, treatment with SU11274 reduced levels of Egr1 and its target MMP9 in resistant cells, suggesting that the activation of Egr1 is mediated by HGF/c‐Met (Fig. 5c). Although more studies are required to investigate the mechanisms of invasive behavior, these results suggest, for the first time, that increased invasive behavior of sorafenib‐resistant cells is regulated in part by HGF/c‐Met‐mediated activation of Egr1 and subsequent induction of MMP9 expression.
Figure 5.
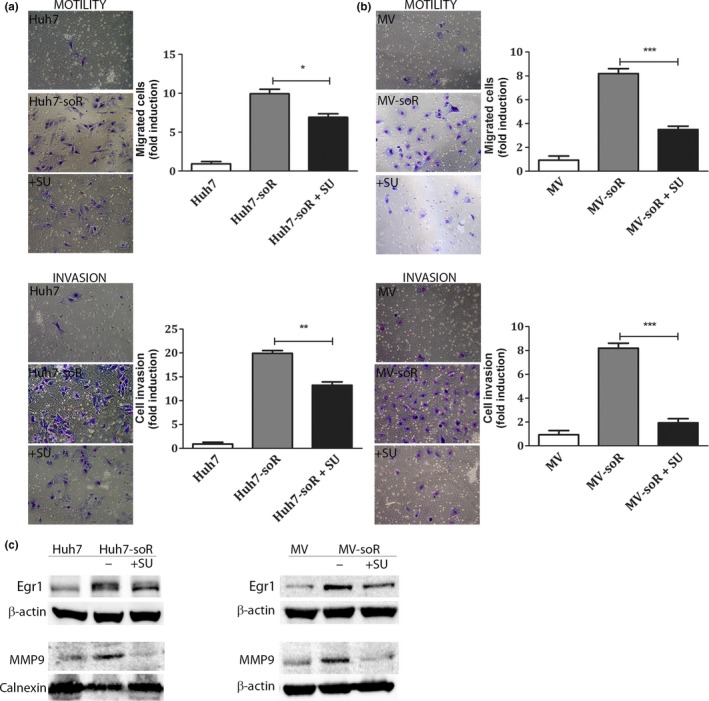
Sorafenib‐resistant (soR) hepatocellular carcinoma cells show greater migration and invasion ability that can be attenuated by the c‐Met inhibitor, SU11274 (SU). Cells were plated in top chambers for migration and invasion assays after treatment with 1.0 μM SU11274; 0.1% DMSO was used to treat control cells. Representative images of migrated and invaded Huh7 and Huh7‐soR (a) and Mahlavu (MV) and MV‐soR (b) cells were captured under a light microscope (magnification, ×20). The average number of stained cells was calculated and fold differences are presented as column graphs on the right. (c) Early growth response factor 1 (Egr1) and MMP9 expressions were analyzed by Western blotting in resistant cells before and after treatment with c‐Met inhibitor (n = 3). *P < 0.05, **P < 0.001, ***P < 0.0001. Error bars indicate SD.
Cotreatment with HGF neutralizing antibody augments the inhibitory effect of SU1124 on cell invasion and induces cell death
The synthesis of HGF by resistant cells suggested the autocrine activation of c‐Met signaling, therefore we tested the effects of HGF‐neutralizing antibody alone or in combination with SU11274 to inhibit c‐Met signaling in resistant cells. Compared to DMSO control, Huh7‐soR cells treated with 1 μg/mL anti‐HGF antibody alone showed a 25% reduction in invasion capacity, whereas Huh7‐soR cells treated with 1 μM SU11274 alone showed a 32% reduction (Fig. 6a, left graph). However, the combined treatment of anti‐HGF antibody and SU11274 achieved the greatest inhibition with a 48% reduction in invasion capacity and appeared to be significantly better than either treatment alone (Fig. 6a, left graph). Similar results were obtained with MV‐soR cells in which single treatments with anti‐HGF antibody and SU11274 caused 30% and 35% reductions in invasion capacity, respectively. However, the condition where both treatments were combined resulted in a significantly greater reduction (67%) when compared to single treatments (Fig. 6a, right graph). Similarly, our data on HGF‐treated parental cells showed that, although treatment with c‐Met inhibitor and HGF neutralizing antibody could decrease the resistance induced by exogenous HGF, only combined treatment with c‐Met inhibitor and HGF neutralizing antibody exerted a significant effect (Fig. S2).
Figure 6.
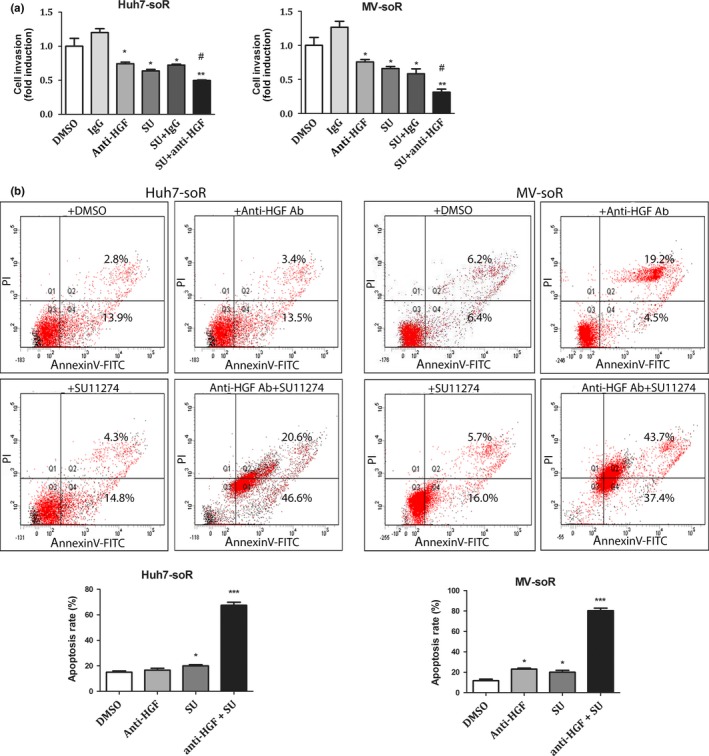
Treatment of sorafenib‐resistant (soR) cells with SU11274 (SU) and hepatocyte growth factor (HGF) neutralizing antibody enhances the inhibitory effect of SU11274 on cell invasion and augments sorafenib‐induced apoptosis. (a) Invasion assay was carried out using Transwell chambers. Huh7‐soR and Mahlavu (MV)‐soR cells were treated with 1.0 μM SU11274 and/or 1.0 μg/mL anti‐HGF antibody for 48 h. Mouse IgG1 and 0.1% DMSO were used to treat control cells. In each condition, invaded cells were visualized under a light microscope, and data are represented as column graphs. #Combination treatment was significantly better than single treatments alone (P < 0.05). (b) Apoptosis was detected using annexin V–FITC/propidium iodide staining. Huh7‐soR and MV‐soR cells were treated with 1.0 μM SU11274 and/or 1.0 μg/mL anti‐HGF antibody for 48 h, then stained with annexin V–FITC and propidium iodide. The rate of apoptosis was determined using a flow cytometer and data are presented as column graphs (n = 3). *P < 0.05, **P < 0.001, ***P < 0.0001. Error bars indicate SD.
As c‐Met signaling is known to promote cell survival, we further analyzed the effect of anti‐HGF and SU1127 treatments alone or in combination on the apoptosis of resistant cells in the presence of sorafenib using annexin V/propidium iodide staining. Compared to DMSO control, anti‐HGF antibody treatment alone did not enhance the apoptosis of Huh7‐soR cells, whereas SU11274 treatment alone significantly increased the apoptosis rate of these cells. Strikingly, the apoptotic rate of Huh7‐soR cells after combined treatment with anti‐HGF antibody and SU11274 was significantly greater and reached to 67.2% (Fig. 6b). The additive effect of anti‐HGF antibody and SU11274 on apoptosis was similar in MV‐soR cells, in which the combination of these agents achieved an 81.1% apoptosis rate. Although single treatments were also able to induce apoptosis in MV‐soR cells, the apoptosis rates achieved were much lower than the combined treatment. Specifically, anti‐HGF antibody was able to induce a 23.7% apoptosis rate while the apoptosis rate induced by SU11274 was 21.7% (Fig. 6b). Taken together, these findings indicate that combined treatment with anti‐HGF antibody and SU11274 has a significant advantage over single agent therapy at these specific concentrations.
Discussion
Target‐based therapies constitute a new promising avenue for the treatment of several malignancies including HCC. However, sorafenib therapy in HCC offers limited survival benefits and is hampered by the occurrence of drug resistance and tumor relapse.
In the present study, we showed that in two different HCC cell lines long‐term sorafenib treatment leads to the establishment of cells with reduced sorafenib sensitivity, increased mesenchymal features, and enhanced tumorigenic behavior. In the established sorafenib‐resistant cells, we detected an increase in c‐Met levels and phosphorylation along with an induction of HGF synthesis and secretion, suggesting autocrine activation of the HGF/c‐Met signaling pathway.
Importantly, an increasing number of studies are suggesting HGF/c‐Met signaling as a general mechanism to anti‐angiogenic therapies. The activation of Met‐dependent signaling pathways as a consequence of c‐Met amplification or upregulation of HGF expression was reported in emergence of resistance to several targeted therapies including EGFR inhibitors,43, 44, 45, 46 anti‐human epidermal growth factor receptor 2 therapies,47, 48 vascular endothelial growth factor receptor inhibitors,49, 50 and BRAF inhibitors.51, 52 However, the role of the HGF/c‐Met pathway in sorafenib resistance is still not clarified. In their study, Xiang et al.53 reported that patient specimens with sorafenib‐resistant HCC revealed higher p‐Met‐positive staining compared to sorafenib‐sensitive HCC specimens, suggesting the activation of c‐Met in these tumors. However, these tumors were not analyzed for c‐Met activation status before sorafenib therapy and thus the question still remains unanswered whether c‐Met active tumors are intrinsically resistant to sorafenib therapy or long‐term treatment of sorafenib causes resistance through c‐Met activation. Also, Jiang et al.54 recently reported that sorafenib and DE605, a novel c‐Met inhibitor, synergistically suppress HCC providing further data to support combinational therapies; however, this study did not investigate the development of acquired resistance following long‐term treatment and thus does not provide insight into the mechanisms of sorafenib resistance.
Importantly, the relationship between HGF expression and sorafenib resistance has not been evaluated in previous studies. However, it has been shown that in various cancers, enhanced autocrine or paracrine c‐Met signaling due to increased expression of HGF in tumor and/or stromal cells creates a compensatory effect leading to incomplete inhibition of this pathway and/or emergence of c‐Met‐targeted drug resistance.55, 56, 57 Our data, for the first time, show compensatory upregulation of HGF expression following long‐term sorafenib treatment without a change in DNA copy number and suggest the importance of evaluation and targeting of HGF expression in optimizing c‐Met‐targeted strategies with or after sorafenib therapy. We also showed the HGF/c‐Met‐mediated induction of Egr1 and MMP9 expressions in resistant cells, suggesting a new mechanism for this pathway in regulating invasion ability. Most importantly, we show that neutralization of HGF in combination with c‐Met kinase inhibition results in induction of apoptosis at significantly higher rates compared to c‐Met kinase inhibition alone, suggesting that neutralization of HGF in combination with c‐Met kinase inhibition could serve as a more powerful approach to increase sorafenib sensitivity.
Although there have been no studies evaluating pharmacological HGF inhibition in combination with sorafenib therapy and the combined use of strategies for the inhibition of HGF/c‐Met signaling needs to be further evaluated for efficacy and safety, this might become essential for tumors with c‐Met activation. Importantly, in addition to advanced HCC, sorafenib has been approved for treatment of two other carcinomas, advanced renal cell carcinoma and progressive differentiated thyroid carcinoma. When we analyzed the related cancer datasets, we found that c‐Met expression is significantly higher in these two carcinomas compared to their normal tissues (Fig. S3). Thus, the autocrine activation of HGF/c‐Met signaling could serve as a shared mechanism in the development of sorafenib resistance among various c‐Met expressing cancers; however, further studies are required to investigate HGF/c‐Met signaling as a general mechanism in sorafenib resistance.
Consequently, designing multitargeted therapies is required to increase patient response in sorafenib therapy and the identification of the emergence of alternative signaling cascades as potential targets for blockade is essential in planning of such therapies. Our results support, for the first time, a substantial role for HGF overexpression and secretion in the development of sorafenib resistance in HCC cells by autocrine c‐Met activation and provide further justification for testing combination therapies with HGF and c‐Met inhibitors in clinical trials.
Disclosure Statement
The authors have no conflict of interest.
Abbreviations
- Akt
protein kinase B
- EGFR
epidermal growth factor receptor
- Egr1
early growth response factor 1
- EMT
epithelial–mesenchymal transition
- HCC
hepatocellular carcinoma
- HGF
hepatocyte growth factor
- PI3K
phosphatidylinositol 3‐kinase
- STAT3
signal transducer and activator of transcription 3
Supporting information
Fig. S1. Hepatocyte growth factor copy number analysis by quantitative PCR.
Fig. S2. Combined treatment of Huh7 and Mahlavu (MV) cells with anti‐hepatocyte growth factor (HGF) and SU11274 could reverse sorafenib resistance induced by exogenous HGF.
Fig. S3. c‐Met expression is increased in three types of cancer for which sorafenib therapy was approved by the FDA.
Doc. S1. Antibodies and primers used in this study.
Acknowledgment
This project was supported by the Turkish Scientific and Technical Research Council (TUBITAK grant no. 110 S 349).
Cancer Sci 107 (2016) 407–416
Funding Information Turkish Scientific and Technical Research Council.
References
- 1. Forner A, Llovet JM, Bruix J. Hepatocellular carcinoma. Lancet 2012; 379: 1245–55. [DOI] [PubMed] [Google Scholar]
- 2. Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin 2011; 61: 69–90. [DOI] [PubMed] [Google Scholar]
- 3. Villanueva A, Hernandez‐Gea V, Llovet JM. Medical therapies for hepatocellular carcinoma: a critical view of the evidence. Nat Rev Gastroenterol Hepatol 2013; 10: 34–42. [DOI] [PubMed] [Google Scholar]
- 4. Thomas MB. Systemic therapy for hepatocellular carcinoma. Cancer J 2008; 14: 123–7. [DOI] [PubMed] [Google Scholar]
- 5. Llovet JM, Ricci S, Mazzaferro V et al Sorafenib in advanced hepatocellular carcinoma. N Engl J Med 2008; 359: 378–90. [DOI] [PubMed] [Google Scholar]
- 6. Mendez‐Sanchez N, Vasquez‐Fernandez F, Zamora‐Valdes D, Uribe M. Sorafenib, a systemic therapy for hepatocellular carcinoma. Ann Hepatol 2008; 7: 46–51. [PubMed] [Google Scholar]
- 7. Gauthier A, Ho M. Role of sorafenib in the treatment of advanced hepatocellular carcinoma: an update. Hepatol Res 2013; 43: 147–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Villanueva A, Llovet JM. Second‐line therapies in hepatocellular carcinoma: emergence of resistance to sorafenib. Clin Cancer Res 2012; 18: 1824–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhai B, Sun XY. Mechanisms of resistance to sorafenib and the corresponding strategies in hepatocellular carcinoma. World J Hepatol 2013; 5: 345–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sun H, Zhu MS, Wu WR, Shi XD, Xu LB. Role of anti‐angiogenesis therapy in the management of hepatocellular carcinoma: the jury is still out. World J Hepatol 2014; 6: 830–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chen KF, Chen HL, Tai WT et al Activation of phosphatidylinositol 3‐kinase/Akt signaling pathway mediates acquired resistance to sorafenib in hepatocellular carcinoma cells. J Pharmacol Exp Ther 2011; 337: 155–61. [DOI] [PubMed] [Google Scholar]
- 12. Tai WT, Cheng AL, Shiau CW et al Dovitinib induces apoptosis and overcomes sorafenib resistance in hepatocellular carcinoma through SHP‐1‐mediated inhibition of STAT3. Mol Cancer Ther 2012; 11: 452–63. [DOI] [PubMed] [Google Scholar]
- 13. Su JC, Tseng PH, Wu SH et al SC‐2001 overcomes STAT3‐mediated sorafenib resistance through RFX‐1/SHP‐1 activation in hepatocellular carcinoma. Neoplasia 2014; 16: 595–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. He AR, Goldenberg AS. Treating hepatocellular carcinoma progression following first‐line sorafenib: therapeutic options and clinical observations. Therap Adv Gastroenterol 2013; 6: 447–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bottaro DP, Rubin JS, Faletto DL et al Identification of the hepatocyte growth factor receptor as the c‐met proto‐oncogene product. Science 1991; 251: 802–4. [DOI] [PubMed] [Google Scholar]
- 16. Cecchi F, Rabe DC, Bottaro DP. Targeting the HGF/Met signalling pathway in cancer. Eur J Cancer 2010; 46: 1260–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Naldini L, Weidner KM, Vigna E et al Scatter factor and hepatocyte growth factor are indistinguishable ligands for the MET receptor. EMBO J 1991; 10: 2867–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Huh CG, Factor VM, Sanchez A, Uchida K, Conner EA, Thorgeirsson SS. Hepatocyte growth factor/c‐met signaling pathway is required for efficient liver regeneration and repair. Proc Natl Acad Sci 2004; 101: 4477–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Borowiak M, Garratt AN, Wustefeld T, Strehle M, Trautwein C, Birchmeier C. Met provides essential signals for liver regeneration. Proc Natl Acad Sci 2004; 101: 10608–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Trusolino L, Bertotti A, Comoglio PM. MET signalling: principles and functions in development, organ regeneration and cancer. Nat Rev Mol Cell Biol 2010; 11: 834–48. [DOI] [PubMed] [Google Scholar]
- 21. Birchmeier C, Birchmeier W, Gherardi E, Vande Woude GF. Met, metastasis, motility and more. Nat Rev Mol Cell Biol 2003; 4: 915–25. [DOI] [PubMed] [Google Scholar]
- 22. Organ SL, Tsao MS. An overview of the c‐MET signaling pathway. Ther Adv Med Oncol 2011; 3: S7–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Christensen JG, Burrows J, Salgia R. c‐Met as a target for human cancer and characterization of inhibitors for therapeutic intervention. Cancer Lett 2005; 225: 1–26. [DOI] [PubMed] [Google Scholar]
- 24. Maroun CR, Rowlands T. The Met receptor tyrosine kinase: a key player in oncogenesis and drug resistance. Pharmacol Ther 2014; 142: 316–38. [DOI] [PubMed] [Google Scholar]
- 25. Noguchi O, Enomoto N, Ikeda T, Kobayashi F, Marumo F, Sato C. Gene expressions of c‐met and hepatocyte growth factor in chronic liver disease and hepatocellular carcinoma. J Hepatol 1996; 24: 286–92. [DOI] [PubMed] [Google Scholar]
- 26. Kondo S, Ojima H, Tsuda H et al Clinical impact of c‐Met expression and its gene amplification in hepatocellular carcinoma. Int J Clin Oncol 2013; 18: 207–13. [DOI] [PubMed] [Google Scholar]
- 27. Kaposi‐Novak P, Lee JS, Gomez‐Quiroz L, Coulouarn C, Factor VM, Thorgeirsson SS. Met‐regulated expression signature defines a subset of human hepatocellular carcinomas with poor prognosis and aggressive phenotype. J Clin Invest 2006; 116: 1582–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Marx‐Stoelting P, Borowiak M, Knorpp T, Birchmeier C, Buchmann A, Schwarz M. Hepatocarcinogenesis in mice with a conditional knockout of the hepatocyte growth factor receptor c‐Met. Int J Cancer 2009; 124: 1767–72. [DOI] [PubMed] [Google Scholar]
- 29. Chow AK, Ng L, Lam CS et al The enhanced metastatic potential of hepatocellular carcinoma (HCC) cells with sorafenib resistance. PLoS ONE 2013; 8: e78675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chen W, Wu J, Shi H et al Hepatic stellate cell coculture enables sorafenib resistance in Huh7 cells through HGF/c‐Met/Akt and Jak2/Stat3 pathways. Biomed Res Int 2014; 2014: 764981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Canadas I, Rojo F, Taus A et al Targeting epithelial‐to‐mesenchymal transition with Met inhibitors reverts chemoresistance in small cell lung cancer. Clin Cancer Res 2014; 20: 938–50. [DOI] [PubMed] [Google Scholar]
- 32. Liska D, Chen CT, Bachleitner‐Hofmann T, Christensen JG, Weiser MR. HGF rescues colorectal cancer cells from EGFR inhibition via MET activation. Clin Cancer Res 2011; 17: 472–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Turke AB, Zejnullahu K, Wu YL et al Preexistence and clonal selection of MET amplification in EGFR mutant NSCLC. Cancer Cell 2010; 17: 77–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cokakli M, Erdal E, Nart D et al Differential expression of Caveolin‐1 in hepatocellular carcinoma: correlation with differentiation state, motility and invasion. BMC Cancer 2009; 9: 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Korhan P, Erdal E, Kandemis E et al Reciprocal activating crosstalk between c‐Met and caveolin 1 promotes invasive phenotype in hepatocellular carcinoma. PLoS ONE 2014; 9: e105278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yuzugullu H, Benhaj K, Ozturk N et al Canonical Wnt signaling is antagonized by noncanonical Wnt5a in hepatocellular carcinoma cells. Mol Cancer 2009; 8: 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bozkaya G, Korhan P, Cokakli M et al Cooperative interaction of MUC1 with the HGF/c‐Met pathway during hepatocarcinogenesis. Mol Cancer 2012; 11: 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Singh A, Settleman J. EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene 2010; 29: 4741–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhai B, Hu F, Jiang X et al Inhibition of Akt reverses the acquired resistance to sorafenib by switching protective autophagy to autophagic cell death in hepatocellular carcinoma. Mol Cancer Ther 2014; 13: 1589–98. [DOI] [PubMed] [Google Scholar]
- 40. Gedaly R, Galuppo R, Musgrave Y et al PKI‐587 and sorafenib alone and in combination on inhibition of liver cancer stem cell proliferation. J Surg Res 2013; 185: 225–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. van Malenstein H, Dekervel J, Verslype C et al Long‐term exposure to sorafenib of liver cancer cells induces resistance with epithelial‐to‐mesenchymal transition, increased invasion and risk of rebound growth. Cancer Lett 2013; 329: 74–83. [DOI] [PubMed] [Google Scholar]
- 42. Ozen E, Gozukizil A, Erdal E, Uren A, Bottaro DP, Atabey N. Heparin inhibits Hepatocyte Growth Factor induced motility and invasion of hepatocellular carcinoma cells through early growth response protein 1. PLoS ONE 2012; 7: e42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bean J, Brennan C, Shih JY et al MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc Natl Acad Sci 2007; 104: 20932–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Engelman JA, Zejnullahu K, Mitsudomi T et al MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007; 316: 1039–43. [DOI] [PubMed] [Google Scholar]
- 45. Yano S, Wang W, Li Q et al Hepatocyte growth factor induces gefitinib resistance of lung adenocarcinoma with epidermal growth factor receptor‐activating mutations. Cancer Res 2008; 68: 9479–87. [DOI] [PubMed] [Google Scholar]
- 46. Yano S, Yamada T, Takeuchi S et al Hepatocyte growth factor expression in EGFR mutant lung cancer with intrinsic and acquired resistance to tyrosine kinase inhibitors in a Japanese cohort. J Thorac Oncol 2011; 6: 2011–7. [DOI] [PubMed] [Google Scholar]
- 47. Shattuck DL, Miller JK, Carraway KL 3rd, Sweeney C. Met receptor contributes to trastuzumab resistance of Her2‐overexpressing breast cancer cells. Cancer Res 2008; 68: 1471–7. [DOI] [PubMed] [Google Scholar]
- 48. Minuti G, Cappuzzo F, Duchnowska R et al Increased MET and HGF gene copy numbers are associated with trastuzumab failure in HER2‐positive metastatic breast cancer. Br J Cancer 2012; 107: 793–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ebos JM, Lee CR, Cruz‐Munoz W, Bjarnason GA, Christensen JG, Kerbel RS. Accelerated metastasis after short‐term treatment with a potent inhibitor of tumor angiogenesis. Cancer Cell 2009; 15: 232–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Paez‐Ribes M, Allen E, Hudock J et al Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell 2009; 15: 220–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Straussman R, Morikawa T, Shee K et al Tumour micro‐environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature 2012; 487: 500–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wilson TR, Fridlyand J, Yan Y et al Widespread potential for growth‐factor‐driven resistance to anticancer kinase inhibitors. Nature 2012; 487: 505–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Xiang Q, Chen W, Ren M et al Cabozantinib suppresses tumor growth and metastasis in hepatocellular carcinoma by a dual blockade of VEGFR2 and MET. Clin Cancer Res 2014; 20: 2959–70. [DOI] [PubMed] [Google Scholar]
- 54. Jiang X, Feng K, Zhang Y et al Sorafenib and DE605, a novel c‐Met inhibitor, synergistically suppress hepatocellular carcinoma. Oncotarget 2015; 6: 12340–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kentsis A, Reed C, Rice KL et al Autocrine activation of the MET receptor tyrosine kinase in acute myeloid leukemia. Nat Med 2012; 18: 1118–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Xie Q, Bradley R, Kang L et al Hepatocyte growth factor (HGF) autocrine activation predicts sensitivity to MET inhibition in glioblastoma. Proc Natl Acad Sci 2012; 109: 570–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Pennacchietti S, Cazzanti M, Bertotti A et al Microenvironment‐derived HGF overcomes genetically determined sensitivity to anti‐MET drugs. Cancer Res 2014; 74: 6598–609. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Hepatocyte growth factor copy number analysis by quantitative PCR.
Fig. S2. Combined treatment of Huh7 and Mahlavu (MV) cells with anti‐hepatocyte growth factor (HGF) and SU11274 could reverse sorafenib resistance induced by exogenous HGF.
Fig. S3. c‐Met expression is increased in three types of cancer for which sorafenib therapy was approved by the FDA.
Doc. S1. Antibodies and primers used in this study.