

Abstract
The epidermal growth factor receptor (EGFR) tyrosine kinase signaling pathways regulate cellular activities. The EGFR tyrosine kinase inhibitors (EGFR‐TKIs) repress the EGFR pathway constitutively activated by somatic EGFR gene mutations and have drastically improved the prognosis of non‐small‐cell lung cancer (NSCLC) patients. However, some problems, including resistance, remain to be solved. Recently, combination therapy with EGFR‐TKIs and cytotoxic agents has been shown to improve the prognosis of NSCLC patients. To enhance the anticancer effects of EGFR‐TKIs, we examined the cross‐talk of the EGFR pathways with ataxia telangiectasia‐mutated (ATM) signaling pathways. ATM is a key protein kinase in the DNA damage response and is known to phosphorylate Akt, an EGFR downstream factor. We found that the combination of an ATM inhibitor, KU55933, and an EGFR‐TKI, gefitinib, resulted in synergistic cell growth inhibition and induction of apoptosis in NSCLC cell lines carrying the sensitive EGFR mutation. We also found that KU55933 enhanced the gefitinib‐dependent repression of the phosphorylation of EGFR and/or its downstream factors. ATM inhibition may facilitate the gefitinib‐dependent repression of the phosphorylation of EGFR and/or its downstream factors, to exert anticancer effects against NSCLC cells with the sensitive EGFR mutation.
Keywords: ATM inhibitor, combination therapy, EGFR mutation, EGFR‐TKI, non‐small‐cell lung cancer
Non‐small‐cell lung cancer is a common cause of cancer‐related deaths worldwide.1 Platinum‐based combination chemotherapy has been applied to patients with advanced stage, metastatic, or recurrent NSCLC, but the effectiveness of this treatment is limited.2 Over the past decade, the development of EGFR‐TKIs, gefitinib and erlotinib, has drastically improved the prognosis of NSCLC patients.3, 4, 5
Epidermal growth factor receptor is an Erb family tyrosine kinase that regulates signaling pathways controlling cellular activities.6 It has been reported that 10% of NSCLC patients carry mutations in the EGFR gene.6 The deletion of exon 19 and the L858R point mutation in exon 21 of EGFR have been found in the histologically normal respiratory epithelia around the lung cancer cells.7 Moreover, the expression of these EGFR gene mutants in mouse type II pneumocytes leads to lung adenocarcinoma.8, 9 Therefore, EGFR mutations are considered to play important roles in the development of lung cancer. These mutations cause EGF‐independent EGFR phosphorylation.10 The EGFR‐TKIs compete with ATP at a critical ATP‐binding site of EGFR, and thus inhibit the kinase activity for its phosphorylation.11 As the EGFR mutations increase the affinity of the receptor to EGFR‐TKIs, NSCLC cells carrying these mutations are highly sensitive to EGFR‐TKIs.12 Therefore, the deletion of exon 19 and the L858R point mutation in exon 21 are referred to as sensitive mutations.13, 14
Despite impressive clinical responses to kinase‐targeted therapy, almost all patients acquire drug resistance to these agents after approximately 1 year.15 One of the most common resistance mechanisms to EGFR‐TKI in NSCLC patients is the T790M point mutation in EGFR exon 20, which decreases the affinity of EGFR to EGFR‐TKIs.16 Therefore, the EGFR T790M point mutation is referred to as a resistant mutation. Second‐generation EGFR‐TKIs, which bind irreversibly to the ATP binding sites of EGFR, were developed to overcome the drug resistance. However, they only showed a partial anticancer effect against the NSCLC cells with the resistant EGFR mutation, and caused more side‐effects than the traditional EGFR‐TKIs, gefitinib and erlotinib.17 Third‐generation EGFR‐TKIs, which target EGFR T790M point mutation, are under development.18 Another approach to overcome the drug resistance of NSCLC cells is the combination of several chemotherapeutic agents with EGFR‐TKIs. In recent clinical trials, favorable outcomes have been observed using combinations of anticancer drugs, such as platinum‐doublet or S‐1 with gefitinib.19, 20, 21, 22
The cross‐talk between signaling pathways reportedly plays a role in the coordination of the cellular responses to various external and internal stresses.23 Ataxia telangiectasia‐mutated, is a key protein kinase involved in the DNA damage response to deleterious DSBs.24 In response to DNA damage or replication stress, ATM kinase is rapidly activated to phosphorylate downstream proteins involved in cell cycle control, DNA repair, and apoptosis, including histone H2AX, Chk2, BRCA1, and p53.25 Therefore, ATM inhibitors could enhance the anticancer effects of radiation or anticancer drugs that induce DNA damage. ATM also reportedly enhances Akt phosphorylation resulting from insulin treatment and IR.26 Akt is a downstream kinase in the IGFR and EGFR pathways. Inhibition of the ATM activity represses Akt activation, leading to reduced cell growth and induction of apoptosis in cancer cells with Akt overphosphorylated by insulin growth factor.25 However, it remains unknown whether ATM is involved in the regulation of the EGFR pathway in NSCLCs.
In this study, we showed that ATM inhibition, along with EGFR inhibition by gefitinib, synergistically represses the growth of NSCLC cells carrying the EGFR gene with the sensitive mutation, but not that of cells carrying the wild‐type allele. We also found that the ATM inhibitor enhanced the EGFR‐TKI‐dependent repression of the phosphorylation of EGFR and/or its downstream factors, in NSCLC cells with the EGFR mutation that confers sensitivity to EGFR‐TKIs. These findings suggest that ATM is involved in the regulation of the EGFR pathway in NSCLC cells that are sensitive to EGFR‐TKIs.
Materials and Methods
Detailed information on human NSCLC cell lines is shown in Table 1.27, 28, 29
Table 1.
Cell lines, epidermal growth factor receptor (EGFR) status, and sensitivity to gefitinib
| Cell line | Histology | EGFR status | Sensitivity for gefitinib |
|---|---|---|---|
| PC‐9 | Adenocarcinoma | Exon 19 deletion (E746–750) | High |
| HCC827 | Adenocarcinoma | Exon 19 deletion (E746–750) | Very high |
| Calu‐6 | Anaplastic | Wild | Low |
| VMRC‐LCD | Adenocarcinoma | Wild | Low |
| A549 | Adenocarcinoma | Wild | Low |
| H441 | Adenocarcinoma | Wild | Low |
| H596 | Adenosquamous carcinoma | Wild | Low |
| RERF‐LC‐OK | Adenocarcinoma | Wild | Low |
| RERF‐LC‐MS | Adenocarcinoma | Wild | Low |
Sensitivity for gefitinib was defined as: low, IC50 > 1 μM; high, IC50 < 100 nM; very high, IC50 < 10 nM.
All experimental procedures are provided in Data S1.
Results
Inhibition of ATM kinase activity enhances the EGFR TKI‐dependent repression of NSCLC cell growth
To explore the possibility that ATM is involved in the regulation of the EGFR pathway, we examined the enhancement of the antitumor effect of EGFR‐TKIs by the inhibition of ATM kinase activity. For this purpose, we first studied the growth inhibition of NSCLC cell lines. We used two lines with sensitive EGFR mutations, PC‐9 and HCC827, and two lines with wild‐type EGFR, Calu‐6 and VMRC‐LCD, and treated them with the EGFR‐TKI gefitinib or the ATM kinase inhibitor KU55933 (Table 1). The growth inhibition by the treatment with gefitinib, even at a low concentration, was confirmed for PC‐9 and HCC827 cells, but not for Calu‐6 and VMRC‐LCD cells, consistent with previous reports (Fig. 1a–d).30 We then examined the effect of KU55933 on the growth of these NSCLC cell lines (Table 1), and found that the growth inhibition rates were ~40% for PC‐9 and Calu‐6 cells, and only ~20% for HCC827 and VMRC‐LCD cells (Fig. S1a). These findings suggest that the ATM kinase plays a limited role in the regulation of NSCLC cell growth under normal conditions, regardless of the EGFR status.
Figure 1.
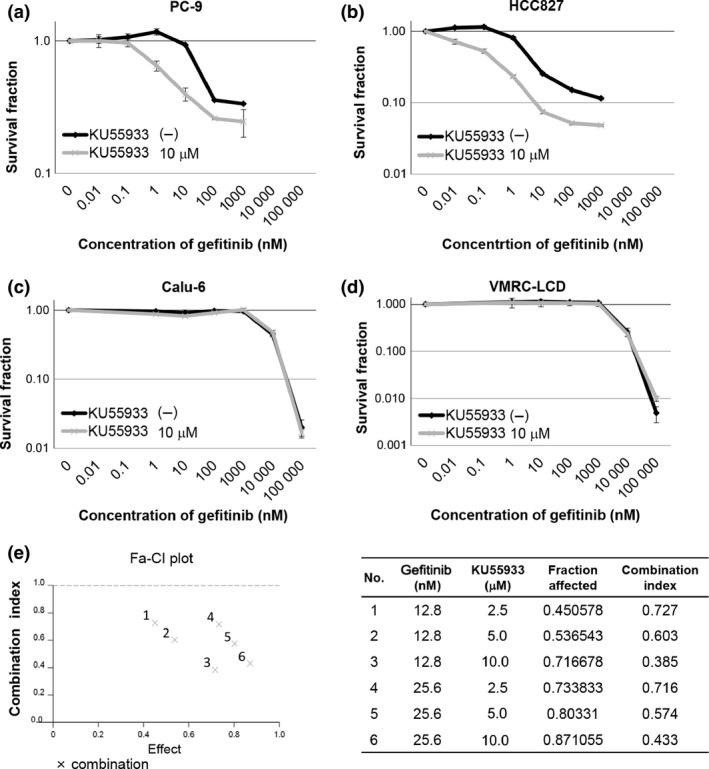
Combined effects of KU55933 and gefitinib on non‐small‐cell lung cancer cell growth. (a–d) Cell growth inhibition rates for four cell lines, PC‐9 (a), HCC827 (b), Calu‐6 (c), and VMRC‐LCD (d), treated with KU55933 and gefitinib or gefitinib alone at the indicated concentrations for 4 days. Each experiment was carried out in three wells and repeated three times. Bars indicate SD. (e) Combination indexes of PC‐9 cells treated with KU55933 and gefitinib at the concentrations shown in the table. The Combination index values are <1, indicating the synergistic inhibition of cell growth.
Next, we examined the effect of the ATM kinase inhibitor on the growth of NSCLC cells treated with gefitinib. The combination of gefitinib and KU55933 showed a significant synergistic effect on cell growth for PC‐9 and HCC827 cells carrying the sensitive EGFR mutation (Fig. 1a,b,e). In contrast, no synergistic effect was observed for Calu‐6 or VMRC‐LCD cells carrying wild‐type EGFR (Fig. 1c,d). The absence of synergistic growth inhibition of KU55933 with gefitinib in cells carrying wild‐type EGFR was confirmed in five additional NSCLC cell lines, using high concentrations of gefitinib (Table 1, Fig. S1b).27, 28, 29 These findings suggest that the ATM kinase activity is involved in the regulation of the gefitinib‐induced growth repression of NSCLC cells carrying the sensitive EGFR mutation.
Effects of ATM inhibition on gefitinib‐mediated repression of phosphorylation of EGFR and its downstream factors
Inhibition of EGFR phosphorylation by EGFR‐TKI leads to blockage of the EGFR downstream pathway and induces cell growth inhibition and apoptosis. Therefore, we considered that the enhancement of gefitinib‐induced cell growth inhibition by the ATM inhibitor could be due to the further repression of the phosphorylation of EGFR and/or its downstream molecules. To gain mechanistic insight into the enhancement of gefitinib‐induced cell growth inhibition by the ATM inhibitor, we examined the phosphorylation status of EGFR in PC‐9 cells. An immunoblotting analysis confirmed the repression of EGFR phosphorylation by gefitinib in PC‐9 cells (Fig. 2a,b). Importantly, KU55933 enhanced the gefitinib‐induced repression of EGFR phosphorylation observed in PC‐9 cells (Fig. 2a,b). Moreover, the combined treatment with KU55933 and gefitinib synergistically repressed the phosphorylation of Akt and ERK, downstream factors in the EGFR pathway, in PC‐9 cells (Fig. 2a). Enhancement of the gefitinib‐induced inhibition of Akt phosphorylation by KU55933 was also observed in HCC827 cells, which are sensitive to gefitinib (Fig. S2a). In contrast, the EGFR phosphorylation levels were significantly lower in Calu‐6 and VMRC‐LCD cells without EGF stimulation, as compared to those in PC‐9 and HCC827 cells (Figs 2c,S2b). Although the phosphorylation at Ser473 of Akt was repressed by KU55933 alone in Calu‐6 and VMRC‐LCD cells with wild‐type EGFR, the combined effect with gefitinib was not observed (Figs 2c,S2b). These findings suggest that the KU55933‐mediated enhancement of gefitinib‐induced cell growth inhibition of PC‐9 and HCC827 cells is due to the synergistic repression of the phosphorylation of EGFR with the sensitive mutation and/or its downstream factors. As the cancer cells with wild‐type EGFR uncontrollably proliferate through signaling mechanisms other than EGFR, the inhibition of Akt and ERK phosphorylation in cells with wild‐type EGFR by the ATM inhibitor might occur through a different mechanism from the repression of the EGFR pathway in cells with a mutated EGFR.
Figure 2.
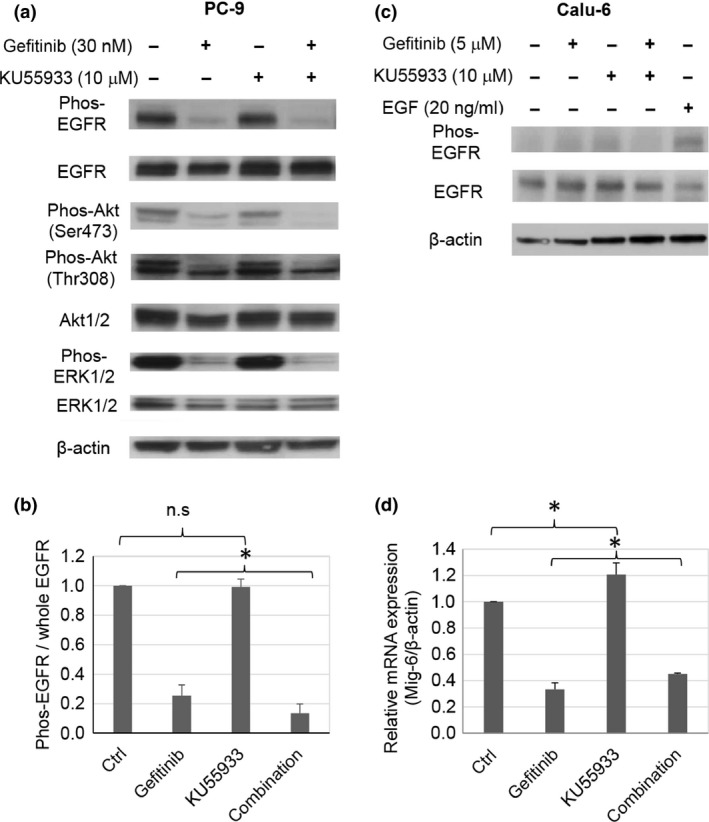
Phosphorylation of epidermal growth factor receptor (EGFR) and its downstream factors after KU55933 and/or gefitinib treatment. (a) Immunoblotting analysis of phosphorylation of EGFR and its downstream factors in PC‐9 cells. The proteins were extracted at 24 h after exposure to the indicated concentrations of KU55933 and/or gefitinib. (b) Quantitative analysis of the immunoblotting shown in (a). All results are the means of three independent experiments. Error bars indicate SD. (c) Immunoblotting analysis of phosphorylation of EGFR and its downstream factors in in Calu‐6 cells. (d) Expression of mig‐6 mRNA in PC‐9 cells treated with KU55933 at 10 μM and/or gefitinib at 30 nM for 24 h. β‐Actin mRNA was used as an internal control. Data are expressed as mean ± SD. *P < 0.05. n.s., not significant.
Next, to investigate the mechanism by which the ATM inhibitor enhances the gefitinib‐induced repression of the EGFR pathway, we examined the expression of Mig‐6, an EGFR negative regulator, after treating PC‐9 and Calu‐6 cells with gefitinib, KU55933, or both. Mig‐6 is reportedly induced by the RAS‐MEK‐MAPK/ERK pathway, and prevents EGFR activation through binding to the C‐lobe in the kinase domain of EGFR.31, 32 In the NSCLC cells with the sensitive EGFR mutation, the overexpression of Mig‐6 enhanced the inhibitory effect of gefitinib.33 In PC‐9 cells harboring the sensitive mutated EGFR, the expression of Mig‐6 was significantly induced by treatment with the ATM inhibitor in combination with gefitinib, as compared to that with gefitinib alone (Figs 2d,S2c). In contrast, in Calu‐6 cells, the expression of Mig‐6 was not affected by KU55933 (Fig. S2d). These results suggest that KU55933 could enhance the antitumor effect of gefitinib by upregulating Mig‐6 expression and the gefitinib‐induced repression of the EGFR pathway.
Gefitinib‐induced apoptosis of NSCLC cells is facilitated by ATM inhibition
Gefitinib reportedly induces apoptosis in cells carrying the sensitive mutation of the EGFR gene.30, 34 As KU55933 enhanced the repression of the phosphorylation of EGFR and its downstream factors in PC‐9 and HCC827 cells, we speculated that the gefitinib‐induced apoptosis of PC‐9 and HCC827 cells is also facilitated by the ATM inhibitor. To test this possibility, we examined the induction of apoptosis after gefitinib and/or KU55933 treatment of NSCLC cells. Flow cytometry analyses revealed that the gefitinib treatment increased the percentage of sub‐G1 fractions in PC‐9 and HCC827 cells carrying the sensitive EGFR mutation. The increase in the sub‐G1 fraction in these cells after gefitinib treatment was significantly enhanced by the addition of KU55993 (Fig. 3a). In contrast, the sub‐G1 fraction was not increased by the combination of gefitinib and KU55933 in Calu‐6 or VMRC‐LCD cells with the wild‐type EGFR gene. These findings suggest that inhibition of ATM facilitates the gefitinib‐induced apoptosis of NSCLC cells carrying the gefitinib‐sensitive EGFR mutation.
Figure 3.
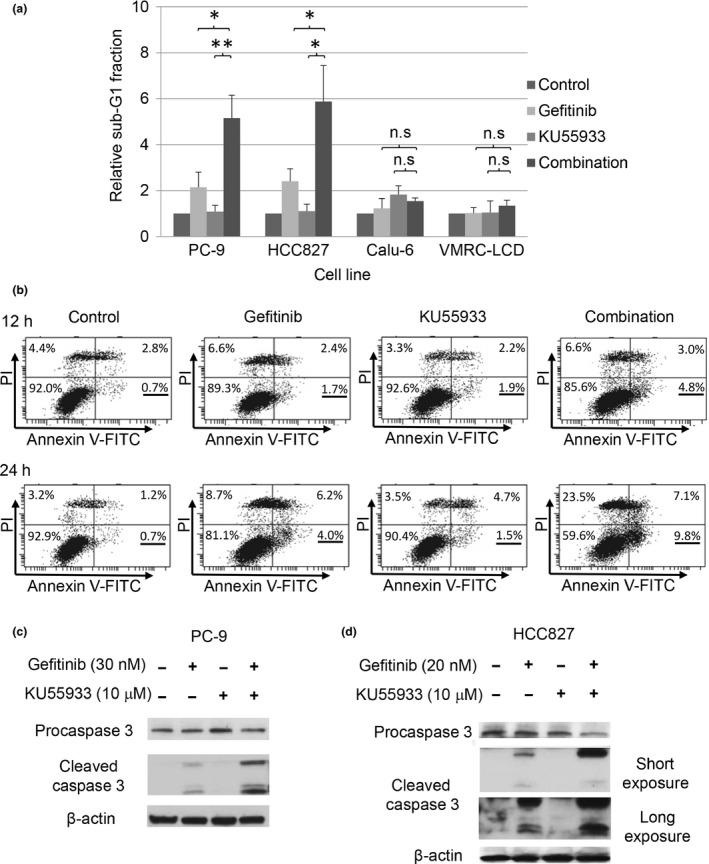
Apoptosis induced by the combination of KU55933 with gefitinib. (a) The sub‐G1 fraction was measured by flow cytometry in PC‐9, HCC827, Calu‐6, and VMRC‐LCD cells, after exposure to 10 μM KU55933 and/or gefitinib (30 nM in PC‐9 and HCC827 cells; 20 μM in Calu‐6 and VMRC‐LCD cells) for 24 h. Each experiment was repeated three times. Bars indicate SD. *P < 0.005; **P < 0.001. n.s., not significant. (b) Annexin V (+) PC‐9 cells were measured by flow cytometry after treatment with 10 μM KU55933 and/or 30 nM gefitinib. The numbers represent the percentage of cells in each region of the quadrant. Left bottom quadrant, viable cells; right bottom quadrant, early apoptotic cells; right top quadrant, late apoptotic cells. (c, d) Immunoblotting analysis of PC‐9 (c) and HCC827 (d) cells using anti‐caspase 3 antibodies. The extracts were prepared from PC‐9 (c) and HCC827 (d) cells after KU55933 and/or gefitinib treatment.
To confirm the enhancement of the gefitinib‐induced apoptosis by KU55993 in cells harboring the sensitive EGFR mutation, we used annexin V binding and caspase 3 assays. The annexin V assay revealed that the percentage of annexin V (+) / propidium iodide (−) early apoptotic cells was significantly higher in PC‐9 cells treated with the combination of KU55933 and gefitinib than in those treated only with gefitinib (Fig. 3b). Furthermore, the anti‐caspase 3 immunoblotting analysis showed that the amount of cleaved caspase 3 was significantly higher in both PC‐9 and HCC827 cells carrying the sensitive EGFR mutation, treated with the combination of KU55933 and gefitinib, compared to those treated with gefitinib only (Fig. 3c,d). Taken together, these findings strongly support the notion that the inhibition of ATM kinase activity and the treatment with gefitinib facilitate the induction of apoptosis in NSCLC cells carrying the sensitive EGFR mutation.
Gefitinib neither disturbs DNA repair nor induces DNA damage
ATM phosphorylates itself and DNA repair‐related factors, such as histone H2AX, in response to DSB induction.35 Therefore, the synergistic inhibitory effects of gefitinib and KU55933 on cell growth in PC‐9 and HCC827 cells might be due to the disturbance of the DNA damage response pathway involving ATM. To investigate this possibility, we examined the phosphorylation status of ATM in gefitinib‐treated cells after IR. Immunoblotting analyses of PC‐9 and HCC827 cells, using antiphosphorylated ATM antibodies, revealed that gefitinib treatment did not affect ATM phosphorylation after IR, suggesting that gefitinib does not affect ATM activation after DNA damage induction (Fig. 4a). We next examined whether gefitinib induces DNA damage by analyzing the focus formation of γH2AX, a marker of DSBs. Immunofluorescence staining of PC‐9 and HCC827 cells revealed that there were no significant differences in the percentages of γH2AX foci‐positive cells before and after treatment with gefitinib (Fig. 4b). Similar results were obtained using HCC827 cells (Fig. S3). Taken together, these findings suggest that gefitinib neither disturbs the DNA damage response nor induces DNA damage in PC‐9 and HCC827 cells.
Figure 4.
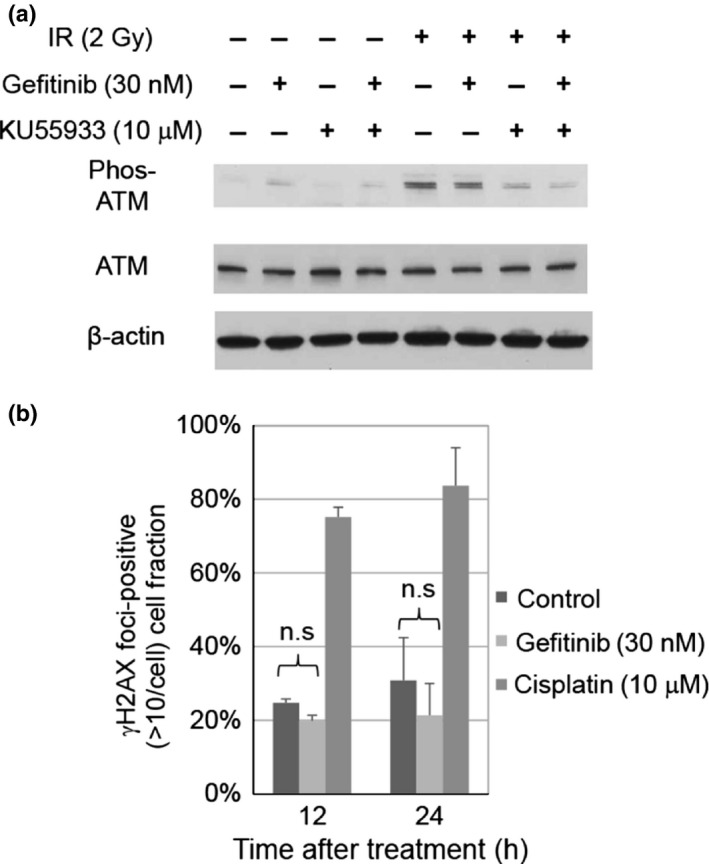
Combination effects of KU55933 with gefitinib on DNA damage response. (a) Effects of gefitinib or the combination treatment on ataxia telangiectasia‐mutated (ATM) phosphorylation in PC‐9 cells. Cells were X‐irradiated 24 h after exposure to KU55933 and/or gefitinib. Proteins were extracted 30 min after ionizing radiation (IR). (b) Kinetics of the phosphorylated H2AX (γH2AX) focus formation in PC‐9 cells after exposure to gefitinib or cisplatin. Results are the means of three independent experiments; error bars show SD. n.s., not significant.
Sensitive EGFR mutation is required for enhanced gefitinib‐induced anticancer effects by ATM inhibition
Enhancement of the gefitinib‐induced apoptosis and cell growth inhibition by KU55933 was observed only in PC‐9 and HCC827 cells carrying the sensitive mutation of the EGFR gene. NIH‐3T3 mouse fibroblast‐like cells exogenously expressing EGFR with the sensitive mutation showed higher levels of EGFR autophosphorylation and greater sensitivity to EGFR‐TKI compared to those expressing exogenous wild‐type EGFR.36 Therefore, we tested whether the enhancement of the antitumor effect of gefitinib by KU55933 is associated with the EGFR mutation. We constructed U2OS cells stably expressing FLAG‐tagged EGFRwt or EGFRmut (Fig. 5a). An immunoblotting analysis using antiphosphorylated EGFR antibodies revealed that the phosphorylation level was significantly higher in EGFRmut than EGFRwt (Fig. 5b). The repression of EGFR phosphorylation by gefitinib was confirmed in EGFRmut cells (Fig. 5b). We next examined the effect of KU55933 on gefitinib‐induced apoptosis in U2OS cells. A flow cytometry analysis revealed that the KU55933 treatment increased the sub‐G1 fraction of gefitinib‐treated EGFRmut cells (Fig. 5c). In contrast, no significant increase in the sub‐G1 fraction by KU55933 treatment was observed in gefitinib‐treated EGFRwt cells. These findings strongly support the notion that the sensitive mutation of the EGFR gene plays a role in the enhancement of gefitinib‐induced apoptosis, through the inhibition of ATM kinase.
Figure 5.
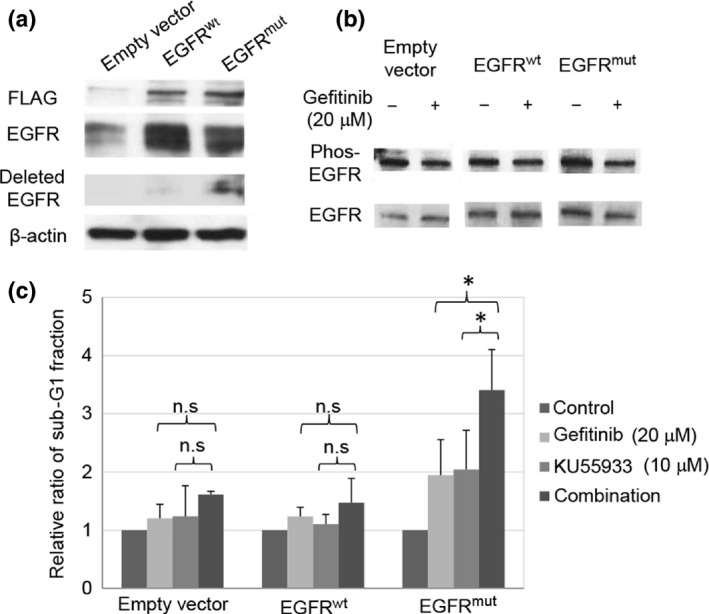
Sensitive EGFR mutation is required for the synergistic effect of an ataxia telangiectasia‐mutated (ATM) inhibitor and gefitinib. (a) Immunoblotting analysis of U2OS cells stably expressing FLAG‐tagged wild‐type (EGFR wt) and mutated EGFR (EGFR mut), using anti‐EGFR‐ and anti‐deleted EGFR‐ (sensitive EGFR mutant) specific antibodies. (b) Immunoblotting analysis of EGFR wt and EGFR mut with gefitinib, using antiphosphorylated EGFR antibodies. Phos, phosphorylation. (c) Flow cytometry measurements of the sub‐G1 fractions of U2OS cells stably expressing FLAG‐tagged EGFR wt or EGFR mut after KU55933 and/or gefitinib treatment at the indicated concentrations. Each experiment was repeated three times. Bars indicate SD. *P < 0.05. n.s., not significant.
Discussion
In this study, we have examined the enhancement of EGFR‐TKI sensitivity by combination therapy and shown the involvement of ATM kinase in the EGFR pathway. The combination of the ATM inhibitor KU55933 and the EGFR‐TKI gefitinib exerted synergistic cell growth inhibition and enhanced apoptosis in NSCLC cells carrying the sensitive EGFR mutation. KU55933 enhanced the gefitinib‐dependent repression of phosphorylation of EGFR and/or its downstream factors in NSCLC cells. These findings suggest that the inhibition of ATM kinase facilitates the anticancer effect of gefitinib, by enhancing the repression of the EGFR pathway in NSCLC cells with the sensitive EGFR mutation.
In NSCLC cells carrying the sensitive EGFR mutation, the ATM inhibitor enhanced the gefitinib‐dependent repression of phosphorylation of EGFR and/or its downstream factors. In contrast, it was difficult to detect the enhancement of the gefitinib‐induced repression of the modification of wild‐type EGFR as it was not overphosphorylated under normal conditions, unlike EGFR with the sensitive mutation. The ATM kinase plays a limited role in the regulation of NSCLC cell growth under normal conditions, regardless of the EGFR status. The ATM inhibitor, however, enhanced the gefitinib‐induced cell growth inhibition and apoptosis only in NSCLC cells carrying the sensitive EGFR mutation. Moreover, the exogenous expression of mutated EGFR synergistically induced apoptosis with the ATM inhibitor and gefitinib. Therefore, the sensitive EGFR mutation could be required to enhance the anticancer effect of gefitinib by the ATM inhibitor. In addition, the expression of Mig‐6, a negative regulator of the EGFR pathway, was upregulated by KU55933 in PC‐9 cells, but not in Calu‐6 cells. Increased Mig‐6 expression effectively inhibits signal transduction to the downstream pathway when EGFR with the sensitive mutation is activated.37 Taken together, the increase of Mig‐6 expression by the ATM inhibitor may enhance the gefitinib sensitivity of NSCLC cells with the sensitive EGFR mutation.
The ATM inhibitor reportedly suppresses the activation of Akt, induces apoptosis, and suppresses cell proliferation under conditions with IGFR overactivation.38 As EGFR and its downstream factors, including Akt, were constantly activated in NSCLC cells carrying the sensitive EGFR mutation, the ATM inhibitor could also repress the overactivation of the downstream factors of EGFR, as in cells with an activated IGFR pathway.10 The ATM inhibitor alone failed to inhibit the phosphorylation of Akt in NSCLC cells with the sensitive EGFR mutation. However, the ATM inhibitor alone repressed PC‐9 cell growth, but failed to do so in HCC827 cells with the EGFR sensitive mutation. These findings suggest that the ATM inhibitor has different targets in these cells. Indeed, the ATM inhibitor enhanced the gefitinib‐induced repression of phosphorylation of EGFR and downstream factors Akt and ERK in PC‐9 cells, whereas only the phosphorylation of Akt was synergistically inhibited with the combined treatment in HCC827 cells. In our study, the ATM inhibitor increased Mig‐6 expression in PC‐9 cells, but not in HCC827 cells (data not shown). The ATM inhibitor might enhance the effect of gefitinib in HCC827 cells through other feedback mechanisms for EGFR downstream factors, including Akt. Further studies are required to clarify the mechanisms underlying the enhanced effects of gefitinib by the ATM inhibitor in NSCLC cells with the sensitive EGFR mutation, as several mechanisms could facilitate the antitumor effect of gefitinib by the ATM inhibitor.
In this study, we have shown the enhanced anticancer effect of an EGFR‐TKI by the ATM inhibitor, KU55933. Improvement of outcomes with combination therapy may either delay or prevent the acquisition of resistance. However, no clinical trials for the treatment of NSCLC have included such ATM inhibitors. Therefore, further studies concerning the combined effects of EGFR‐TKIs with various ATM inhibitors, such as caffeine or telomelysin, may provide new insights for the development of treatment strategies for NSCLC cases with sensitive EGFR mutations.39, 40, 41
Disclosure Statement
The authors have no conflict of interest.
Abbreviations
- Akt
protein kinase B
- ATM
ataxia telangiectasia‐mutated
- DSB
double‐strand break
- EGFR
epidermal growth factor receptor
- EGFRwt
wild‐type EGFR
- EGFRmut
sensitive mutant EGFR
- γH2AX
phosphorylated H2AX
- IGFR
insulin‐like growth factor receptor
- IR
ionizing radiation
- NSCLC
non‐small‐cell lung cancer
- TKI
tyrosine kinase inhibitor
Supporting information
Fig. S1. Cell growth inhibition rate with KU55933 and the combination of KU55933 and gefitinib.
Fig. S2. Phosphorylation of EGFR and its downstream factors in HCC827 and VMRC‐LCD cells, and Mig‐6 expression in PC‐9 cells and Calu‐6 cells after KU55933 and/or gefitinib treatment.
Fig. S3. Combinational effect of KU55933 with gefitinib on DNA repair in HCC827 cells.
Data S1. Supporting data and experimental procedures.
Acknowledgments
We thank Hidehiko Kawai for operating the IN Cell Analyzer 2000. This work was supported by the Japan Society for the Promotion of Science (Kakenhi Grant Nos. 26116514, 15H02821, and 26430114), the Platform Project for Supporting in Drug Discovery and Life Science Research (Platform for Dynamic Approaches to Living System) from the Ministry of Education, Culture, Sports, Science, and Technology of Japan, and the Japan Agency for Medical Research and Development.
Cancer Sci 107 (2016) 444–451
Funding Information
Japan Society for the Promotion of Science; Ministry of Education, Culture, Sports, Science, and Technology of Japan; Japan Agency for Medical Research and Development.
References
- 1. World Health Organization . Fact sheet N°297. [Cited 11 September 2014.] Available from URL: http://www.who.int/mediacentre/factsheets/fs297/en/
- 2. Schiller JH, Harrington D, Belani CP et al Comparison of four chemotherapy regimens for advanced non‐small‐cell lung cancer. N Engl J Med 2002; 346: 92–8. [DOI] [PubMed] [Google Scholar]
- 3. Maemondo M, Inoue A, Kobayashi K et al Gefitinib or chemotherapy for non‐small‐cell lung cancer with mutated EGFR. N Engl J Med 2010; 362: 2380–8. [DOI] [PubMed] [Google Scholar]
- 4. Mitsudomi T, Morita S, Yatabe Y et al Gefitinib versus cisplatin plus docetaxel in patients with non‐small‐cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): an open label, randomised phase 3 trial. Lancet Oncol 2010; 11: 121–8. [DOI] [PubMed] [Google Scholar]
- 5. Mok TS, Wu YL, Thongprasert S et al Gefitinib or carboplatin‐paclitaxel in pulmonary adenocarcinoma. N Engl J Med 2009; 361: 947–57. [DOI] [PubMed] [Google Scholar]
- 6. Pao W, Miller V, Zakowski M et al EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci USA 2004; 101: 13306–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tang X, Shigematsu H, Bekele BN et al EGFR tyrosine kinase domain mutations are detected in histologically normal respiratory epithelium in lung cancer patients. Cancer Res 2005; 65: 7568–72. [DOI] [PubMed] [Google Scholar]
- 8. Ji H, Li D, Chen L et al The impact of human EGFR kinase domain mutations on lung tumorigenesis and in vivo sensitivity to EGFR‐targeted therapies. Cancer Cell 2006; 9: 485–95. [DOI] [PubMed] [Google Scholar]
- 9. Politi K, Zakowski MF, Fan PD, Schonfeld EA, Pao W, Varmus HE. Lung adenocarcinomas induced in mice by mutant EGF receptors found in human lung cancers respond to a tyrosine kinase inhibitor or to down‐regulation of the receptors. Genes Dev 2006; 20: 1496–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Choi SH, Mendrola JM, Lemmon MA. EGF‐independent activation of cell‐surface EGF receptors harboring mutations found in gefitinib‐sensitive lung cancer. Oncogene 2007; 26: 1567–76. [DOI] [PubMed] [Google Scholar]
- 11. Wakeling AE, Guy SP, Woodburn JR et al ZD1839 (Iressa): an orally active inhibitor of epidermal growth factor signaling with potential for cancer therapy. Cancer Res 2002; 62: 5749–54. [PubMed] [Google Scholar]
- 12. Carey KD, Garton AJ, Romero MS et al Kinetic analysis of epidermal growth factor receptor somatic mutant proteins shows increased sensitivity to the epidermal growth factor receptor tyrosine kinase inhibitor, erlotinib. Cancer Res 2006; 66: 8163–71. [DOI] [PubMed] [Google Scholar]
- 13. Ciardiello F, Tortora G. EGFR antagonists in cancer treatment. N Engl J Med 2008; 358: 1160–74. [DOI] [PubMed] [Google Scholar]
- 14. Mitsudomi T, Yatabe Y. Mutations of the epidermal growth factor receptor gene and related genes as determinants of epidermal growth factor receptor tyrosine kinase inhibitors sensitivity in lung cancer. Cancer Sci 2007; 98: 1817–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Morgillo F, Bareschino MA, Bianco R, Tortora G, Ciardiello F. Primary and acquired resistance to anti‐EGFR targeted drugs in cancer therapy. Differentiation 2007; 75: 788–99. [DOI] [PubMed] [Google Scholar]
- 16. Kobayashi S, Boggon TJ, Dayaram T et al EGFR mutation and resistance of non‐small‐cell lung cancer to gefitinib. N Engl J Med 2005; 352: 786–92. [DOI] [PubMed] [Google Scholar]
- 17. Katakami N, Atagi S, Goto K et al LUX‐Lung 4: a phase II trial of afatinib in patients with advanced non‐small‐cell lung cancer who progressed during prior treatment with erlotinib, gefitinib, or both. J Clin Oncol 2013; 31: 3335–41. [DOI] [PubMed] [Google Scholar]
- 18. Tan CS, Gilligan D, Pacey S. Treatment approaches for EGFR‐inhibitor‐resistant patients with non‐small‐cell lung cancer. Lancet Oncol 2015; 16: e447–59. [DOI] [PubMed] [Google Scholar]
- 19. Chen CY, Chang YL, Shih JY et al Thymidylate synthase and dihydrofolate reductase expression in non‐small cell lung carcinoma: the association with treatment efficacy of pemetrexed. Lung Cancer 2011; 74: 132–8. [DOI] [PubMed] [Google Scholar]
- 20. Okabe T, Okamoto I, Tsukioka S et al Synergistic antitumor effect of S‐1 and the epidermal growth factor receptor inhibitor gefitinib in non‐small cell lung cancer cell lines: role of gefitinib‐induced down‐regulation of thymidylate synthase. Mol Cancer Ther 2008; 7: 599–606. [DOI] [PubMed] [Google Scholar]
- 21. Wu Y‐L, Lee JS, Thongprasert S et al Intercalated combination of chemotherapy and erlotinib for patients with advanced stage non‐small‐cell lung cancer (FASTACT‐2): a randomised, double‐blind trial. Lancet Oncol 2013; 14: 777–86. [DOI] [PubMed] [Google Scholar]
- 22. Sugawara S, Oizumi S, Minato K et al Randomized phase II study of concurrent versus sequential alternating gefitinib and chemotherapy in previously untreated non‐small cell lung cancer with sensitive EGFR mutations: NEJ005/TCOG0902. Ann Oncol 2015; 26: 888–94. [DOI] [PubMed] [Google Scholar]
- 23. Katanasaka Y, Kodera Y, Yunokawa M, Kitamura Y, Tamura T, Koizumi F. Synergistic anti‐tumor effects of a novel phosphatidyl inositol‐3 kinase/mammalian target of rapamycin dual inhibitor BGT226 and gefitinib in non‐small cell lung cancer cell lines. Cancer Lett 2014; 347: 196–203. [DOI] [PubMed] [Google Scholar]
- 24. Savitsky K, Bar‐Shira A, Gilad S et al A single ataxia telangiectasia gene with a product similar to PI‐3 kinase. Science 1995; 268: 1749–53. [DOI] [PubMed] [Google Scholar]
- 25. Cremona CA, Behrens A. ATM signalling and cancer. Oncogene 2014; 33: 3351–60. [DOI] [PubMed] [Google Scholar]
- 26. Viniegra JG, Martinez N, Modirassari P et al Full activation of PKB/Akt in response to insulin or ionizing radiation is mediated through ATM. J Biol Chem 2005; 280: 4029–36. [DOI] [PubMed] [Google Scholar]
- 27. Blanco R, Iwakawa R, Tang M et al A gene‐alteration profile of human lung cancer cell lines. Hum Mutat 2009; 30: 1199–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Helfrich BA, Raben D, Varella‐Garcia M et al Antitumor activity of the epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor gefitinib (ZD1839, Iressa) in non‐small cell lung cancer cell lines correlates with gene copy number and EGFR mutations but not EGFR protein levels. Clin Cancer Res 2006; 12: 7117–25. [DOI] [PubMed] [Google Scholar]
- 29. Van Schaeybroeck S, Kyula J, Kelly DM et al Chemotherapy‐induced epidermal growth factor receptor activation determines response to combined gefitinib/chemotherapy treatment in non‐small cell lung cancer cells. Mol Cancer Ther 2006; 5: 1154–65. [DOI] [PubMed] [Google Scholar]
- 30. Sordella R, Bell DW, Haber DA, Settleman J. Gefitinib‐sensitizing EGFR mutations in lung cancer activate anti‐apoptotic pathways. Science 2004; 305: 1163–7. [DOI] [PubMed] [Google Scholar]
- 31. Zhang Y‐W, Vande Woude GF. Mig‐6, signal transduction, stress response and cancer. Cell Cycle 2007; 6: 507–13. [DOI] [PubMed] [Google Scholar]
- 32. Zhang X, Pickin KA, Bose R, Jura N, Cole PA, Kuriyan J. Inhibition of the EGF receptor by binding of MIG6 to an activating kinase domain interface. Nature 2007; 450: 741–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Naruo Y, Nagashima T, Ushikoshi‐Nakayama R et al Epidermal growth factor receptor mutation in combination with expression of MIG6 alters gefitinib sensitivity. BMC Syst Biol 2011; 5: 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tracy S, Mukohara T, Hansen M, Meyerson M, Johnson BE, Janne PA. Gefitinib induces apoptosis in the EGFRL858R non‐small‐cell lung cancer cell line H3255. Cancer Res 2004; 64: 7241–4. [DOI] [PubMed] [Google Scholar]
- 35. Park SY, Kim YM, Pyo H. Gefitinib radiosensitizes non‐small cell lung cancer cells through inhibition of ataxia telangiectasia mutated. Mol Cancer 2010; 9: 222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Greulich H, Chen TH, Feng W et al Oncogenic transformation by inhibitor‐sensitive and ‐resistant EGFR mutants. PLoS Med 2005; 2: e313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nagashima T, Ushikoshi‐Nakayama R, Suenaga A et al Mutation of epidermal growth factor receptor is associated with MIG6 expression. FEBS J 2009; 276: 5239–51. [DOI] [PubMed] [Google Scholar]
- 38. Li Y, Yang DQ. The ATM inhibitor KU‐55933 suppresses cell proliferation and induces apoptosis by blocking Akt in cancer cells with overactivated Akt. Mol Cancer Ther 2010; 9: 113–25. [DOI] [PubMed] [Google Scholar]
- 39. Blasina A, Price BD, Turenne GA, McGowan CH. Caffeine inhibits the checkpoint kinase ATM. Curr Biol 1999; 9: 1135–8. [DOI] [PubMed] [Google Scholar]
- 40. Hosoya N, Miyagawa K. Targeting DNA damage response in cancer therapy. Cancer Sci 2014; 105: 370–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kuroda S, Urata Y, Fujiwara T. Ataxia‐telangiectasia mutated and the Mre11‐Rad50‐NBS1 complex: promising targets for radiosensitization. Acta Med Okayama 2012; 66: 83–92. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Cell growth inhibition rate with KU55933 and the combination of KU55933 and gefitinib.
Fig. S2. Phosphorylation of EGFR and its downstream factors in HCC827 and VMRC‐LCD cells, and Mig‐6 expression in PC‐9 cells and Calu‐6 cells after KU55933 and/or gefitinib treatment.
Fig. S3. Combinational effect of KU55933 with gefitinib on DNA repair in HCC827 cells.
Data S1. Supporting data and experimental procedures.