

Abstract
Protein methylation is one of the important post‐translational modifications. Although its biological and physiological functions were unknown for a long time, we and others have characterized a number of protein methyltransferases, which have unveiled the critical functions of protein methylation in various cellular processes, in particular, in epigenetic regulation. In addition, it had been believed that protein methylation is an irreversible phenomenon, but through identification of a variety of protein demethylases, protein methylation is now considered to be dynamically regulated similar to protein phosphorylation. A large amount of evidence indicated that protein methylation has a pivotal role in post‐translational modification of histone proteins as well as non‐histone proteins and is involved in various processes of cancer development and progression. As dysregulation of this modification has been observed frequently in various types of cancer, small‐molecule inhibitors targeting protein methyltransferases and demethylases have been actively developed as anticancer drugs; clinical trials for some of these drugs have already begun. In this review, we discuss the biological and physiological importance of protein methylation in human cancer, especially focusing on the significance of protein methyltransferases as emerging targets for anticancer therapy.
Keywords: Anticancer drug, arginine methylation, epigenetics, lysine methylation, post‐translational modification
Protein methylation is a prevalent post‐translational modification, which is principally observed in lysine and arginine residues. Although the first ε‐N‐methyl‐lysine in the flagella protein of Salmonella typhimurium was reported in 1959,1 biological and physiological functions of protein methylation remained unknown for a long time. In the 21st century, we and other researchers characterized a number of protein methyltransferases and elucidated their functions, in particular focusing on their epigenetic regulation through histone methylation.1, 2, 3 The accumulated knowledge clearly indicates that histone methylation plays a pivotal role in transcriptional regulation; for instance, methylation of histone H3K9 is associated with silenced chromatin (heterochromatin), whereas methylation of histone H3K4 is an important mark of actively transcribed genes.
To date, lysine and arginine are considered to be target amino acids for methyltransferase reaction. Regarding lysine methylation, there are three different forms, which are monomethyl‐, dimethyl‐ and trimethyl‐lysines.1 Each form of lysine methylation is sophisticatedly produced by certain specific protein lysine methyltransferases; for example, histone H4K20 monomethylation and di/trimethylation are generated by SETD8 and SUV420H1/SUV420H2, respectively. There are also three primary methylated forms of an arginine residue: monomethyl‐arginine, asymmetric dimethyl‐arginine, and symmetric dimethyl‐arginine. Protein arginine methyltransferases are classified into type I or type II according to modification types. Although all PRMTs catalyze the formation of an monomethyl‐arginine intermediate, type I PRMTs (PRMT1, 2, 3, 4, 5, and 8) can catalyze the production of asymmetric dimethyl‐arginine, and type II PRMTs (PRMT5 and 7) are able to catalyze the production of symmetric dimethyl‐arginine.4
Previously, methyl groups were believed to turn over more slowly than many other post‐translational modifications. Furthermore, protein methylation had been thought to be irreversible until the first protein lysine demethylase LSD1/KDM1 was reported in 2004.5 Since then, JmjC‐domain containing protein family members have been reported to have protein lysine demethylase activity,6 suggesting that lysine methylation is dynamically regulated by protein lysine methyltransferases and demethylases. Moreover, most of the studies regarding protein methylation initially highlighted its importance of epigenetic regulation through histone methylation, but dozens of reports recently described the significance of non‐histone substrates, which shows that a variety of biological processes including cell cycle regulation, DNA repair, and apoptosis are regulated by protein methylation.1, 4 Hence, now methylation is widely recognized as a fundamental post‐translational modification of protein, as important as phosphorylation.
Dysregulation of protein methylation is involved in many disease conditions including cancer and, indeed, there are a large number of reports describing abnormal states of protein methyltransferases and demethylases such as aberrant expression and somatic mutations in human cancer.1, 4, 7, 8, 9 Furthermore, small molecular inhibitors targeting protein methyltransferases and demethylases have been actively developed as anticancer drugs, and clinical trials have already been started.1 In this review article, we summarize the biological significance of protein methylation and discuss the importance of protein methyltransferases as targets for development of anticancer drugs.
Functions of protein methylation
Epigenetic regulation through histone methylation
Epigenetic regulation by protein methyltransferases and demethylases through histone methylation has been well characterized. Histone methylation is now widely known to play a crucial role in the regulation of chromatin functions, mainly transcriptional regulation (Fig. 1). Among the core histones, most of the methylation sites reported so far were observed in histone H3 and H4 (Fig. 2), and each histone mark occurring at each methylation site is indicated to have a unique function.
Figure 1.
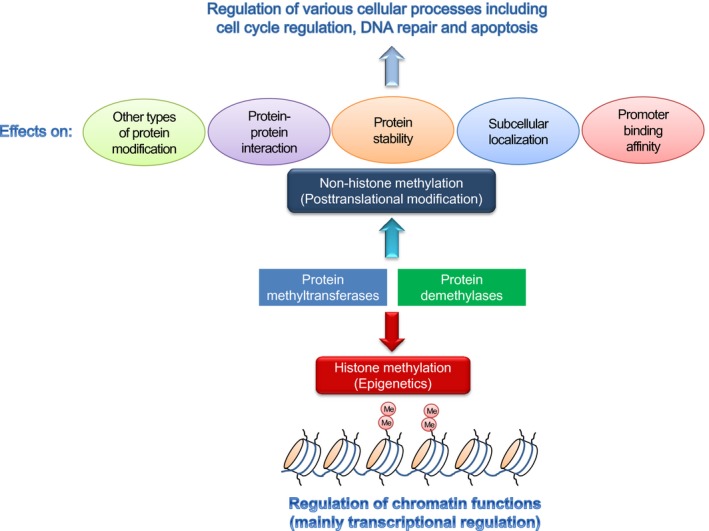
Protein methyltransferases and demethylases principally regulate biological processes in two ways. One is regulation of transcription for target downstream genes through methylation (Me) of histone proteins. The other is non‐histone methylation as one of the post‐translational modifications.
Figure 2.
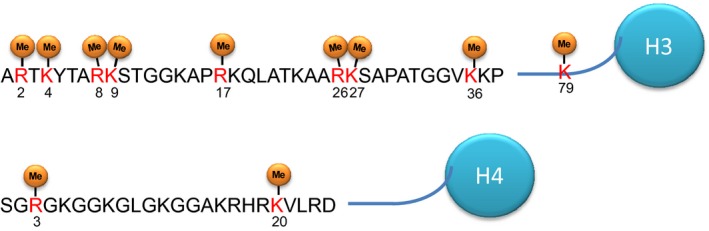
Identified methylation sites (Me) on histones H3 and H4. Each histone mark occurred at each methylation site is indicated to have a unique function.
Among various histone lysine methylations, methylation of H3K4 is described as a transcriptional active mark and monomethylation of H3K4 (H3K4me1) is enriched at the enhancer regions.10 Histone H3K4 dimethylation (H3K4me2) is found at both enhancer regions and promoter regions as well as in bodies of actively transcribed genes.11 Histone H3K4 trimethylation (H3K4me3) is known as a prominent feature in the promoter regions of actively transcribed genes.12 In contrast, the methylation of nucleosomal histone H3K9 is required for the assembly of constitutive heterochromatin. Dimethylation and trimethylation of histone H3K9 (H3K9me2/me3) provides binding sites for the heterochromatin protein HP1, which recruits additional silencing factors and locks in the repressed state. In addition to H3K9 methylation, dimethylation and trimethylation of H3K27 (H3K27me2/me3), associated with transcriptional repression, are characteristically observed in Polycomb group target genes.13 Moreover, trimethylation of H4K20 (H4K20me3) is a hallmark of silenced heterochromatic regions, whereas monomethylation and dimethylation of H4K20 (H4K20me1/me2) are involved in DNA replication and DNA damage repair.14 On the contrary, trimethylation of H3K36 (H3K36me3) was enriched through coding regions, peaking near the 3′‐ends of transcription units, which is thought to be associated with transcriptional elongation.15 In addition, the histone lysine methyltransferase SETD2‐dependent H3K36 trimethylation is considered to play an important role in homologous recombination repair and genome stability.16 Dimethylation and trimethylation of H3K79 (H3K79me2/me3) are associated with the proximal transcribed region of active genes, and there are several similarities between pattering of H3K4 methylation and that of H3K79 in mammalian chromatin.17
As for histone arginine methylation, asymmetric dimethylation of H3R2 by PRMT6 counteracts the trimethylation of H3K4, which results in transcriptional repression.18 Symmetric dimethylation of H3R8 by PRMT5 is linked to transcriptional repression and is tightly associated with symmetric dimethylation of H4R3, which is also a transcriptional repression mark and generated by PRMT5.19 Asymmetric dimethylation of both H3R17 and H3R26 is methylated by CARM1, considered as a transcriptional activation mark.19 Interestingly, although symmetric dimethylation of H4R3 is a transcriptional repression mark, asymmetric dimethylation of H4R3 is a transcriptional activation mark,19 implying that symmetric dimethylation and asymmetric dimethylation are functionally different.
Taken together, the aforementioned knowledge suggests that the position and modification status (the number of methyl group, or which isomer) defines the functions of histone methylation.
Regulation of various pathways through non‐histone methylation
The accumulated evidence indicates that methylation of non‐histone proteins also plays a critical role in the regulation of various signaling pathways. For instance, the functions of p53 and RB1, two important tumor suppressor proteins, are sophisticatedly regulated by lysine methylation.1 As the detailed molecular mechanisms of non‐histone methylation were described in another review article,1 we here comment on several key points relevant to methylation of non‐histone proteins. There are at least five principal functions of methylation on non‐histone proteins as follows: (i) it affects other types of modifications such as phosphorylation on substrates; (ii) it influences protein–protein interactions; (iii) it regulates stability of substrate proteins; (iv) it defines subcellular localization of substrates; and (v) it affects the promoter binding affinity of substrate proteins (Fig. 1). On the basis of these characteristics, methylation of non‐histone proteins is involved in various biological processes in the cell.
Dysregulation of protein lysine methyltransferases in human cancer
It has been reported that a number of protein lysine methyltransferases are involved in human cancers as shown in Table 1. We selected several pivotal enzymes as targets for anticancer therapy developed, and detail their characteristics below.
Table 1.
Protein lysine methyltransferases dysregulated in cancer
| Family name | Enzyme name | Substrate | Changes in cancer | Cancer type | Specific inhibitors |
|---|---|---|---|---|---|
| SET and MYND domain‐containing proteins (SMYD) | SMYD2 (KMT3C) | Histone H3, p53, RB1, PARP1, HSP90AB1, PTEN, ER‐α | Overexpression DNA amplification | Bladder cancer, breast cancer, cervical cancer, esophageal cancer, CRC, HCC, head and neck cancer, lymphoma, ovarian cancer, pancreatic cancer, RCC | AZ505 (preclinical) LLY‐507 (preclinical) A‐893 (preclinical) |
| SMYD3 (KMT3E) | Histone H3, histone H4, VEGFR1, MAP3K2 | Overexpression DNA amplification | Breast cancer, CCC, cervical cancer, CRC, esophageal cancer, gastric cancer, HCC, lung cancer, MTC, pancreatic cancer, prostate cancer | BCI‐121 (preclinical) EPZ031686 (preclinical) | |
| Polycomb complex | EZH2 (KMT6) | Histone H2B, histone H3, RORα, STAT3 | Overexpression DNA amplification GOF missense mutations (Y641, A677, A687) LOF mutations | AML, bladder cancer, breast cancer, CCC, CML, CRC, esophageal cancer, glioblastoma, lymphoma, NSCLC, SCLC, T‐ALL, osteosarcoma, RCC | GSK126 (preclinical) EPZ005687 (preclinical) EPZ‐6438 (phase I/II)† |
| Nuclear receptor‐binding SET‐domain proteins (NSD) | NSD1 (KMT3B) | Histone H3, NF‐κB | Chromosomal translocation (NUP98‐NSD1: t(5;11)(q35;p15)) DNA amplification | AML, glioblastoma, lung cancer, multiple myeloma | – |
| WHSC1 (MMSET and NSD2) | Histone H3 | Chromosomal translocation (IGH‐WHSC1: t(4;14)(p16;q32)) Overexpression DNA amplification | Bladder cancer, breast cancer, CCC, CML, esophageal cancer, HCC, multiple myeloma, NSCLC, SCLC, osteosarcoma, prostate cancer and RCC | MCTP39 (preclinical) LEM‐06 (preclinical) | |
| WHSC1L1 (NSD3) | Histone H3 | Chromosomal translocation (NUP98‐WHSC1L1: t(8;11)(p11.2;p15), WHSC1L1‐NUT: t(8;15)(p11.2;q14)), Overexpression, DNA amplification | AML, bladder cancer, breast cancer, NUT, SCLC, lymphoma | – | |
| SET‐domain containing proteins (SETD) | SETD1A (KMT2F) | Histone H3, HSP70 | Overexpression | Bladder cancer, breast cancer, CRC, HCC, lung cancer, RCC | – |
| SETD8 (KMT5A) | Histone H4, PCNA | Overexpression | Bladder cancer, CML, HCC, NSCLC, prostate cancer, SCLC | UNC0379 (preclinical) | |
| Suppressor of Variegation 3–9 Homolog | SUV39H2 (KMT1B) | Histone H2AX, histone H3, LSD1 | Overexpression | ALL, Bladder cancer, cervical cancer, esophageal cancer, NSCLC, osteosarcoma, prostate cancer, STT | – |
| Euchromatic histone‐lysine N‐methyltransferase | EHMT2 (KMT1C, G9a) | Histone H3, C/EBPβ | Overexpression | AML, bladder cancer, breast cancer, CCC, CML, esophageal cancer, NSCLC, SCLC, prostate cancer | BIX‐01294 (preclinical) UNC0638 (preclinical) |
| DOT1‐like histone H3K79 methyltransferase | DOT1L (KMT4) | Histone H3 | DOT1L physically interacts with MLL fusion proteins | MLL | EPZ004777 (preclinical) EPZ‐5676 (phase I)† |
| MLL family | MLL (KMT2A) | Histone H3 | Chromosomal translocation | AML | – |
| MLL2 (KMT2D) | Histone H3 | Overexpression (mRNA) Mutations | Bladder cancer, breast cancer, CRC, lung cancer, melanoma, MLL | – | |
| MLL3 (KMT2C) | Histone H3 | Point mutations Small insertions/deletions | Breast cancer, esophagus cancer, glioblastoma, melanoma, MLL, pancreas cancer, stomach cancer | – |
†Inhibitors currently undergoing clinical trials. –, not particular. ALL, acute lymphoblastic leukemia; AML; acute myeloid leukemia; CCC, cholangiocarcinoma; C/EBPβ, CCAAT/enhancer binding protein; CML, chronic myelogenous leukemia; CRC, colorectal cancer; DOT1L, disruptor of telomeric silencing 1‐like; ERα, estrogen receptor α; GOF, gain‐of‐function; HCC, hepatocellular carcinoma; HSP, heat shock protein; LOF, loss‐of‐function; MLL, mixed‐lineage leukemia; MTC, medullary thyroid cancer; NF‐κB, nuclear factor‐κB; NSCLC, non‐small‐cell lung carcinoma; NUT, NUT midline carcinoma; PARP, poly(ADP‐ribose) polymerase; PTEN, phosphatase and tensin homolog; RB, retinoblastoma; RCC, renal cell carcinoma; RORα, retinoid‐related orphan receptor α; SCLC, small‐cell lung carcinoma; STAT, signal transducer and activator of transcription; STT, soft tissue tumors; T‐ALL, T‐cell acute lymphoblastic leukemia; VEGFR, vascular endothelial growth factor receptor.
SET and MYND domain‐containing proteins
We previously reported that SMYD3 is overexpressed in colorectal cancer, hepatocellular carcinoma, and breast cancer, and possesses histone lysine methyltransferase activity.2, 20, 21 Since then, multiple reports have shown that dysregulation of SMYD3 is involved in many types of cancer.1 Reduction of SMYD3 expression leads to suppression of cancer cell growth and induction of apoptosis.2, 20 Hence, SMYD3 is now considered as one of the important targets for anticancer therapy. In addition to histone proteins, vascular endothelial growth factor receptor 1 and MAP3K2 were reported as substrates of SMYD3.22, 23 Two specific inhibitors targeting enzyme activity of SMYD3 were reported recently; one is BCI‐121, which could suppress the growth of various types of cancer cells overexpressing SMYD3.24 The other is EPZ031686, which showed good bioavailability following oral dosing in mice.25
We also reported that SMYD2, a family member of SMYD methyltransferases, is overexpressed in various types of cancer.26 Given that knockdown of SMYD2 induces suppression of cancer cell growth,26, 27 it is also considered a critical target for anticancer therapy. We and others have reported a variety of substrates of SMYD2 including histone H3, p53, RB1, heat shock protein 90AB1, poly (ADP‐ribose) polymerase 1, and phosphatase and tensin homolog.1, 28, 29, 30 In particular, as SMYD2 was reported to inactivate functions of tumor suppressor proteins p53 and RB1 through lysine methylation, it appears to serve as an oncogenic protein. Therefore, inhibitors targeting SMYD2 enzyme activity have been actively developed. AZ‐505, the first reported SMYD2 specific inhibitor, showed an IC50 value of 120 nM (enzyme inhibition); in this development process, p53 peptide was used as a substrate.31 Later, Nguyen et al.32 reported that LLY‐507 worked as a specific inhibitor of SMYD2, which showed an IC50 of <15 nM (enzyme inhibition). LLY‐507 also inhibited SMYD2‐mediated p53 methylation in U2OS cells with an IC50 of 0.6 μM, implying that LLY‐507 is a selective and cell‐active small molecule inhibitor of SMYD2. Sweis et al.33 recently reported that A‐893, a benzoxazinone derivative, specifically inhibits SMYD2 enzyme activity (IC50, 2.8 nM). This inhibitor clearly suppressed p53K370 methylation mediated by SMYD2 in lung carcinoma A549 cells.
Because both SMYD2 and SMYD3 are principally localized in the cytoplasm, cytoplasmic proteins should serve as substrates of these enzymes. For the better understanding of functions of these two proteins and for effective drug development, their localization must be considered.
Polycomb complex
EZH2, a protein lysine methyltransferase and a component of the Polycomb repressive complex 2, plays an essential role in the epigenetic maintenance of the repressive chromatin mark, H3K27me3. We previously reported dysregulation of EZH2 in many types of malignancies.34 EZH2 also methylates histone H2BK120 and this methylation inhibits ubiquitination of H2B.35 Anticancer drugs targeting mutant‐type or wild‐type of EZH2 have been actively developed;1 for example, McCabe et al.36 showed that GSK126, a small molecular inhibitor of EZH2 methyltransferase activity, inhibits the proliferation of several EZH2 mutant lymphoma cells. Additionally, a phase I/II clinical trial of EPZ‐6438, which is a specific inhibitor against EZH2, is currently ongoing for patients with relapsed or refractory B‐cell non‐Hodgkin's lymphoma or advanced solid tumors.
Nuclear receptor‐binding SET‐domain proteins
The NSD protein lysine methyltransferase family is comprised of three members, NSD1, WHSC1 (NSD2/MMSET), and WHSC1L1 (NSD3), which methylate histone H3K36. We and others reported frequent dysregulation of NSD family enzymes in various types of cancer. Among them, it is important that chromosome translocations involving this family member are often observed; the cryptic t(5;11)(q35;p15.5) translocation creating a fusion gene of NUP98 and NSD1 is mainly identified in pediatric AML. The expression of the NUP98–NSD1 fusion protein is strongly associated with a poor prognosis in this disease.37 The translocation t(4; 14)(p16; q32), one of the most commonly observed translocations in multiple myeloma, accounts for 15% of patients, and is associated with very poor prognosis.38 The t(4; 14) translocation leads to the simultaneous overexpression of two genes, WHSC1 and FGFR3. Although overexpression of WHSC1 isoforms is a universal feature of t(4; 14) of cases, approximately 30% of t(4; 14) patients do not express FGFR3.39 Additionally, the poor prognosis of t(4; 14) persists irrespective of FGFR3 expression.40 These data imply WHSC1 to have oncogenic activity. Moreover, the NUP98–WHSC1L1 fusion gene, which was identified in AML or therapy‐related myelodysplastic syndrome, is considered to be related to leukemogenesis,41 and to be required for the blockade of differentiation as well as the persistent proliferation of NUT midline carcinoma cells.42 Furthermore, elevated expression of WHSC1 and WHSC1L1 is often observed in many types of human cancers, and these enzymes are essential for the growth of cancer cells.43, 44, 45, 46
Suppressor of variegation 3–9 homolog
SUV39H1 and SUV39H2 were reported as histone methyltransferases, which could selectively methylate histone H3K9, and are associated with heterochromatin formation and transcription repression. We previously reported that SUV39H2 is involved in multiple types of human malignancies.47, 48 As attenuation of SUV39H2 effectively suppresses the growth of cancer cells and its expression is hardly detectable in normal tissues except for testis,47, 48 SUV39H2 seems to be an ideal target for the development of anticancer drugs. In addition to histone H3, we identified histone H2AX as a substrate of SUV39H2. Through methylation of histone H2AX at Lys 134, SUV39H2 regulates γ‐H2AX levels after DNA double‐strand breaks; attenuation of this methylation enhances radiosensitivity and chemosensitivity of cancer cells.47 Moreover, we also discovered the protein lysine demethylase LSD1, which is overexpressed in a variety of human cancers, to be methylated by SUV39H2.49 SUV39H2‐mediated methylation on LSD1 at Lys 322 inhibits polyubiquitination and subsequent degradation, which results in stabilizing LSD1 protein in cancer cells.49
DOT1‐like histone H3K79 methyltransferase
Dot1, also called Kmt4, was first identified during the screening of yeast genes that disrupt telomeric silencing.50 Dot1 and its mammalian homolog, DOT1L, possess histone methyltransferase activity toward histone H3K79, which is associated with active transcription, whereas this family of enzyme does not possess the SET domain. DOT1L is implicated in the development of MLL‐rearranged leukemia, where chromosomal translocations between the MLL (encoding lysine‐specific methyltransferase 2A and officially known as KMT2A) gene and various fusion partners were observed.51 Several of these fusion partners interact directly or indirectly with DOT1L, which results in inappropriate recruitment of DOT1L to gene targets of these MLL fusion proteins including HoxA cluster and the homeobox gene Meis1.51 Hence, although DOT1L itself is not genetically altered in the disease, its mislocation of enzymatic activity causes a direct consequence of the chromosomal translocation affecting MLL patients.52 Studies in model systems suggested that DOT1L is required for the transforming activity of MLL fusion proteins; DOT1L has therefore been proposed to be a catalytic driver of leukemogenesis in this disease.52 Given these kinds of evidence, inhibition of DOT1L is an appropriate strategy to treat MLL. Daigle et al.52 reported the DOT1L‐specific inhibitor EPZ004777, which showed an IC50 of 0.4 nM (enzyme inhibition), and that in vivo treatment with EPZ004777 extended survival in a mouse MLL xenograft model. Recently, the same group also developed a new DOT1L inhibitor called EPZ‐5676, which showed high potency and selectivity.53 EPZ‐5676 is currently under clinical investigation for acute leukemias bearing MLL rearrangement.
Dysregulation of protein arginine methyltransferases in human cancer
Several protein arginine methyltransferases are also dysregulated in human cancer as shown in Table 2. We below discuss the significance of protein arginine methyltransferases in cancer.
Table 2.
Protein arginine methyltransferases dysregulated in cancer
| Family name | Enzyme name (type) | Substrate | Changes in cancer | Cancer type | Specific inhibitors |
|---|---|---|---|---|---|
| PRMT family (protein arginine methyltransferase family) | PRMT1 (type I) | Histone H4, MRE11, 53BP1, SAM68, INCENP | Overexpression | Bladder cancer, breast cancer, CRC, esophageal cancer, gastric cancer, lymphoma, NSCLC, pancreatic cancer, testicular cancer, SCLC | DCLX069 (preclinical) DCLX078 (preclinical) |
| PRMT4, CARM1 (type I) | Histone H3, AIB1, p300, CBP, RNA PII CTD | Overexpression | Breast cancer, CRC, prostate cancer | TBBD (preclinical) | |
| PRMT5 (type II) | Histone H3, histone H4, E2F1, p53, CRAF, SmD3 | Overexpression | CRC, gastric cancer, leukemia, lung cancer, MCL | EPZ015666 (preclinical) | |
| PRMT6 (type I) | Histone H2A, histone H3, p21CDKN1A | Overexpression | Bladder cancer, breast cancer, cervical cancer, CML, CRC, esophageal cancer, gastric cancer, lymphoma, NSCLC, osteosarcoma, prostate cancer, SCLC | EPZ020411 (preclinical) |
CBP, CREB binding protein; CML, chronic myelogenous leukemia; CRAF, C‐Raf Proto‐Oncogene; CRC, colorectal cancer; CTD, carboxy terminal domain; INCENP, inner centromere protein; MCL, mantle cell lymphoma; MRE11, meiotic recombination 11; NSCLC, non‐small‐cell lung carcinoma; SAM68, Src‐associated substrate in mitosis of 68 kDa; SCLC, small‐cell lung carcinoma.
PRMT1
We previously reported elevated expression of PRMT1 in various types of human malignancies.54 PRMT1 is a type I protein arginine methyltransferase and catalyzes methylation of histone H4R3.4 PRMT1 is the first eukaryotic protein arginine methyltransferase to be cloned and has been shown to act as a coactivator of nuclear receptor‐mediated gene transcription together with p300/CBP, a histone acetyltransferase, and PRMT4/CARM1. We recently reported that PRMT1 methylates INCENP at Arg 887 within an AURKB‐binding site. PRMT1‐mediated INCENP methylation plays a critical role in the chromosomal passenger complex function and appropriate chromosomal alignment and segregation in cancer cells.55 Moreover, other non‐histone substrates for PRMT1, including MRE11, 53BP1, and SAM68, have also been reported by others.4 Xie et al.56 reported identification of DCLX069 and DCLX078 as PRMT1‐specific inhibitors through the combination of virtual screening and bioassays. These inhibitors effectively blocked proliferation of breast cancer, liver cancer, and AML cells.56
PRMT5
PRMT5 is a type II arginine methyltransferase and upregulated in several human malignancies, including lymphomas, lung cancer, breast cancer, colorectal cancer, and MCL.4, 57 Chan‐Penebre et al. recently reported that EPZ015666 is an orally available inhibitor of PRMT5 with an IC50 of 22 nM (enzyme inhibition).57 Treatment of MCL cell lines with EPZ01566 resulted in repressing methylation of SmD3, a substrate of PRMT5, and inducing cell death with IC50 values in the nanomolar range.57 Furthermore, oral administration of EPZ015666 showed dose‐dependent growth‐suppressive effects in multiple MCL xenograft models,57 implying the importance of PRMT5 inhibition for treatment of MCL.
PRMT6
We also reported that PRMT6 with type I enzymatic activity was dysregulated in various types of cancer.54 PRMT6 is the prevalent protein arginine methyltransferase responsible for the methylation of histone H3R2, which antagonizes the MLL complex‐mediated methylation of histone H3K4. PRMT6 localizes mainly in the nucleus, and appears to methylate glycine‐ and arginine‐rich sequences in proteins. Interestingly, we and others showed that PRMT6 regulates expression and subcellular localization of p21CDKN1A, a potent inhibitor of CDK, through histone methylation and direct methylation of p21CDKN1A at arginine 156.29, 58 In addition, the PRMT6‐specific inhibitor EPZ020411, which is a novel aryl pyrazole, was reported recently.59 The IC50 of EPZ020411 for PRMT6 enzyme inhibition is 10 nM, and treatment with EPZ020411 resulted in a dose‐dependent decrease in H3R2 methylation in A375 human melanoma cells exogenously overexpressing PRMT6.59
Conclusion
In the 21st century, discoveries of protein methyltransferases have accelerate research in this field, which has elucidated a variety of functions regulated by protein methylation. Meanwhile, the history of research relevant to protein methylation is relatively short compared to that of protein phosphorylation, whose functions have been actively investigated since the 1970s. Therefore, there are still several unelucidated issues we need to clarify to better understand the roles of protein methylation. For example, although we principally focused on SET‐domain proteins as protein lysine methyltransferases so far, non‐SET protein lysine methyltransferases such as METTL10 and METTL21A have also been reported to possess lysine methyltransferase activity.1 This implies that our genome may possess additional uncharacterized proteins with lysine methyltransferase activity. In addition, to date, histone proteins have mainly been investigated as substrates of protein methyltransferases, but numerous non‐histone substrates may still remain to be elucidated.1 For the development of effective anticancer drugs, it is essential to clarify the biological and physiological significance of methylation on unidentified non‐histone substrates in cancer. Furthermore, current advancement in next‐generation sequencing technology discovered a large number of somatic mutations in protein methyltransferases, some of which are considered to significantly affect methyltransferase activity.1 However, the biological functions and clinical relevance of most mutations have not been analyzed yet. For the development of personalized medicine using anticancer drugs targeting protein methyltransferases, further biological analysis of individual mutations should be undertaken.
In summary, protein methyltransferases are currently attracting considerable attention as new targets for development of anticancer therapy. Due to the fact that protein methyltransferases are surely a key target class for next‐generation targeted cancer therapy, further detailed molecular analysis may explore the biological diversity of protein methylation and accelerate development of anticancer drugs targeting protein methyltransferases.
Disclosure Statement
Y. Nakamura is a stock holder and a scientific advisor of OncoTherapy Science and also has research grants from OncoTherapy Science, Inc. R. Hamamoto has no conflict of interest to declare.
Abbreviations
- CDK
cyclin‐dependent kinase
- Dot1
disruptor of telomeric silencing 1
- DOT1L
DOT1‐like
- EZH2
enhancer of Zeste homolog 2
- INCENP
inner centromere protein
- LSD
Lysine‐specific demethylase
- MCL
mantle cell lymphoma
- MLL
mixed lineage leukemia
- NSD
nuclear receptor‐binding SET‐domain
- PRMT
protein arginine methyltransferase
- RB
retinoblastoma
- SMYD
SET and MYND domain‐containing
- SUV39H
suppressor of variegation 3‐9 homolog
Acknowledgments
We thank members of Nakamura Laboratory for their kind support and helpful discussion.
Cancer Sci 107 (2016) 377–384
Funding Information
No sources of funding were declared for this study.
References
- 1. Hamamoto R, Saloura V, Nakamura Y. Critical roles of non‐histone protein lysine methylation in human tumorigenesis. Nat Rev Cancer 2015; 15: 110–24. [DOI] [PubMed] [Google Scholar]
- 2. Hamamoto R, Furukawa Y, Morita M et al SMYD3 encodes a histone methyltransferase involved in the proliferation of cancer cells. Nat Cell Biol 2004; 6: 731–40. [DOI] [PubMed] [Google Scholar]
- 3. Rea S, Eisenhaber F, O'Carroll D et al Regulation of chromatin structure by site‐specific histone H3 methyltransferases. Nature 2000; 406: 593–9. [DOI] [PubMed] [Google Scholar]
- 4. Yang Y, Bedford MT. Protein arginine methyltransferases and cancer. Nat Rev Cancer 2013; 13: 37–50. [DOI] [PubMed] [Google Scholar]
- 5. Shi Y, Lan F, Matson C et al Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 2004; 119: 941–53. [DOI] [PubMed] [Google Scholar]
- 6. Klose RJ, Kallin EM, Zhang Y. JmjC‐domain‐containing proteins and histone demethylation. Nat Rev Genet 2006; 7: 715–27. [DOI] [PubMed] [Google Scholar]
- 7. Cho HS, Shimazu T, Toyokawa G et al Enhanced HSP70 lysine methylation promotes proliferation of cancer cells through activation of Aurora kinase B. Nat Commun 2012; 3: 1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hayami S, Kelly JD, Cho HS et al Overexpression of LSD1 contributes to human carcinogenesis through chromatin regulation in various cancers. Int J Cancer 2011; 128: 574–86. [DOI] [PubMed] [Google Scholar]
- 9. Hayami S, Yoshimatsu M, Veerakumarasivam A et al Overexpression of the JmjC histone demethylase KDM5B in human carcinogenesis: involvement in the proliferation of cancer cells through the E2F/RB pathway. Mol Cancer 2010; 9: 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Heintzman ND, Stuart RK, Hon G et al Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet 2007; 39: 311–8. [DOI] [PubMed] [Google Scholar]
- 11. Hu D, Gao X, Morgan MA, Herz HM, Smith ER, Shilatifard A. The MLL3/MLL4 branches of the COMPASS family function as major histone H3K4 monomethylases at enhancers. Mol Cell Biol 2013; 33: 4745–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Barski A, Cuddapah S, Cui K et al High‐resolution profiling of histone methylations in the human genome. Cell 2007; 129: 823–37. [DOI] [PubMed] [Google Scholar]
- 13. Schwartz YB, Pirrotta V. Polycomb silencing mechanisms and the management of genomic programmes. Nat Rev Genet 2007; 8: 9–22. [DOI] [PubMed] [Google Scholar]
- 14. Jorgensen S, Schotta G, Sorensen CS. Histone H4 lysine 20 methylation: key player in epigenetic regulation of genomic integrity. Nucleic Acids Res 2013; 41: 2797–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Edmunds JW, Mahadevan LC, Clayton AL. Dynamic histone H3 methylation during gene induction: HYPB/Setd2 mediates all H3K36 trimethylation. EMBO J 2008; 27: 406–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pfister SX, Ahrabi S, Zalmas LP et al SETD2‐dependent histone H3K36 trimethylation is required for homologous recombination repair and genome stability. Cell Rep 2014; 7: 2006–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Steger DJ, Lefterova MI, Ying L et al DOT1L/KMT4 recruitment and H3K79 methylation are ubiquitously coupled with gene transcription in mammalian cells. Mol Cell Biol 2008; 28: 2825–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hyllus D, Stein C, Schnabel K et al PRMT6‐mediated methylation of R2 in histone H3 antagonizes H3 K4 trimethylation. Genes Dev 2007; 21: 3369–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Di Lorenzo A, Bedford MT. Histone arginine methylation. FEBS Lett 2011; 585: 2024–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hamamoto R, Silva FP, Tsuge M et al Enhanced SMYD3 expression is essential for the growth of breast cancer cells. Cancer Sci 2006; 97: 113–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tsuge M, Hamamoto R, Silva FP et al A variable number of tandem repeats polymorphism in an E2F‐1 binding element in the 5′ flanking region of SMYD3 is a risk factor for human cancers. Nat Genet 2005; 37: 1104–7. [DOI] [PubMed] [Google Scholar]
- 22. Kunizaki M, Hamamoto R, Silva FP et al The lysine 831 of vascular endothelial growth factor receptor 1 is a novel target of methylation by SMYD3. Cancer Res 2007; 67: 10759–65. [DOI] [PubMed] [Google Scholar]
- 23. Mazur PK, Reynoird N, Khatri P et al SMYD3 links lysine methylation of MAP3K2 to Ras‐driven cancer. Nature 2014; 510: 283–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Peserico A, Germani A, Sanese P et al A SMYD3 small‐molecule inhibitor impairing cancer cell growth. J Cell Physiol 2015; 230: 2447–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mitchell LM, Boriack‐Sjodin PA, Smith S et al Novel oxindole sulfonamides and sulfamides: EPZ031686, the first orally bioavailable small molecule SMYD3 inhibitor. ACS Med Chem Lett 2015; in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cho HS, Hayami S, Toyokawa G et al RB1 methylation by SMYD2 enhances cell cycle progression through an increase of RB1 phosphorylation. Neoplasia 2012; 14: 476–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Komatsu S, Imoto I, Tsuda H et al Overexpression of SMYD2 relates to tumor cell proliferation and malignant outcome of esophageal squamous cell carcinoma. Carcinogenesis 2009; 30: 1139–46. [DOI] [PubMed] [Google Scholar]
- 28. Hamamoto R, Toyokawa G, Nakakido M, Ueda K, Nakamura Y. SMYD2‐dependent HSP90 methylation promotes cancer cell proliferation by regulating the chaperone complex formation. Cancer Lett 2014; 351: 126–33. [DOI] [PubMed] [Google Scholar]
- 29. Nakakido M, Deng Z, Suzuki T, Dohmae N, Nakamura Y, Hamamoto R. PRMT6 increases cytoplasmic localization of p21CDKN1A in cancer cells through arginine methylation and makes more resistant to cytotoxic agents. Oncotarget 2015; 6: 30957–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Piao L, Kang D, Suzuki T et al The histone methyltransferase SMYD2 methylates PARP1 and promotes poly(ADP‐ribosyl)ation activity in cancer cells. Neoplasia 2014; 16: 257–64, 64 e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ferguson AD, Larsen NA, Howard T et al Structural basis of substrate methylation and inhibition of SMYD2. Structure 2011; 19: 1262–73. [DOI] [PubMed] [Google Scholar]
- 32. Nguyen H, Allali‐Hassani A, Antonysamy S et al LLY‐507, a cell‐active, potent, and selective inhibitor of protein‐lysine methyltransferase SMYD2. J Biol Chem 2015; 290: 13641–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sweis RF, Wang Z, Algire M et al Discovery of A‐893, a new cell‐active benzoxazinone inhibitor of lysine methyltransferase SMYD2. ACS Med Chem Lett 2015; 6: 695–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Takawa M, Masuda K, Kunizaki M et al Validation of the histone methyltransferase EZH2 as a therapeutic target for various types of human cancer and as a prognostic marker. Cancer Sci 2011; 102: 1298–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kogure M, Takawa M, Saloura V et al The oncogenic polycomb histone methyltransferase EZH2 methylates lysine 120 on histone H2B and competes ubiquitination. Neoplasia 2013; 15: 1251–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. McCabe MT, Ott HM, Ganji G et al EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2‐activating mutations. Nature 2012; 492: 108–12. [DOI] [PubMed] [Google Scholar]
- 37. Shiba N, Ichikawa H, Taki T et al NUP98‐NSD1 gene fusion and its related gene expression signature are strongly associated with a poor prognosis in pediatric acute myeloid leukemia. Genes Chromosom Cancer 2013; 52: 683–93. [DOI] [PubMed] [Google Scholar]
- 38. Xie Z, Chng WJ. MMSET: role and therapeutic opportunities in multiple myeloma. Biomed Res Int 2014; 2014: 636514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Santra M, Zhan F, Tian E, Barlogie B, Shaughnessy J Jr. A subset of multiple myeloma harboring the t(4;14)(p16;q32) translocation lacks FGFR3 expression but maintains an IGH/MMSET fusion transcript. Blood 2003; 101: 2374–6. [DOI] [PubMed] [Google Scholar]
- 40. Keats JJ, Maxwell CA, Taylor BJ et al Overexpression of transcripts originating from the MMSET locus characterizes all t(4;14)(p16;q32)‐positive multiple myeloma patients. Blood 2005; 105: 4060–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Taketani T, Taki T, Nakamura H, Taniwaki M, Masuda J, Hayashi Y. NUP98‐NSD3 fusion gene in radiation‐associated myelodysplastic syndrome with t(8;11)(p11;p15) and expression pattern of NSD family genes. Cancer Genet Cytogenet 2009; 190: 108–12. [DOI] [PubMed] [Google Scholar]
- 42. French CA, Rahman S, Walsh EM et al NSD3‐NUT fusion oncoprotein in NUT midline carcinoma: implications for a novel oncogenic mechanism. Cancer Discov 2014; 4: 928–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kang D, Cho HS, Toyokawa G et al The histone methyltransferase Wolf‐Hirschhorn syndrome candidate 1‐like 1 (WHSC1L1) is involved in human carcinogenesis. Genes Chromosom Cancer 2013; 52: 126–39. [DOI] [PubMed] [Google Scholar]
- 44. Toyokawa G, Cho HS, Masuda K et al Histone lysine methyltransferase Wolf‐Hirschhorn syndrome candidate 1 is involved in human carcinogenesis through regulation of the Wnt pathway. Neoplasia 2011; 13: 887–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Vougiouklakis T, Hamamoto R, Nakamura Y, Saloura V. The NSD family of protein methyltransferases in human cancer. Epigenomics 2015; 7: 863–74. [DOI] [PubMed] [Google Scholar]
- 46. Saloura V, Cho HS, Kyiotani K et al WHSC1 promotes oncogenesis through regulation of NIMA‐related‐kinase‐7 in squamous cell carcinoma of the head and neck. Mol Cancer Res 2015; 13: 293–304. [DOI] [PubMed] [Google Scholar]
- 47. Sone K, Piao L, Nakakido M et al Critical role of lysine 134 methylation on histone H2AX for gamma‐H2AX production and DNA repair. Nat Commun 2014; 5: 5691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mutonga M, Tamura K, Malnassy G et al Targeting suppressor of variegation 3‐9 homologue 2 (SUV39H2) in acute lymphoblastic leukemia (ALL). Transl Oncol 2015; 8: 368–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Piao L, Suzuki T, Dohmae N, Nakamura Y, Hamamoto R. SUV39H2 methylates and stabilizes LSD1 by inhibiting polyubiquitination in human cancer cells. Oncotarget 2015; 6: 16939–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Singer MS, Kahana A, Wolf AJ et al Identification of high‐copy disruptors of telomeric silencing in Saccharomyces cerevisiae . Genetics 1998; 150: 613–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. McLean CM, Karemaker ID, van Leeuwen F. The emerging roles of DOT1L in leukemia and normal development. Leukemia 2014; 28: 2131–8. [DOI] [PubMed] [Google Scholar]
- 52. Daigle SR, Olhava EJ, Therkelsen CA et al Selective killing of mixed lineage leukemia cells by a potent small‐molecule DOT1L inhibitor. Cancer Cell 2011; 20: 53–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Klaus CR, Iwanowicz D, Johnston D et al DOT1L inhibitor EPZ‐5676 displays synergistic antiproliferative activity in combination with standard of care drugs and hypomethylating agents in MLL‐rearranged leukemia cells. J Pharmacol Exp Ther 2014; 350: 646–56. [DOI] [PubMed] [Google Scholar]
- 54. Yoshimatsu M, Toyokawa G, Hayami S et al Dysregulation of PRMT1 and PRMT6, Type I arginine methyltransferases, is involved in various types of human cancers. Int J Cancer 2011; 128: 562–73. [DOI] [PubMed] [Google Scholar]
- 55. Deng X, Von Keudell G, Suzuki T et al PRMT1 promotes mitosis of cancer cells through arginine methylation of INCENP. Oncotarget 2015; 6: 35173–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Xie Y, Zhou R, Lian F et al Virtual screening and biological evaluation of novel small molecular inhibitors against protein arginine methyltransferase 1 (PRMT1). Org Biomol Chem 2014; 12: 9665–73. [DOI] [PubMed] [Google Scholar]
- 57. Chan‐Penebre E, Kuplast KG, Majer CR et al A selective inhibitor of PRMT5 with in vivo and in vitro potency in MCL models. Nat Chem Biol 2015; 11: 432–7. [DOI] [PubMed] [Google Scholar]
- 58. Stein C, Riedl S, Ruthnick D, Notzold RR, Bauer UM. The arginine methyltransferase PRMT6 regulates cell proliferation and senescence through transcriptional repression of tumor suppressor genes. Nucleic Acids Res 2012; 40: 9522–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Mitchell LH, Drew AE, Ribich SA et al Aryl pyrazoles as potent inhibitors of arginine methyltransferases: identification of the first PRMT6 tool compound. ACS Med Chem Lett 2015; 6: 655–9. [DOI] [PMC free article] [PubMed] [Google Scholar]