

Abstract
Context:
Bioavailability of conventional tablet of erythromycin stearate is low as it is unstable at acidic pH and also shows a low dissolution rate.
Objective:
It was proposed to protect it from the acidic condition of the stomach along with an increase in dissolution rate by formulating pH sensitive nanoparticles.
Materials and Methods:
The nanoparticles were prepared by the solvent evaporation technique using different quantities of Eudragit L100-55 and polyvinyl alcohol (PVA). Size reduction was achieved by high speed homogenization technique using Digital Ultra Turrax homogenizer. The formulation was optimized using 32 factorial design, keeping drug polymer ratio and surfactant concentration as independent variables. Particle size, entrapment efficiency, and drug-release (DR) were studied as dependent variables.
Results:
Optimized batch containing 1:0.3 erythromycin stearate: Eudragit L100-55 ratio and 1.0% PVA showed 8.24 ± 0.71% DR in pH 1.2 in 1-h and 90.38 ± 5.97% in pH 5.5 and pH 6.8 within 2-h, respectively.
Discussion:
The optimized batch exhibited lower release in acidic pH and faster release in higher pH compared to the marketed preparation.
Conclusion:
Thus the present study concludes that pH sensitive nanoparticles of erythromycin stearate increases the dissolution of the drug in intestinal pH and also protect it from acidic pH, which may help in improving the bioavailability of erythromycin.
KEY WORDS: Eudragit, factorial design, high-shear, optimization, polyvinyl alcohol
Erythromycin stearate is used in the treatment of infections caused by the susceptible strains of microorganisms such as respiratory tract infections, pertussis, in the treatment of infections caused by Corynebacterium minutissimum.[1] It has low bioavailability due to the low solubility and inactivation of the drug by gastric acid.[2] Protection from the acidic environment of the stomach by enteric coating of the tablet is predicted to improve the bioavailability of erythromycin.[3] But erythromycin stearate has not been formulated with an acid resistant coating because stearate derivative is less water soluble than erythromycin base.[2] Bioavailability studies performed by DiSanto and Chodos demonstrated that the film-coated preparations of erythromycin stearate were 43–59% less well absorbed than the enteric coated base formulation. The dissolution specification of erythromycin stearate tablet (USP) states that NLT 75% drug should dissolve in 2-h in phosphate buffer pH 6.8 at 100 rpm.[4] Therefore, an increase in dissolution rate of erythromycin stearate along with the enteric coating may help in improving its bioavailability. A study conducted by Somogyi et al.[5] also proves that improving solubility increases the bioavailability of erythromycin base. The current formulation that is, pH sensitive nanoparticles offer protection of drug in the acidic pH[6] of the stomach and being nano size also improves the dissolution rate of erythromycin stearate.
Nanoparticles are solid particles ranging in size from 10 nm to 1000 nm. They consist of macromolecular materials in which the active principle drug or biologically active material is dissolved, entrapped or encapsulated, and or to which the active material is adsorbed or attached.[7] Nanotechnology is leading to useful advances in medical diagnosis and treatment and has a considerable commercial potential out with medicine.[8] Materials of nano size have been promoted as a revolutionary technology for cell and tissue engineering, medical device development, and the encapsulation and delivery of drugs, diagnostics, and genes. The drug is loaded in the form of dissolved, entrapped, encapsulated or attached to nanoparticle matrix. There are many advantages of nanoparticulate drug delivery system including the fact that the particle and surface characteristics of nanoparticles can be easily manipulated to achieve both passive and active drug targeting after the parenteral administration. Site specific targeting can be achieved by attaching targeting ligands on the surface of particles or use of magnetic guidance. The system can be used for drug delivery by various routes of administration including oral, nasal, parenteral, intra-ocular, etc.[9]
Nanoparticles are prepared from a variety of materials such as proteins, polysaccharides, and synthetic biodegradable polymers. Polymer selection may depend on size of desired nanoparticles, properties of the drug (solubility, stability) to be encapsulated, surface character and functionality, and the desired drug-release (DR) profile from the final product.
pH sensitive polymeric nanoparticles are nanoparticles where the pH sensitive polymer is used for the entrapment of drug. The basic purpose of preparing such nanoparticles remains same that is, to increase the solubility and bioavailability of drug along with the protection from the acidic condition of the stomach. Most carriers have been used as enteric-coating for a long time and their safety has been approved. The carriers dissolve rapidly at specific pH and specific sites, which result in quick release and high drug concentration gradient, this phenomenon is important for drug absorption. Nanoparticles are an effective platform for delivery of hydrophobic and hydrophilic drugs since the drugs are protected from possible degradation due to the harsh microenvironment.[10,11,12,13,14]
Materials and Methods
Materials
Erythromycin stearate was a gift sample from Swiss Pharma Pvt., Ltd., Ahmedabad, India. Eudragit L100-55 was procured by Vikram-Thermo (India) Ltd., Ahmedabad. Polyvinyl alcohol (PVA) (partially hydrolyzed), lactose, and sodium hydroxide were purchased from Sulab laboratories, Vadodara, India. Solvents such as ethanol and methanol were of analytical grade and obtained from by Fishers scientific Pvt. Ltd., Mumbai, India.
Method of preparation
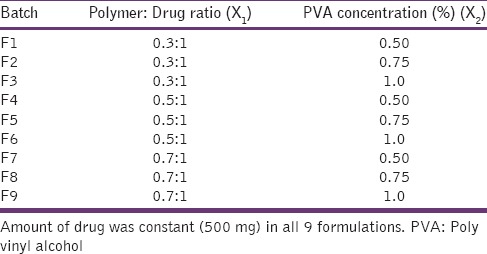
Nanoparticles were prepared by the solvent evaporation technique.[15] Briefly, weighed the quantity of drug and polymer of different ratio [Table 1] were dissolved in 5 ml ethanol. The solution was added slowly using syringe fitted with 22 gauge needle, to 50 ml aqueous solution of PVA under homogenization using Digital Ultra Turrax homogenizer (Germany) at 15000 rpm for 15 min. This suspension was continuously stirred on a magnetic stirrer for 5–6 h to remove the alcohol.
Table 1.
Composition of batch F1-F9
Optimization of formulation
Optimization of the formulation was carried out to study the effect of formulation variables on the product characteristics and get an optimum composition for the nanoparticles showing desired properties (i.e., the CQAs or Critical Quality Attributes). CQAs of pH sensitive nanoparticles were decided to be the particle size, entrapment efficiency (EE), and release profile. Critical formulation parameters for such nanoparticles were identified to be the polymer (Eudragit L100-55) fraction and concentration of surfactant (PVA). Hence, they were chosen as independent variables for optimization studies. It was decided to perform the optimization studies at 3 levels of these 2 variables. Nine batches [Table 1] were planned as per 32 factorial designs. Preliminary studies showed that the suspension prepared using polymer: drug ratio 1.5:1 was in the micrometric size range. Therefore, the polymer: drug ratio was restricted to not more than 1:1. The approved limit of PVA is 1.4% according to IIG database of USFDA,[16] thus it was not used above 1.5% in the present work. Process parameters were kept constant at homogenization speed 15000 rpm for time 15-min.
Experimental trials were performed for all 9 combinations. Mean particle size (Y1), percentage EE (Y2), and in-vitro DR (Y3) were selected as dependent variables. Statistical treatment of dependent variables was carried out on the experimental data obtained for optimization batches using design expert DX7 Stat-Ease software.
Evaluation Parameters for Optimization
Particle size and size distribution
Particle size (diameter) of different formulations were measured with the help of Malvern Zetasizer.[17] The size distribution was studied in terms of polydispersity index (PDI).
Percentage entrapment efficiency
The nanoparticles were washed with ethyl acetate and the drug content in the residual nanoparticles was determined by solubilizing the nanoparticles in ethanol. Concentration of drug in the solution was analyzed spectrophotometrically at 482 nm, after treating with 1 mL conc. sulfuric acid for 30 min. and suitable dilution.[18,19] Percentage EE of the nanoparticles was calculated by the following equation:
%EE=(ED/TD)×100
Where, ED = Amount of drug in nanoparticles; TD = Total drug taken.
In-vitro drug release
In-vitro DR studies were carried out in modified USP type 2 dissolution test apparatus by tying a Hi-Media dialysis membrane 110 (molecular weight cut-off 12,000–14,000 kDa) bag containing 1 ml formulation to the shaft. The shaft was immersed in 900 ml dissolution medium at dissolution apparatus was operated at 100 rpm at 37°C ± 0.5°C.[20] Sample aliquots (5 ml each) were withdrawn at different predetermined time intervals. Required treatment and dilutions were made with dissolution medium and the solution was analyzed spectrophotometrically, at 482 nm against a suitable blank. The same volume of dissolution medium was replaced in the vessel after each withdrawal to maintain sink condition. Dissolution medium used were 0.1 N hydrochloric acid for initial 2-h, phosphate buffer pH 5.5 for next 30 min, and phosphate buffer pH 6.8 for remaining 1.5-h as suggested by Crison for study of release profile of modified release formulation.[21] For dissolution profile of nanoparticles result of percentage, DR was plotted against the time to analyze release pattern.
In-vitro drug release kinetic
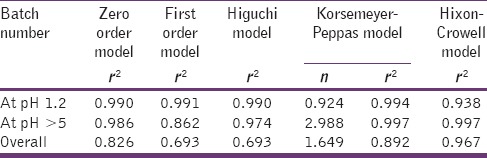
In order to understand the kinetic and mechanism of DR, the result of in-vitro DR study of optimized batch was fitted into various kinetic equations such as zero order, first order Higuchi's model, Korsmeyer–Peppas equation, and Hixson–Crowell equation. Regression coefficient (r2) and n-value were calculated for the linear curve obtained by regression analysis of the plots.[22,23]
Comparison with marketed preparation
The optimized nanoparticulate formulation was compared with the marketed formulation (enteric coated tablet, 250 mg) in terms of in-vitro DR study. The release study of each formulation was carried out for 6 times. The release was calculated in percentage DR and reported as a mean ± standard deviation.
Drug content
Drug content of the optimized nanoparticles was determined in order to determine the amount of formulation needed to deliver the required dose of erythromycin stearate. Procedure for estimation of drug content was similar to that followed in determining percentage EE. Briefly, the amount of entrapped drug was determined and the drug content was calculated using the equation:
%Drug content=(Entrapped drug/Total weight on nanoparticle)×100
Zeta potential
Zeta potential of the optimized nano-suspension was measured for predicting the physical stability of the formulation. The nano-suspension containing 10 mg/mL erythromycin was inserted into the cuvette to measure zeta potential using Malvern Zeta-sizer at 25 ± 0.5°C.
Result and Discussion
Mean particle size
Wide differences in particle size of different formulations were found due to the change in composition. The nanoparticles formed by different formulation combination were ranging between 500 and 2556 nm [Table 2]. With an increase in the concentration of stabilizer, a decrease in particle size was observed. It is evident from the particle size analysis of different batches that particle size of the formulation is a contribution of both energy inputs by the ultra turrax as well as the formulation composition, specially the surfactant concentration. Formulation F6 containing 1:0.5 drug: polymer and 1% PVA showed lowest particle size 540.0 nm whereas the formulation F7 contain 1:0.7 drug: polymer and 0.5% PVA shows highest particle size 2556 nm. With the increase in polymer fraction, the mean particle size decreases but with further increase in polymer fraction, an increase in particle size was observed. This may be due to the increasing in viscosity of dispersion due to high polymer concentration, which may reduce the size reduction potential of the homogenizer.
Table 2.
Characterization of batch F1-F9
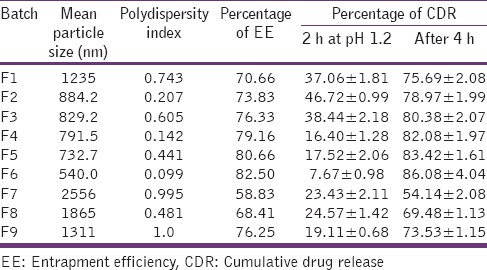
PDI <0.5 is considered to be good for any dispersion. It is observed that the particle size distribution range is wide in high polymer concentration. This may be the nonuniform distribution of energy due to high viscosity.
Statistical analysis was carried out using Microsoft Excel 2007 and Design Expert (DX7) software (Stat-Ease, Inc. Minneapolis, USA). Surface quadratic model was suggested by DX7 software, which indicates that both polymer fraction and surfactant concentration have a significant effect on particle size. This is also evident from their P values [Table 3].
Table 3.
Summary of multiple regression analysis for particle size and EE
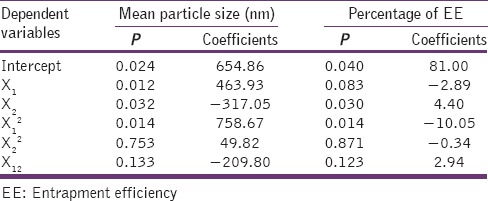
Mean particle size = 654.86 + 463.93 × polymer fraction − 317.05 × concentration of PVA − 209.80 × polymer fraction2
Positive sign of coefficient of polymer fraction (X1) alone in regression equation indicates that the particle size increases as the polymer fraction increases, similar to the effect of polymer concentration on particle size of nanoparticles reported by many researchers such as Mainardes and Evangelista[24] and Quintanar-Guerrero et al.[25] But polymer fraction does not show the linear effect on particle size. As expressed in the above equation, the effect of polymer fraction on mean particle size is the cumulation of the positive effect of a linear term and the negative effect of the square term. Whereas, the concentration of PVA (X2) showed a negative impact on particle size, that is, the particle size decreases with increase in surfactant conc. as the surfactant reduces the inter-particulate attraction during precipitation. This was similar to the effect of stabilizer reported by Matteucci et al.[26] Cold water soluble PVA is partially hydrolyzed form. The residual vinyl acetate groups from partial hydrolysis give the PVA amphiphilic character, as the vinyl acetate groups interact favorably with oils and the vinyl alcohol groups interact favorably with water. In addition to the physical adsorption, PVA can become covalently grafted to the surface of polymer particles during the dispersion polymerization. It is expected that the particles with adsorbed or grafted PVA will display the significantly enhanced colloidal stability compared to electrostatically stabilized particles.[27]
Entrapment efficiency
Formulations were rinsed with ethyl acetate to wash off the un-entrapped drug because the drug is soluble in ethyl acetate while the polymer is insoluble. Thus, the un-entrapped drug was solubilized in ethyl acetate and removed by filtration. The nanoparticulate residue was then dissolved in methanol, which solubilizes the polymeric matrix as well as a drug.
EE of the optimization batches was found to be in between 55 and 85%. Formulation F6 showed the highest percentage EE, whereas the formulation F7 yields nanoparticles with the lowest percentage EE [Table 2]. The percentage EE increases with the polymer fraction and concentration of PVA.
Two-factor interaction model was predicted to be applicable on the effect of independent factors on EE by design expert software (DX7). The P value, it can be concluded that drug to the polymer ratio, concentration of PVA and interaction of these two factors have a significant effect (P < 0.05) on the EE [Table 3].
%EE = 88.28 + 0.82 × Polymer fraction + 2.47 × concentration of PVA + 0.67 × polymer fraction × concentration of PVA
All these significant factors showed a positive effect on percentage EE, but the concentration of PVA showed the greater effect on EE. The increase in EE with an increase in PVA concentration may be due to the fact that viscosity of PVA solution reduces diffusion of ethanol, thereby reducing the mobility of drug into the aqueous phase. Also, the positive interaction of independent factors was observed on EE.
Percentage in-vitro drug release
In-vitro drug release study was carried out to study the percentage in-vitro release profile of the drug in a different medium. Minimum (5–10%) DR was observed in pH 1.2 (acidic pH) which indicates the drug was protected from pH 1.2 to maximum (75–91%) DR was in the pH range of intestine. All formulation showed <10% of DR in pH 1.2 in 120-min and more than 80% DR was observed at pH 5.5 and pH 6.8 within 120-min [Table 2]. The drastic increase in-vitro DR in pH 5.5 and pH 6.8 may be due to the presence of pH-sensitive polymer and also due to the decrease in particle size. Polymer protects the drug from the acidic condition and decreases in particle size leads to increase in dissolution of the drug.
As the concentration of PVA increases, percentage in-vitro DR increases. This may be due to the fact that PVA, being hydrophilic in nature, assisted in the dissolution of the insoluble drug. A slight increase in release rate was observed with initial increase in polymer fraction. But, the increasing polymer fraction after a point resulted in decrease in percentage of in-vitro DR, which is an expected phenomenon that the release rate decreases with an increase in polymer density in the matrix. This factor is further accompanied by the fact that with the increase in polymer fraction leads to the decrease in concentration of PVA at the surface of nanoparticle to assist dissolution.
% In-vitro DR (after 4 h) = 85.18 − 6.31 × Polymer fraction + 4.68 × Concentration of PVA (X2) − 1.97× (concentration of PVA)2
From the P value [Table 4], it can be concluded that the polymer fraction (X1) and the concentration of PVA (X2) have prominent effect (P < 0.05) on in-vitro release. Polymer fraction showed a negative effect on percentage DR, that is, the percentage DR decreased with polymer concentration. Whereas, the percentage DR increased with the concentration of PVA.
Table 4.
Summary of multiple regression analysis for in-vitro drug release
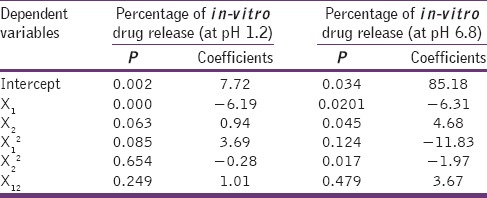
Thus, it can be concluded that the nanoparticles made up of Eudragit remained as drug-reservoir in a gastric environment having acidic pH in which the polymer is insoluble, releasing the drug slowly. These nanoparticles are converted into nano-suspension in the intestinal pH (pH >5) where the polymer is soluble, thereby increasing the dissolution rate.
Characterization of optimized formulation
By overlaying the required response factors, the DoE software Stat-Ease Inc., Minneapolis, USA indicated that composition is containing polymer: drug weight-ratio 0.3:1 and 1% surfactant concentration is the optimized batch. Responses of dependent variables were predicted using DoE software. The optimized batch was prepared and evaluated for the characteristic properties. Particle size and EE of the optimized formulation were 513.0 nm with PDI 0.102, and 80.33%, respectively. This optimized batch was also evaluated for the drug content, zeta potential, and release profile [Table 5].
Table 5.
Characterization of optimized batch

Zeta potential is a measure of the charge of particles as such as larger the value of the zeta potential large the amount of charge on the surface. It represents an index for particle stability. In case of charged particles, as the zeta potential increases, the repulsive interactions are larger leading to the formulation of more stable particles with more uniform size distribution. Hans and Lowman[9] suggested that the high value of zeta potential indicates the electrostatic repulsion between particles, zeta potential between ± 30 mV show good physical stability. Zeta potential of optimized formulation, measured using Malvern Zeta-sizer was found to be 22.4 mV. Thus, it can be predicted that the electrostatic repulsion between the particles will help in preventing their aggregation, indicating a physically stable suspension formulation. Though, it is a fact that the more is the charge, more is the physical stability. But the more is the electronegativity, more will be the irritation to the biological membranes is caused.[28] So the charge −22 mV can be considered acceptable. The physical stability can be increased by other techniques also, like lyophilization.
As desired, the optimized formulation released <10% in acidic pH and >90% in alkaline pH. The release kinetics data [Table 6] indicates that in the first 1 h in acidic medium, the nanoparticles followed anomalous release mechanism. Whereas, in alkaline medium, the pH sensitive nanoparticles showed dissolution based release profile, that is, Hixon–Crowell kinetics (r2 = 0.997). The overall release of drug from optimized batch follows Hixon–Crowell model, having correlation coefficient 0.967. This proves that once in a basic medium, the polymer dissolves completely and the dissolution rate is guided by the size of the nanoparticle.
Table 6.
Release kinetic of optimized batch
Comparison with marketed product
The marketed tablet of erythromycin stearate released <60% drug in a period of 4-h, out of which approximately >15% drug was released at pH 1.2 in first 1-h [Figure 1]. Whereas, the formulated pH-sensitive polymeric nanoparticles showed maximum DR at pH 5.5 and 6.8 and <10% DR in pH 1.2 (acidic pH), which is as per the desired criteria of enteric coating.[29,30] This fulfills the objective of the present research work to protect the drug from acidic pH and maximum release in the intestinal pH.
Figure 1.
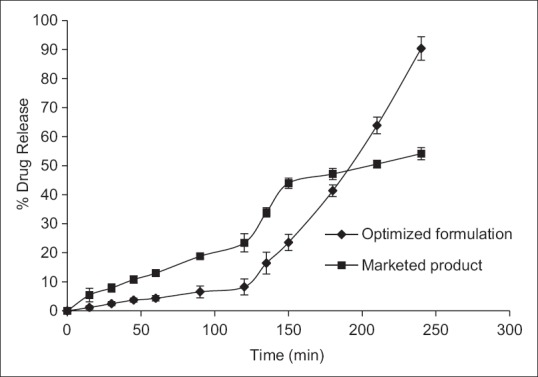
Comparison of in-vitro release profile with marketed product (release reported as mean ± standard deviation, n = 6)
Drug content of the formulation is approximately 50% [Table 5]. This suggests that to administer the usual dose of 250 mg erythromycin stearate, 500 mg nanoparticle is needed to be taken. Additionally, the release profile shows that approximately 30% increase in percentage cumulative DR can be achieved by the nanoparticles. This may further help to reduce the dose if the in-vivo increase in bioavailability could be proved.
Conclusion
Erythromycin stearate has low bioavailability due to low solubility and inactivation of the drug by gastric acid. Moreover, erythromycin stearate has narrow absorption window. Thus, the present study concludes that pH sensitive nanoparticles of erythromycin stearate increases the dissolution of the drug in intestinal pH and also protect it from acidic pH, which may help in improving the bioavailability of erythromycin. This was confirmed from the analysis of optimized batch, which exhibited <10% release in acidic pH and faster release in higher pH compared to the marketed preparation. Further in-vivo studies are needed to be done to reflect the correlation of in-vitro results in the in-vivo performance of the formulation.
Financial support and sponsorship
This research work was solely conceptualized, funded and conducted in Parul Institute of Pharmacy.
Conflicts of interest
There are no conflicts of interest.
References
- 1.Schlossberg D, Samuel R. Antibiotics Manual. USA: People's Medical Publishing House; 2010. p. 182. [Google Scholar]
- 2.DiSanto AR, Chodos DJ. Influence of study design in assessing food effects on absorption of erythromycin base and erythromycin stearate. Antimicrob Agents Chemother. 1981;20:190–6. doi: 10.1128/aac.20.2.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Piscitelli SC, Rodvold KA, Pai MP, editors. Drug Interactions in Infectious Diseases. 3rd ed. London: Humana Press, Springer Science and Business Media; 2011. p. 246. [Google Scholar]
- 4.32nd ed. Rockville: United States Pharmacopeial Convention; 2009. Monograph of erythromycin stearate tablets. United States Pharmacopoeia; p. 2297. [Google Scholar]
- 5.Somogyi AA, Bochner F, Hetzel D, Williams DB. Evaluation of the intestinal absorption of erythromycin in man: Absolute bioavailability and comparison with enteric coated erythromycin. Pharm Res. 1995;12:149–54. doi: 10.1023/a:1016215510223. [DOI] [PubMed] [Google Scholar]
- 6.Wang XQ, Zhang Q. pH-sensitive polymeric nanoparticles to improve oral bioavailability of peptide/protein drugs and poorly water-soluble drugs. Eur J Pharm Biopharm. 2012;82:219–29. doi: 10.1016/j.ejpb.2012.07.014. [DOI] [PubMed] [Google Scholar]
- 7.Kreuter J. Nanoparticles and microparticles for drug and vaccine delivery. J Anat. 1996;189(Pt 3):503–5. [PMC free article] [PubMed] [Google Scholar]
- 8.Moghimi SM, Hunter AC, Murray JC. Nanomedicine: Current status and future prospects. FASEB J. 2005;19:311–30. doi: 10.1096/fj.04-2747rev. [DOI] [PubMed] [Google Scholar]
- 9.Kulshreshtha AK, Singh ON, Wall GM. New York: Springer Science and Business Media; 2009. Pharmaceutical Suspensions: From Formulation Development to Manufacturing; pp. 24–5. [Google Scholar]
- 10.Hans ML, Lowman AM. Biodegradable nanoparticles for drug delivery and targeting. Curr Opin Solid State Mater Sci. 2002;6:319–27. [Google Scholar]
- 11.Felber AE, Dufresne MH, Leroux JC. pH-sensitive vesicles, polymeric micelles, and nanospheres prepared with polycarboxylates. Adv Drug Deliv Rev. 2012;64:979–92. doi: 10.1016/j.addr.2011.09.006. [DOI] [PubMed] [Google Scholar]
- 12.Alishahi A, Mirvaghefi A, Tehrani MR, Farahmand H, Koshio S, Dorkoosh FA, et al. Chitosan nanoparticle to carry vitamin C through the gastrointestinal tract and induce the non-specific immunity system of rainbow trout (Oncorhynchus mykiss) Carbohydr Polym. 2011;86:142–6. [Google Scholar]
- 13.des Rieux A, Fievez V, Garinot M, Schneider YJ, Préat V. Nanoparticles as potential oral delivery systems of proteins and vaccines: A mechanistic approach. J Control Release. 2006;116:1–27. doi: 10.1016/j.jconrel.2006.08.013. [DOI] [PubMed] [Google Scholar]
- 14.Wu TH, Yen FL, Lin LT, Tsai TR, Lin CC, Cham TM. Preparation, physicochemical characterization, and antioxidant effects of quercetin nanoparticles. Int J Pharm. 2008;346:160–8. doi: 10.1016/j.ijpharm.2007.06.036. [DOI] [PubMed] [Google Scholar]
- 15.Rao JP, Geckeler KE. Polymer nanoparticles: Preparation techniques and size control parameters. Prog Polym Sci. 2011;36:887–913. [Google Scholar]
- 16.Inactive Ingredient ‘Polyvinyl Alcohol’. [Last accessed on 2015 Jan]. Available from: http://www.accessdata.fda.gov/scripts/cder/iig/getiigWEB.cfm .
- 17.Vaculikova E, Grunwaldova V, Kral V, Dohnal J, Jampilek J. Preparation of candesartan and atorvastatin nanoparticles by solvent evaporation. Molecules. 2012;17:13221–34. doi: 10.3390/molecules171113221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wankhade R, Bhalerao S, Panchory H, Pundir A, Pradhan R. Analysis of erythromycin and benzoyl peroxide in combined dosage form by UV-visible spectrophotometry. Int J Pharm Pharm Sci. 2012;4:527–31. [Google Scholar]
- 19.Danielson ND, Holeman JA, Bristol DC, Kirzner DH. Simple methods for the qualitative identification and quantitative determination of macrolide antibiotics. J Pharm Biomed Anal. 1993;11:121–30. doi: 10.1016/0731-7085(93)80132-k. [DOI] [PubMed] [Google Scholar]
- 20.Costa P, Sousa Lobo JM. Modeling and comparison of dissolution profiles. Eur J Pharm Sci. 2001;13:123–33. doi: 10.1016/s0928-0987(01)00095-1. [DOI] [PubMed] [Google Scholar]
- 21.Crison JR. Developing dissolution test for modified release dosage forms: General consideration. Dissolution Technologies. 1999. [Last cited on 2015 Aug 18]. p. 6. dx.doi.org/10.14227/DT060199P5. Available from: http://www.dissolutiontech.com/DTresour/299articles/299Crison.htm .
- 22.Pagar K, Vavia P. Rivastigmine-loaded L-lactide-depsipeptide polymeric nanoparticles: Decisive formulation variable optimization. Sci Pharm. 2013;81:865–85. doi: 10.3797/scipharm.1211-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dash S, Murthy PN, Nath L, Chowdhury P. Kinetic modeling on drug release from controlled drug delivery systems. Acta Pol Pharm. 2010;67:217–23. [PubMed] [Google Scholar]
- 24.Mainardes RM, Evangelista RC. PLGA nanoparticles containing praziquantel: Effect of formulation variables on size distribution. Int J Pharm. 2005;290:137–44. doi: 10.1016/j.ijpharm.2004.11.027. [DOI] [PubMed] [Google Scholar]
- 25.Quintanar-Guerrero D, Fessi H, Allemann E, Doelker E. Influence of stabilizing agents and preparatives variables on the formation of poly (D, L-lactic acid) nanoparticles by an emulsification-diffusion technique. Int J Pharm. 1996;143:133–41. [Google Scholar]
- 26.Matteucci ME, Hotze MA, Johnston KP, Williams RO., 3rd Drug nanoparticles by antisolvent precipitation: Mixing energy versus surfactant stabilization. Langmuir. 2006;22:8951–9. doi: 10.1021/la061122t. [DOI] [PubMed] [Google Scholar]
- 27.Lee A, Tsai HY, Yates MZ. Steric stabilization of thermally responsive N-isopropylacrylamide particles by poly (vinyl alcohol) Langmuir. 2010;26:18055–60. doi: 10.1021/la1039128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Clogston JD, Patri AK. Zeta potential measurement. Methods Mol Biol. 2011;697:63–70. doi: 10.1007/978-1-60327-198-1_6. [DOI] [PubMed] [Google Scholar]
- 29.Zhao H, Cafiero S, Williams Z, Bynum KC. Practical considerations for the development of a robust two-step dissolution test for enteric-coated immediate- and extended-release solid oral dosage formulations. Dissolution Technol. 2011;18:6–10. [Google Scholar]
- 30.Sharma M, Sharma V, Panda AK, Majumdar DK. Development of enteric submicron particle formulation of papain for oral delivery. Int J Nanomedicine. 2011;6:2097–111. doi: 10.2147/IJN.S23985. [DOI] [PMC free article] [PubMed] [Google Scholar]