

Abstract
Reactions of indene and various substituted indoles with [(diimine)PtII(Me)(TFE)]+ cations have been studied (diimine = ArN = C(Me) − C(Me) = NAr; TFE = 2,2,2-trifluoroethanol). Indene displaces the TFE ligand from platinum to form a stable π coordination complex that, upon heating, undergoes C-H activation with first order kinetics, ΔH‡ = 29 kcal/mol, ΔS‡ = 10 eu, and a kinetic isotope effect of 1.1 at 60 °C. Indoles also initially form coordination complexes through the C2=C3 olefin, but these undergo rearrangement to the corresponding N-bound complexes. The relative rates of initial coordination and rearrangement are affected by excess acid or methyl substitution on indole.
Introduction
Selective C-H bond activation is a potentially valuable approach to synthetic problems in areas ranging from fuels and bulk chemicals to fine chemicals and pharmaceutical synthesis.1 Studies of C-H activation in our laboratory have focused on models of the Shilov system,2 particularly [(diimine)PtII(Me)(solv)]+ (2, diimine = ArN = C(Me) − C(Me) = NAr; solv = 2,2,2-trifluoroethanol (TFE), H2O).3 These cations are capable of activating a variety of carbon-hydrogen bonds.4 Cations 2 can be generated by protonolysis of (diimine)PtIIMe2 species 1 in TFE with aqueous HBF43, 4ab or BX3 (X = C6F5,4cd F5), the latter producing H+ by boron coordination to TFE.4c In deuterated solvent 2 is formed as a mixture of two isotopologs. Reaction of 2 with a C-H group then results in liberation of methane as a mixture of CH4 and CH3D along with the formation of a new alkyl or aryl platinum(II) complex [(diimine)PtII(R)(solv)]+ (Scheme 1).6
Scheme 1.

The presence of donor heteroatoms (N, O, S, etc.) in the substrate may inhibit C-H activation because these heteroatoms tend to bind tightly to electrophilic metal centers and block C-H bond coordination. In the cases of methanol and dimethyl ether,4e stable complexes of the organic substrate with platinum are observed, but kinetics studies indicate that substrate-platinum binding is rapid and reversible, and that re-binding (or rearrangement) of the substrate to a C-H bond-coordinated intermediate is rate-determining. Heteroatom binding can be used to advantage, however, in coordinatively directed cyclometallation reactions,7 the basis of many synthetic methods. Several recent examples of these involving nitrogen-containing heterocycles have appeared.8 Specifically regarding indole, a functionality found frequently in natural products and pharmaceutical agents, several groups have identified conditions for cyclization of indoles with pendant olefins,9 and both Sames10 and Sanford11 have reported selective arylation of simple indoles via palladium-catalyzed C-H activation.
Prior literature reports of stable group 10 metal-indole complexes are few in number. Platinum-coordinated tryptophan and tryptamine derivatives have been studied,12 but only a single platinum complex, cis-Pt(indole)2Cl2, having a neutral, simple L-type donor indole ligand13 has been reported. Unfortunately, it was characterized only by elemental analysis and infrared, and neither offers insight into the binding of the indole ligand.14 Moreover, although (1-indolyl)metal amides (functioning as X-type ligands) are common,15 and indole-metal arene complexes have been described for several transition metals,16 we are aware of no examples of transition metal complexes having indole C2=C3 olefin ligation.
The present work examines the reactions of indoles with cations 2. Towards this end, we describe the reaction of these cations with indene, indole’s hydrocarbon analog, as well as with various methyl-substituted indoles. We find that indene forms a stable olefin coordination complex that undergoes C-H activation upon heating. Analogously, indoles initially form olefin coordination complexes, but rather than undergoing C-H activation, these complexes rearrange to stable N-bound 3H-indole species. We expect that C-H activation of nitrogen-containing unsaturated heterocycles is more likely to proceed from a metal-heterocycle π complex than an N-complex, so strategies to extend the lifetime of platinum(II)-indoleπ complexes have been pursued. We find that the rate of rearrangement of indole π complexes to N-complexes can be diminished by introducing steric bulk on the indole or by increasing [H+].
Results and Discussion
Reactions of Indene with Cations 2
Treatment of platinum cations 2 with one equivalent of indene results in the formation of stable olefin complexes, 3 (Scheme 2). In each case, complexation is efficient and rapid. Exchange of coordinated indene with 1 equivalent (15 mM) of added indene-d3 under the same conditions is slower, with kobs ~ 5 × 10−3 s−1, t1/2 ~ 1 min (from McKay analysis).17 The resulting products 3 can be characterized by NMR and electrospray MS methods. The 1H NMR spectrum for 3a is sharp at ambient temperature, and 195Pt-H coupling constants of 74 and 79 Hz are observed for the C2 and C3 protons of indene. The C1 methylene protons appear as a pair of doublets (2JH-H = 23 Hz) with weak 195Pt side bands (JPt-H = 41 Hz). By contrast, ambient temperature 1H NMR spectra of 3b, 3c, and 3d give broad signals for the C1, C2, and C3 indenyl C—H protons near 3.2, 6.5, and 5.6 ppm, respectively. In all cases, Pt-CH3 signals were shifted significantly upfield from the parent cation 2: whereas 2 typically displays δ(Pt-CH3) in the range 0.6 – 1.3 ppm, indene complexes 3 have δ(Pt-CH3) between −0.8 and −0.3 ppm. This shift is similar to that for a previously-reported η2-benzene adduct, which has δ(Pt-CH3) = −1.3 ppm at −33 °C.4a Upon gentle heating, complexes 3 react to form η3-indenyl complex 4 and methane (Scheme 2). In the case of 3a, the reaction is clean (> 95% yield) as determined by 1H NMR. By contrast, heating complexes 3b and 3c results in formation of a complex mixture of products, including 4.
Scheme 2.

Mechanism of Indene Activation
First-order rate constants were measured for the conversion of indene complex 3a to indenyl complex 4a (Table 1). Importantly, two trials at 60 °C with 1 and 5 equivalents of indene gave the same rate constants (1.24 (9) × 10−4 s−1 and 1.37 (9) × 10−4 s−1) within error, indicating a zero order rate dependence on [indene]. Activation parameters for the conversion of 3a to 4a, measured by Eyring analysis for kinetic runs conducted at temperatures ranging from 50 to 90 °C (Figure 1), are ΔH‡ = 28.7 ± 0.7 kcal·mol−1 and ΔS‡ = 9.7 ± 2.2 e.u.18 A kinetic isotope effect was determined for the conversion indene-d3 to deuterated indenyl complex 4a–d2 (Scheme 3). 1,1,3-Trideuteroindene was prepared according to a previously reported procedure19 and was complexed to 2a as above. Conversion of 3a-d3 to 4a-d2 proceeded smoothly to afford a selectively deuterated product. Comparisons of measured rate constants for parallel runs gave a kinetic isotope effect of 1.1.
Table 1.
First-order rate constants for the conversion of indene π complex 3a to η3-indenyl complex 4a.
| Entry | T (°C) | k (s−1) | t(1/2) (min) |
|---|---|---|---|
| 1 | 50 | 3.38 (11) × 10−5 | 342(11) |
| 2 | 60 | 1.31 (9) × 10−4 | 88.5(58) |
| 3 | 70 | 4.60 (18) × 10−4 | 25.1(10) |
| 4 | 80 | 1.84 (10) × 10−3 | 6.28(32) |
| 5 | 90 | 5.10 (10) × 10−3 | 2.28(38) |
Scheme 3.

The small, positive value for ΔS‡ and the small KIE are suggestive of an intramolecular 3a-to-4a process not involving rate-determining C-H oxidative addition. A mechanistic scenario that is in accord with these features involves rate-determining rearrangement of π complex 3a to C-H σ complex 5, followed by more rapid C-H oxidative cleavage and reductive elimination of methane (Scheme 4). Analogous reaction of 2e (solv = CF3CD2OD) with p-xylene was found to afford a mixture of products resulting from competitive aryl (kH/kD ≈ 4) and benzylic (kH/kD ≈ 1) C-H activation processes, the latter affording an η3-benzylic product. 4c Rearrangement of 3a to 4a thus resembles the benzylic activation process, and suggests that benzylic C-H bond activation by 2e also proceeds via an initially formed arene π complex that undergoes rate-determining rearrangement to a C-H σ complex, followed by C-H oxidative cleavage.
Scheme 4.

Reactions of Cations 2 with Indoles
Indole reacts rapidly with platinum cations 2, ultimately to give N-complexation products 8 (Scheme 5). Analogously to reaction with indene, the initially formed adduct is a C2=C3 π complex 6; however, N-ligation, likely via transient 7, inhibits C-H activation, and tautomer 8 is the stable adduct that is generated.20 Reaction of 2a with indole at 40 °C begins with a buildup of 6a-d2 (1H NMR), which converts to 8a in minutes. H/D exchange of N1-H with solvent is rapid relative to complexation, but exchange of C3-H may occur before or after π complexation.21 Although the participation of platinum in the H/D exchange cannot be excluded, this exchange is rapid in the presence of 0.2 mole percent BF3-TFE solution (not containing 1a) and is complete in minutes at room temperature. NMR spectra are consistent with assignment of the 3D tautomer 8: in all three complexes (8a, 8b, 8c) the 13C signal for the C3 methylene carbon appears as a multiplet at ~44 ppm, and the 1H signal for the C2 hydrogen appears as a singlet with 195Pt satellites (JPt-H ~ 40 Hz) at ~ 8 ppm.
Scheme 5.

Reactions of methyl-substituted indoles were studied with cation 2a. Reactions of 2a with 1-methylindole and 2-methylindole proceed through the intermediacy of observable and more stable π complexes (9, 11; Scheme 6).22 1H NMR spectra for 9 and 11 share important features with indene complex 3a: each shows a significantly upfield Pt-CH3 chemical shift relative to cation 2a. Upon rearrangement to the corresponding N-bound complexes 10 and 12, the Pt-CH3 signal returns downfield (Table 2). Other heterocycles (pyrrole, carbazole, benzofuran) did not afford clean results under these conditions; most notably, N-protected indoles gave rise to complex mixtures of products. 23
Scheme 6.

Table 2.
NMR data for Pt-Me groups of TFE adduct 2a, indene π complex 3a, and the π and N-indole adducts.
| Complex | 1H δa (ppm) | 13C δ (ppm) | JPt-H b (Hz) |
|---|---|---|---|
| 2a (TFE)c | +0.66 | 21.5 | 72 |
| 3a (indene) | −0.74 | 4.7 | 72 |
| 6a (indole π) | −1.02 | −d | 62a |
| 8a (indole N) | 0.74 | −12.1 | 78 |
| 9 | −0.75 | −3.6 | 73 |
| 10 | 0.53 | −13.7 | 78 |
| 11 | −1.16 | −3.0 | 73 |
| 12 | 0.26 | −16.8 | 78 |
Data recorded at 500 or 600 MHz.
Recorded at 300 MHz.
Notes 4bc.
The lifetime of this species is too short to record 13C NMR.
The mechanism of conversion of π-bound indole complexes to final N-bound complexes 8, 10, and 12 likely involves direct (intramolecular) slippage of platinum from the C2 = C3 π bond to N1 because the rate of π to N1 conversion is independent of [indole]. Moreover, in a simple crossover experiment, a solution of the 2-methyl indole adduct 9 (~90% conversion from 2a) was treated with 1 equivalent of 1-methylindole. Upon further reaction a distribution of 81(1)% 9, 10(1)% 10, 9(1)% 11 was observed, and finally a mixture containing 91(1)% 10 and 9(1)% 12 was observed. Thus little crossover is observed, excluding a mechanism involving indole displacement from the metal center.
Rearrangements of complexes 9 and 11 to complexes 10 and 12 are spontaneous at room temperature (also zero-order in indole). For the conversion of 2a to 10 at 30 °C, the rate of rearrangement of 9 to 10 is close to the rate for the binding of 2-methylindole to cation 2a, thus enabling NMR observation of all three species simultaneously in a single experiment (Figure 2). First order rate constants for the rearrangements of π complexes 9 and 11 were measured at 40 °C in the presence of excess indole (Table 3). Strikingly, rearrangement of these π complexes requires hours at 40 °C, whereas N-ligated complexes 8 are fully formed at room temperature in only minutes, indicating that methylation of the indole in the 1 or 2 position substantially retards the rate of rearrangement of indole π complexes.
Table 3.
First order rate constants for rearrangement of π complexes at 40 °C.
| Entry π | Complex | product | k (s−1) | T1/2 (min) | |
|---|---|---|---|---|---|
| 1 | 6a | 8a | 1.24(16) × 10−2 | 0.94(12) | π → N |
| 2 | 9 | 10 | 3.05(7) × 10−4 | 37.9(86) | π → N |
| 3 | 11 | 12 | 7.87(37) × 10−5 | 147(71) | π → N |
| 4 | 3a | 4a | 8.4(6) × 10−6a | 1380(98) | C-H activation |
Extrapolated from Eyring plot.
The rate of rearrangement of indole π complex 6a to N-ligated complex 8a is significantly diminished by added acid. When 2a is mixed with one equivalent of indole in the presence of a small amount (15 mole percent) of excess acid at room temperature, 6a is completely converted to 8a in minutes at room temperature, but conversion takes hours in the presence of 50 mole percent excess acid. In a more systematic set of experiments, 2a is prepared from 1a with a variable excess of acid, then five equivalents of indole is added, and reaction progress monitored by NMR at 40 °C; apparent rate constants for rearrangement of 6a are shown in Table 4. Only modest reductions of rate are observed up to about three-fold excess acid, but when the excess acid becomes comparable to the amount of excess indole the rate slows dramatically (entry 4). Under these conditions indole complexation to form 6a is also slowed, to only approximately three times the rate of rearrangement (compared to at least two orders of magnitude faster in the absence of acid). Furthermore, the initial product of π to N1 rearrangement is not 8a but a different N–adduct (presumably 7a); the rate of tautomerization of the first-formed N–adduct to the ultimate product 8a is slow under these acidic conditions.24 Thus protonation of the nitrogen lone pair slows both coordination of platinum to the C=C double bond and subsequent migration of platinum to nitrogen; the inhibition of [1D] to [3D] indole tautomerization suggests that it is mediated by free (unprotonated) indole.
Table 4.
Acid dependence of rate constant for rearrangement 6a.
| Entry | BF3 | k (s−1) | t1/2 (min) |
|---|---|---|---|
| 1 | 1.1 equiv. | 1.24(16) × 10−2 | 0.94(12) |
| 2 | 2.5 equiv. | 1.09(21) × 10−2 | 1.09(19) |
| 3 | 4.0 equiv. | 3.51(12) × 10−3 | 3.29(11) |
| 4a | 5.0 equiv. | 1.48(5) × 10−4 | 78.3(27) |
Conditions: BF3 is added to 1a in TFE. 5 equivalents of indole in TFE is then injected.
Indole binding and tautomerization are slow relative to kRAR.
Conclusions
The new indole complexes generated by addition of indole, 1-methylindole and 2-methylindole to [(diimine)Pt(Me)(solv)]+ cations are very rare examples of stable complexes of this class of heterocycle with late transition metals. The initial interaction is not with the nitrogen lone pair, but rather indole C2=C3 olefin ligation, the first examples of such complexation. Indene forms a more stable π complex with cation 2a that undergoes subsequent activation of the indenyl C-H group in hours at 60 °C to afford methane and an η3-indenyl adduct. By contrast, indole π complexes of cation 2a rearrange to N-ligated species. The rate of rearrangement can be controlled by substitution of the indole or by added H+. Presumably, productive C-H activation chemistry on the indole nucleus is more likely to proceed from a complex of the indole C2=C3 olefin rather than N1; therefore strategies to prevent the formation of apparently inert N complexes from the corresponding π complexes are of interest to the continued development of synthetic strategies for functionalization of indoles by metal-mediated C-H activation, although we have not yet achieved the latter. Progress toward these objectives is ongoing in our laboratories.
Experimental Section
General Considerations
1H NMR and 13C NMR spectra were recorded at ambient temperature using a Varian Inova 500, 600, or Mercury 300 spectrometer. The data are reported by chemical shift (ppm) from tetramethylsilane, multiplicity (s, singlet; d, doublet; t, triplet; m, multiplet; dd, double doublet; dt, double triplet), coupling constants (Hz), and integration. All 13C NMR data were collected proton-decoupled (13C{1H}), except for those for 4a, as specified. Mass spectra were acquired on a Finnigan LCQ ion trap or Agilent 5973 Network mass selective detector and were obtained by peak matching. All reactions were carried out under an atmosphere of nitrogen in glassware that had been oven-dried. Unless otherwise noted, all reagents were commercially obtained and, where appropriate, purified prior to use. Tris(pentafluorophenyl)borane [B(C6F5)3] was purified by sublimation (90 °C, 0.5 Torr). 2,2,2-Trifluoroethanol-d3 (TFE-d3) was dried over 3 Å molecular sieves for at least 5 days and then vacuum distilled onto B(C6F5)3. After 6 h, the trifluoroethanol-d3 was vacuum distilled and stored in a Teflon needle-valved vessel. Boron trifluoride was purified according to Brown’s procedure25 and stored as a solution in TFE-d3. The platinum dimethyl complexes were synthesized following earlier reported procedures, as noted. 2,2,2-Trifluoroethanol-d3, B(C6F5)3, and platinum dimethyl complexes were stored in a Vacuum Atmospheres dinitrogen atmosphere drybox.
Indene complex 3a
Platinum dimethyl complex 1a4a (19.2 μmol, 10.5 mg) was weighed out in an oven-dried 4 mL vial in the drybox. TFE-d3 (700 μL) and BF3 (0.477 M in TFE-d3, 18 μmol, 38 μL) were then added and the suspension was stirred until it became a homogeneous orange solution. Indene (19.2 μmol, 2.24 mg, 2.25 μL) was added, and the solution was transferred to an oven-dried J-Young NMR tube. After incubation at room temperature (24 hours in this case) the solution was analyzed by NMR and found to contain 3a in > 95% yield.
1H NMR (600 MHz, TFE-d3) δ: 7.51 (dt, JH-H = 6.8 Hz (doublet), 1.5 Hz (triplet), 1H), 7.26-7.23 (m, 3H), 7.15 (s, 1H), 7.10 (s, 1H), 6.99 (s, 1H), 6.96 (s, 1H), 6.16 (d, JH-H = 4 Hz, JPt-H = 79 Hz, 1H), 5.24 (apparent t, JH-H ~ 6.4 Hz, JPt-H = 74 Hz, 1H), 3.30 (d, JH-H = 23 Hz, JPt-H = 41 Hz, 1H), 3.07 (d, JH-H = 23 Hz, JPt-H = 38 Hz, 1H), 2.32 (s, 6H), 2.25 (s, 3H), 2.20 (s, 3H), 2.13 (s, 3H), 2.12 (s, 3H), 2.07 (s, 3H), 1.97 (s, 3H), −0.74 (s, JPt-H = 53 Hz, 3H). 13C NMR (150 MHz, TFE-d3) δ: 185.6, 178.7, 148.4, 142.7, 141.5, 141.0, 140.9, 139.7, 131.8, 131.6, 131.0, 131.0, 130.8, 130.5, 130.4, 129.5, 129.2, 128.9, 126.2, 125.5, 103.1, 92.4, 40.7, 21.1, 21.1, 20.4, 20.2, 17.9, 17.9, 17.9, 17.8, 4.7. FTIR (neat): 2920 (w), 1476 (w), 1384 (w), 1237 (w), 1180 (m), 1160 (m), 1051 (s), 847 (w), 766 (w), 718 (w) cm−1. FAB+ MS, calcd for C32H40N2Pt ([M + H]+): 647.2840, found 647.2822. Calcd for C31H36N2Pt ([M-Me]+): 631.2526, found 631.2302.
Indene complex 3a-d3 was prepared as above. 1H NMR (500 MHz, TFE-d3) matches the data above, except that signals at δ = 6.16, 3.30, and 3.07 ppm are absent.
Indene complexes 3b, 3c, 3d
Complexes 3b, 3c, 3d were prepared by direct analogy to complex 3a above, starting from platinum dimethyl species 1b, 1d4a and 1c.3 3b: 1H NMR (300 MHz, TFE-d3) δ: 7.51 (d, 7.23 Hz, 1H), 7.29-7.19 (m, 3H), 7.02 (broad s, 2H), 6.57 (broad s, 4H), 6.46 (broad s, 1H), 5.64 (broad s, 1H), 3.20 (broad s, 2H), 2.32 (broad s, 12H), 2.07 (broad s, 6H), −0.59 (s, JPt-H = 71 Hz, 3H). 3c: 1H NMR (300 MHz, TFE-d3) δ: 7.96 (s, 2H), 7.56-7.52 (m, 1H), 7.53 (s, 4H), 7.29-7.11 (m, 3H), 6.53 (s, 1H), 5.68 (s, 1H), 3.22 (s, 2H), 2.12 (s, 6H), −0.34 (s, = 69 Hz, 3H). 3d: 1H NMR (300 MHz, TFE-JPt-H d3) δ: 7.52-7.47 (m, 3H), 7.31-7.20 (m, 3H), 6.84 (broad s, 4H), 6.33 (broad s, 1H), 5.54 (broad s, 1H), 3.13 (broad s, 2H), 2.11 (broad s, 6H), 1.33 (broad s, 36H), −0.66 (s, JPt-H = 69 Hz, 3H).
Eyring Analysis for the conversion of 3a to 4a
3a was prepared as a solution in TFE-d3 as described above, with the exception that 2.50 eq of B(C6F5)3 was used as the acid source. Conversion and rate were then recorded as described in the main text.
Indenyl complex 4a
The solution of 3a prepared above was heated in its J-Young tube to the temperature and time described in the main text. 1H NMR (500 MHz, TFE-d3) δ: 7.12-7.10 (m, 6H), 6.79 (dd JH-H = 5.7, 3.1 Hz, ~3H), 5.01 (s,26 2H), 2.40 (s, 6H), 2.12 (s, 6H), 1.97 (s, 12H). 13C NMR (150 MHz, TFE-d3) δ: 172.6 (2C), 147.6 (2C), 140.8 (2C), 134.6, 131.2 (4C), 129.9 (4C), 129.3 (4C), 120.0 (2C), 71.6 (JPt-C = 130 Hz, 2C), 21.2 (2C), 18.0 (2C), 17.6 (4C). 13C NMR (125 MHz, not {1H}, TFE-d3) δ: 172.5 (s, 2C), 147.6 (s, 2C), 140.8 (d, JC-H = 6 Hz, 2C), 134.6, 132-130 (m, J1,C-H ~ 150 Hz, 2C), 131.2 (d, JC-H = 152 Hz, 2C), 129.9 (d, JC-H = 154 Hz, 4C), 129.3 (s, 4C), 120.0 (d, JC-H = 163 Hz, 2C), 71.6 (d, JC-H = 181 Hz, 2C), 21.2 (q, JCH = 126 Hz, 2C), 18.0 (q, JC-H = 132 Hz, 2C), 17.6 (q, JC-H = 126 Hz, 4C). FTIR (neat): 2919 (w), 1476 (w), 1331 (w), 1385, 1244 (w), 1182 (m), 1157 (m), 1054 (s), 851, 754 (w) cm−1. FAB+ MS, calcd for C31H36N2Pt ([M + H]+): 631.2526, found 631.2530 l. 2H NMR (76.7 MHz, TFE-h3) for this compound is shown in the Supporting Information.
Indenyl complex 4a-d2 was prepared as above. 1H NMR (500 MHz, TFE-d3) matches the data above, except that the signal at δ = 5.01 ppm is absent. FAB+ MS, calcd for C31H34D2N2Pt ([M-d2 + H]+): 633.2652, and for C31H32D3N2Pt ([M-d3]+): 633.2637; found 33.2631. 2H NMR (76.7 MHz, TFE-h3) for this compound is shown in the Supporting Information.
Indole π-complex 6a
Complex 6a is observed as an intermediate in kinetic runs (Table 4, entry 4). 1H NMR (500 MHz, TFE-d3) δ: 7.62 (s, 1H), 7.60 (d, JH-H = 7.8 Hz, 1H), 7.25-7.22 (m, 2H), 7.17 (t, JH-H = 8.0 Hz, 1H), 7.14 (s, 1H), 7.01 (s, 1H), 6.97 (s, 1H), 6.94 (s, 1H), 5.40 (s, JH-H = 94 Hz, ~0H),27 2.39 (s, 3H), 2.31 (s, 3H), 2.25 (s, 3H), 2.18 (s, 3H), 2.13 (s, 3H), 2.03 (s, 3H), 1.83 (s, 3H), 1.81 (s, 3H), −1.02 (s, JPt-H = 62 Hz, 3H).
Indole N-complex 8a
Platinum dimethyl complex 1a4a (46.6 μmol, 25.4 mg) was weighed out in an oven-dried 4 mL vial in the drybox. TFE-d3 (350 μL) and BF3 (0.455 M in TFE-d3, 53.5 μmol, 118 μL) were then added and the suspension was stirred until it became a homogeneous orange solution. Indole (47 μmol, 5.5 mg) was added, and the solution was transferred, rinsing with TFE-d3 (350 μL) to an oven-dried J-Young NMR tube. The solution was analyzed by NMR and found to contain 8a in > 95% yield. 1H NMR (500 MHz, TFE-d3) δ: 7.89 (s, JPt-H = 34 Hz, 1H), 7.73 (d, JH-H = 7.3 Hz, 1H), 7.40-7.35 (m, 3H), 7.12 (s, 1H), 7.09 (s, 1H), 6.97 (s, 1H), 6.50 (s, 1H), 2.35 (s, 3H), 2.34 (s, 3H), 2.27 (s, 3H), 2.20 (s, 3H), 2.16 (s, 3H), 1.88 (s, 3H), 1.87 (s, 3H), 1.80 (s, 3H), 0.61 (s, JPt-H = 73 Hz, 3H). 13C NMR (125 MHz, TFE-d3) δ: 182.1, 175.6, 175.1, 153.9, 142.7, 140.9, 140.4, 139.8, 134.5, 130.9, 130.9, 130.8, 130.7, 130.6, 130.5, 130.1, 130.0, 129.3, 128.9, 125.8, 122.3, ~44 (m, 1C), 21.1, 20.8, 20.0, 19.5, 17.9, 17.8 (2C), 17.7, −12.1. FTIR (neat): 2921 (m), 2094 (w), 1474 (m), 1449 (m), 1382 (m), 1313 (m), 1240 (m), 1184 (s), 1128 (s), 1069 (s), 1018 (s), 852 (m), 818 (m), 769 (m), 644 (m), 534 (m) cm−1. FAB+ MS, calc’d for C31H35N3Pt2H2 ([M-H]+): 649.2839, found 649.2847.
Indole complex 8b
Platinum dimethyl complex 1b4a (32.5 μmol, 16.8 mg) was weighed out in an oven-dried 4 mL vial in the drybox. TFE-d3 (700 μL) and BF3 (0.478 M in TFE-d3, 37.3 μmol, 78.1 μL) were then added and the suspension was stirred until it became a homogeneous orange solution. Indole (33 μmol, 3.8 mg) was added, and the solution was transferred to an oven-dried screw-capped NMR tube. The solution was analyzed by NMR and found to contain 8b in > 95% yield.
1H NMR (500 MHz, TFE-d3) δ: 8.01 (s, JPt-H = 36 Hz, 1H), 7.87 (d, JH-H = 7.9 Hz, 1H), 7.46-7.41 (m, 2H), 7.38-7.35 (m, 1H), 7.06 (s, 1H), 6.75 (s, 1H), 6.71 (s, 1H), 6.66 (s, 1H), 6.49 (s, broad, 1H), 6.06 (s, broad, 1H), 2.39 (s, 3H), 2.37 (s, 3H), 2.25 (s, broad, 3H), 1.96 (s, 3H), 1.95 (s, 3H), 1.69 (s, 3H), 0.74 (s, JPt-H = 73 Hz, 3H). 13C NMR (125 MHz, TFE-d3) δ: 182.3, 174.6, 175.1, 153.9, 147.8, 145.7, 141.9, 141.8, 141.9-141.5 (m, broad, 2C), 134.5, 131.2, 130.5, 130.2, 129.4, 125.9, 122.7, 121.0, 121.0, 120.7 (broad, 1C), 119.5 (broad, 1C), ~44 (m, 1C), 21.6, 21.5, 21.2, 21.9-21.8 (m, 2C), −11.3. FTIR (neat): 3014 (w), 2921 (w), 2875 (w), 1609 (w), 1593 (w), 1468 (w), 1453 (w), 1384 (w), 1308 (w), 1185 (m), 1130 (m), 1060 (s), 863 (w), 771 (w), 685 (w) cm−1. FAB+ MS, calcd for C29H31N3Pt2H2 ([M-H]+): 620.2448, found 620.2459.
Indole complex 8c
Platinum dimethyl complex 1c3 (36.0 μmol, 26.4 mg) was weighed out in an oven-dried 4 mL vial in the drybox. TFE-d3 (700 μL) and BF3 (0.478 M in TFE-d3, 41.4 μmol, 86.6 μL) were then added and the suspension was stirred until it became a homogeneous orange solution. Indole (36 μmol, 9.2 mg) was added, and the solution was transferred to an oven-dried screw-capped NMR tube. The solution was analyzed by NMR and found to contain 8c in > 95% yield.
1H NMR (500 MHz, TFE-d3) δ: 8.27 (s, JPt-H ~ 40 Hz, 1H), 8.02 (s, 1H), 7.78 (d, JH-H = 8.5 Hz, 1H), 7.73 (s, 1H), 7.68 (s, 1H), 7.55 (S, 1H), 7.45-7.27 (m, 5H), 2.04 (s, 3H), 2.03 (s, 3H), 0.76 (s, JPt-H ~ 60 Hz, 3H). 13C NMR (125 MHz, TFE-d3) δ: 184.5, 177.7, 175.6 (m, 1C), 152.9 (m, 1C), 148.5, 147.0, 136.1-134.1 (m, apparent 4C), 134.1, 130.7, 129.4, 126.2, 124.6 (apparent 2C), 124-122 (m, apparent 4C), 122.3 (q, JC-F = 227 Hz), 122.0, ~44 (m, 1C), 21.9, 20.5, −9.4. Three CF3 quartets cannot be identified because of coincidences with the solvent CF3; each has δ ~ 126 ppm, JC-F ~280 Hz. FTIR (neat): 3065 (w), 2932 (w), 2882 (w), 1622 (w), 1460 (m), 1373 (s), 1281 (s), 1182 (s), 1141 (s), 1067 (m), 903 (m), 848 (w), 771 (w), 705 (w), 684 (m) cm−1. FAB+ MS, calcd for C29H19F12N3Pt2H2 ([M-H]+): 836.1317, found 836.1333.
2-Methylindole-π-complex 9
Platinum dimethyl complex 1a4a (33.4 μmol, 18.2 mg) was weighed out in an oven-dried 4 mL vial in the drybox. TFE-d3 (700 μL) and BF3 (0.455 M in TFE-d3, 38.4 μmol, 84.3 μL) were then added and the suspension was stirred until it became a homogeneous orange solution. 2-Methylindole (33 μmol, 4.4 mg) was added, and the solution was transferred to an oven-dried screw-capped NMR tube. The solution was analyzed by NMR and found to contain 9 in > 95% yield.
1H NMR (500 MHz, TFE-d3) δ: 7.35 (d, JH-H = 7.5 Hz, 1H), 7.29 (d, JH-H = 7.5 Hz, 1H), 7.25 (s, 1H), 7.20-7.14 (m, 3H), 6.97 (s, 1H), 6.90 (s, 1H), 2.53 (s, 3H), 2.43 (s, 3H), 2.35 (s, 3H), 2.29 (s, 3H), 2.25 (s, 3H), 2.10 (s, 3H), 1.90 (s, 3H), 1.83 (s, 3H), 1.63 (s, 3H), −0.75 (s, JPt-H = 67, 3H). 13C NMR (125 MHz, TFE-d3) δ: 178.0, 176.8, 143.3, 142.7, 141.9, 141.5, 140.2, 139.7, 131.5, 131.4, 130.7, 130.6, 130.5, 130.4, 130.1, 129.4, 126.7, 126.1, 123.9, 114.1, 21.1, 21.0, 20.2, 19.9, 18.1, 18.0, 17.5 (2C), −3.6. (3 C cannot be uniquely identified because of dispersity in the aryl region and the transitory nature of 9.) FTIR (neat): 2918 (w), 1612 (w), 1505 (w), 1479 (w), 1458 (w), 1383 (w), 1329 (w), 1238 (m), 1157 (m), 1059 (s), 848 (w), 742 (m), 571 (w), 534 (w) cm−1. 9 is not sufficiently long-lived for HRMS.
2-Methylindole-N-complex 10
The crude solution of 9 prepared above was incubated at room temperature for 50 hours to give 10 in > 95% yield.
1H NMR (500 MHz, TFE-d3) δ: 7.74 (d, JH-H = 7.8 Hz, 1H), 7.32 (d, JH-H = 7.9 Hz, 1H), 7.29-7.27 (m, 2H), 7.11 (s, 1H), 7.10 (s, 1H), 6.87 (s, 1H), 6.44 (s, 1H), 2.41 (s, 3H), 2.35 (s, 3H), 2.33 (s, 3H), 2.24 (s, 3H), 2.19 (s, 3H), 2.12 (s, 3H), 1.86 (s, 3H), 1.85 (s, 3H), 1.83 (s, 3H), 0.53 (s, JPt-H = 74, 3H). 13C NMR (125 MHz, TFE-d3) δ: 186.0, 181.7, 175.8, 154.9, 142.8, 142.0, 140.3, 139.6, 135.0, 130.9, 130.7, 130.4, 129.9, 128.9, 128.8, 128.7, 122.5, 121.6, 121.1, 121.0, 112.4, ~45 (m, 1C), 22.1, 21.1, 20.8, 20.1, 19.5, 18.4, 17.9, 17.8, 13.5, −13.7. FTIR (neat): 3512 (broad, w), 3399 (broad, w), 2921 (broad, w), 1613 (w), 1573 (w), 1476 (m), 1456 (m), 1384 (m), 1316 (m), 1240 (m), 1187 (s), 1130 (s), 1069 (s), 852 (w), 756 (w), 646 (w), 535 (w) cm−1. FAB+ MS, calc’d for C32H38N3Pt2H2 ([M]+): 663.2996, found 663.2980.
1-Methylindole- π-complex 11
Platinum dimethyl complex 1a4a (46.9 μmol, 25.6 mg) was weighed out in an oven-dried 4 mL vial in the drybox. TFE-d3 (700 μL) and BF3 (0.455 M in TFE-d3, 54 μmol, 119 μL) were then added and the suspension was stirred until it became a homogeneous orange solution. 1-Methylindole (46.9 μmol, 6.16 mg, 6.00 μL) was added, and the solution was transferred to an oven-dried screw-capped NMR tube. The solution was analyzed by NMR and found to contain 11 in > 95% yield.
1H NMR (500 MHz, TFE-d3) δ: 7.57 (d, JH-H = 8.7 Hz, 1H), 7.50 (s, JPt-H = 49 Hz, 1H), 7.28 (t, JH-H = 7.8 Hz, 1H), 7.22-7.17 (m, 2H), 7.16 (s, 1H), 7.07 (s, 1H), 6.97 (s, 1H), 6.91 (s, 1H), 5.95 (s, JH-H = 88 Hz, ~0H),27 3.65 (s, 3H), 2.41 (s, 3H), 2.32 (s, 3H), 2.24 (s, 6H), 2.14 (s, 3H), 2.00 (s, 3H), 1.86 (s, 3H), 1.80 (s, 3H), −1.16 (s, JPt-H = 70 Hz, 3H). 13C NMR (125 MHz, TFE-d3) δ: 179.4, 177.1, 144.9, 142.6, 141.5, 140.4, 140.1, 137.3, 131.7, 131.5, 130.8, 130.7, 130.6, 130.2, 130.1, 129.8, 125.2, 125.0, 128.9, 128.3, 111.9, 34.2, 21.1, 21.0, 20.0, 19.8, 18.0, 17.7 (2C), 17.6, −3.0. Because of dispersion in the 130 ppm region, 1C cannot be located. FTIR (neat): 2919 (w), 1477 (w), 1454 (w), 1383 (w), 1327 (w), 1238 (w), 1184 (m), 1128 (m), 1054 (s), 847 (w), 753 (w), 679 (w) cm−1. FAB+ MS, calc’d for C32H39N3Pt2H1 ([M]+): 662.2933, found 662.2932.
1-Methylindole-N-complex 12
The crude solution of 11 prepared above was incubated at room temperature for 2 days, then heated to 40 °C for 4 hours to give 12 in ~ 78% yield by NMR.
1H NMR (500 MHz, TFE-d3) δ: 7.32 (t, JH-H = 7.6 Hz, 1H), 7.28 (d, JH-H = 7.4 Hz, 1H), 7.22 (t, JH-H = 7.4 Hz, 1H), 7.19 (d, JH-H = 8.4 Hz, 1H), 7.08 (s, 1H), 7.07 (s, 1H), 6.91 (s, 1H), 6.69 (s, 1H), 3.83 (s, 3H), 2.33 (s, 3H), 2.30 (s, 3H), 2.24 (s, 3H), 2.18 (s, 3H), 2.13 (s, 3H), 2.01 (s, 3H), 1.98 (s, 3H), 1.85 (s, 3H), 0.26 (s, JPt-H = 77 Hz, 3H). 13C NMR (125 MHz, TFE-d3) δ: 180.1, 176.3, 148.4, 144.6, 142.0, 139.9, 139.7, 137.0, 136.9, 130.9, 130.7, 130.7, 130.4, 130.2, 129.3, 128.8, 128.8, 128.6, 127.6, 125.6, 125.5, 112.7, 39.9 (JPt-C = 81 Hz), 21.1, 20.7, 20.0, 19.8, 17.9, 17.9, 17.9, 17.8, −16.8. FTIR (neat): 2923 (m, broad), 2100 (w), 1609 (w), 1515 (m), 1463 (s), 1384 (m), 1312 (m), 1238 (m), 1186 (s), 1130 (s), 1069 (s, broad), 860 (w), 818 (w), 759 (m), 646 (w), 535 (w) cm−1. FAB+ MS, calcd for C32H39N3Pt2H ([M]+): 662.2933, found 663.2939.
Crossover experiment between 9 and 1-methylindole
Platinum dimethyl complex 1a (22.4 μmol, 12.2 mg) was weighed out in an oven-dried screw-capped NMR tube in the drybox. TFE-d3 (511 μL) and BF3 (0.477 M in TFE-d3, 24.6 μmol, 51.6 μL) were then added. The suspension was removed from the drybox and sonicated until it became a homogeneous orange solution. The reaction was then thermally equilibrated to 30 °C in a Varian mercury 300 MHz NMR, and 2-methylindole (0.183 M in TFE-d3, 22.4 μmol, 122 μL) was then injected. [9] was monitored, and reached a maximum in ca. 2 min. at which conversion of 2a to 9 was ~90% complete. The tube was then removed from the instrument, 1-methylindole was injected, and the sample was re-inserted (~ 30 sec.) An initial ratio of 81(1)% 9, 10(1)% 10, 9(1)% 11 was observed with 0(1)% 2a. Kinetic data were recorded for 9.7 hours. After an additional 2 days at RT, 9 was completely converted to 10 (kRAR = 9.1(3) × 10−5 sec−1) and 11 was completely converted to 12. A ratio of 91(1)% 10 to 9(1)% 12 was observed.
Conversion of 6a to 8a at high [H+]
Platinum dimethyl complex 1a (22.7 μmol, 12.4 mg) was weighed out in a 1 mL graduated cylinder in the drybox. BF3 (0.477 M in TFE-d3, 56.8 μmol, 119 μL) and TFE-d3 (to make 1.00 mL) were then added. 480 μL of this solution (10.9 μmol Pt, 2.5 equiv. BF3) was then distributed into each of 2 screw-capped NMR tubes. Separately, indole (14.5 mg, 124 μmol), BF3 (0.477 M in TFE-d3, 62.0 μmol, 130 μL). and TFE-d3 (356 μL) were mixed in a 10 mL culture tube. 220 μL of this solution (5 equiv. indole to Pt, 2.5 equiv. BF3 to Pt) was then distributed into each of two 1 mL syrenges. The NMR tubes and syrenges were removed from the drybox. One NMR tube was then thermally equilibrated to 40 °C in a Varian inova 500 MHz NMR spectrometer. It was then ejected, and the content of 1 syrenge was added to the NMR tube (to make 10.9 μmol Pt, 5 equiv. BF3, 5 equiv. indole, and 700 μL TFE-d3). The tube was shaken and re-inserted. After rapid re-shimming, kinetic data were recorded for 7.5 hours. A second trial was then conducted with the other NMR tube and syrenge. Reaction progress was monitored by NMR. Rate constants for this experiment are kbinding = 5.35_10−4 s−1 and kRAR = 1.44_10−4 s−1, where kbinding is the pseudo first order rate constant for the binding of indole to 2a.24
Comment regarding H/D Exchange and Methane Isotopologs Formed in the Conversion of 3 to 4
In the conversion of 3a to 4a the methane that is formed is 65% CH4, 25% CH3D due to D+ incorporation in the formation of 2a from 1a. A distribution of 53% CH4, 47% CH3D is observed in the conversion of 3a-d3 to 4a-d2. 2H NMR spectra of 4a and 4a-d2 were recorded to track 2H incorporation in these compounds (see Supporting Information). 2H incorporation is observed in C2-H (6.8 ppm) in both 4a and, 4a-d2. Little 2H incorporation is observed in C1,3-H (5.0 ppm) in 4a. As expected, 2H is observed in this position in 4a-d2. A small amount of H/D exchange with solvent is observed in the diimine backbone (2.4 ppm), but none is observed in the mesityl methyl groups. Accordingly, the high level of CH4 relative to CH3D observed in the formation of 4a-d2 is attributed to some H/D exchange with C2—H in the indenyl fragment prior to methane liberation and a low level of decomposition of the diimine ligand.
Supplementary Material
Spectral data for the crossover experiment between 9 and 1-methylindole, spectral and kinetic data for the conversion of 6a to 8a in the presence of excess acid, 2H NMR spectra for 4a and 4a-d2, and graphical 1H NMR spectra for 3a, 3a-d3, 4a, 4a-d2, 8a, 8b, 8c, 9, 10, 11, and 12 are available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
This work was supported by the BP MC2 program and by the NIH (NRSA fellowship GM075691 to T.J.W.). These sponsors are gratefully acknowledged. We thank Dr. Tom G. Driver for helpful discussions, especially regarding the preparation and use of BF3-TFE-d3.
References and Notes
- 1.For recent reviews on C-H activation and functionalization, see: Arndtsen BA, Bergman RG, Mobley TA, Peterson TH. Acc Chem Res. 1995;28:154–162.Erker G. Angew Chem Int Ed. 1999;38:1699–1712.Davies HML, Antoulinakis EG. J Organomet Chem. 2001;617–618:47–55.Crabtree RH. J Chem Soc Dalton Trans. 2001;17:2437–2450.Labinger JA, Bercaw JE. Nature. 2002;417:507–514. doi: 10.1038/417507a.Fekl U, Goldberg KI. Adv Inorg Chem. 2003;54:259–320.
- 2.Shilov AE, Shul’pin GB. Chem Rev. 1997;97:2879–2932. doi: 10.1021/cr9411886. [DOI] [PubMed] [Google Scholar]
- 3.Johansson L, Ryan OB, Tilset MJ. Am Chem Soc. 1999;121:1974–1975. [Google Scholar]; Johansson L, Tilset M. J Am Chem Soc. 2001;123:739–740. doi: 10.1021/ja002505v. [DOI] [PubMed] [Google Scholar]
- 4.(a) Johansson L, Tilset M, Labinger JA, Bercaw JE. J Am Chem Soc. 2000;122:10846–10855. [Google Scholar]; (b) Zhong AH, Labinger JA, Bercaw JE. J Am Chem Soc. 2002;124:1378–1399. doi: 10.1021/ja011189x. [DOI] [PubMed] [Google Scholar]; (c) Heyduk AF, Driver TG, Labinger JA, Bercaw JE. J Am Chem Soc. 2004;126:15034–15035. doi: 10.1021/ja045078k. [DOI] [PubMed] [Google Scholar]; (d) Driver TG, Day MW, Labinger JA, Bercaw JE. Organometallics. 2005;24:3644–3654. [Google Scholar]; (e) Owen JS, Labinger JA, Bercaw JE. J Am Chem Soc. 2006;128:2005–2016. doi: 10.1021/ja056387t. [DOI] [PubMed] [Google Scholar]
- 5.Driver TG, Labinger JA, Bercaw JE. California Institute of Technology; Unpublished Results. [Google Scholar]
- 6.Johansson L, Tilset M. J Am Chem Soc. 2001;123:739–740. doi: 10.1021/ja002505v. [DOI] [PubMed] [Google Scholar]
- 7.(a) Dunina VV, Zalevskaya OA, Potapov VM. Russian Chem Rev. 1988;57:250–269. [Google Scholar]; (b) Ryabov AD. Chem Rev. 1990;90:403–424. [Google Scholar]
- 8.Some examples include Dick AR, Hull KL, Sanford MS. J Am Chem Soc. 2004;126:2300–2301. doi: 10.1021/ja031543m.Desai LV, Hull KL, Sanford MS. J Am Chem Soc. 2004;126:9542–9543. doi: 10.1021/ja046831c.Tan KL, Bergman RG, Ellman JA. J Am Chem Soc. 2001;123:2685–2686. doi: 10.1021/ja0058738.Tan KL, Bergman RG, Ellman JA. J Am Chem Soc. 2002;124:13964–13965. doi: 10.1021/ja0281129.Dangel BD, Godula K, Youn SW, Sezen B, Sames D. J Am Chem Soc. 2002;124:11856–11857. doi: 10.1021/ja027311p.
- 9.Recent examples include Pd: Ferreira EM, Stoltz BM. J Am Chem Soc. 2003;125:9578–9579. doi: 10.1021/ja035054y.Abbiati G, Beccalli EM, Broggini G, Zoni C. J Org Chem. 2003;68:7625–7628. doi: 10.1021/jo034636v.Pt: Liu C, Han X, Wang X, Widenhoefer RA. J Am Chem Soc. 2004;126:3700–3701. doi: 10.1021/ja031814t.
- 10.Lane BS, Brown MA, Sames D. J Am Chem Soc. 2005;127:8050–8057. doi: 10.1021/ja043273t. [DOI] [PubMed] [Google Scholar]
- 11.Deprez NR, Kalyni D, Krause A, Sanford MS. J Am Chem Soc. 2006;128:4972–4973. doi: 10.1021/ja060809x. [DOI] [PubMed] [Google Scholar]
- 12.Kaminskaia NV, Ullmann GM, Fulton DB, Kostic’ NM. Inorg Chem. 2000;39:5004–5013. doi: 10.1021/ic000254l. [DOI] [PubMed] [Google Scholar]
- 13.Osa T, Hino H, Fujieda S, Shiio T, Kono T. Chem Pharm Bull. 1986;34:3563–3572. doi: 10.1248/cpb.34.3563. [DOI] [PubMed] [Google Scholar]
- 14.We believe that indole is bound through N1 as suggested by the authors, but is in the 3H-tautomer. 1H NMR (300 MHz, dichloromethane-d2) δ: 8.37 (s, JPt-H = 30 Hz, 2H, C2-H), 8.58 (d, JPt-H = 8.2 Hz, 2H), 7.62-7.08 (m, 6H), 4.03 (s, 4H, C3-H2).
- 15.For example: indole + Pt0(PEt3)4 → trans-[(PEt3)2PtII(H)(1-indolyl)] + 2 PEt3. Chantson JT, Lotz S. J Organomet Chem. 2004;689:1315–1324.
- 16.[(η6-C8H7N)MLn] complexes are known for several metals. Common examples include MLn = Cr(CO)3, see Fischer EO, Goodwin HA, Kreiter CG, Simmons HD, Sonogashira K, Wild SB. J Organomet Chem. 1968;14:359–374.Nesmeyanov AN, Ustynyuk NA, Thoma T, Prostakov NS, Soldatenkov AT, Pleshakov VG, Urga K, Ustynyuk YA, Trifonova OI, Oprunenko YF. J Organomet Chem. 1982;231:5–24.MLn = CpRu+, see Moriarty RM, Ku YY, Gill US. Organometallics. 1988;7:660–665.Other examples include M = MnI, CoIII, RhI, RhIII, IrI, and IrIII. [(η5-C8H6N)MLn] complexes are known for MLn = Mn(CO)3+, see Ji LN, Kershner DL, Rerek ME, Basolo F. J Organomet Chem. 1985;296:83–94.for MLn = Cp*Ir2+, see White C, Thompson SJ, Maitlis PMJ. Chem Soc Dalton Trans. 1977:1654–1661.Other examples include M = TiIV, MoII, and FeII.
- 17.Analyzed as in Romeo R, Monsu Scolaro L, Nastasi N, Arena G. Inorg Chem. 1996;35:5087–5096. doi: 10.1021/ic960107g.
- 18.Error determined by established methods. See Morse PM, Spencer MD, Wilson SR, Girolami GS. Organometallics. 1994;13:1646–1655.
- 19.Yasuda M, Pak C, Sakurai H. Bull Chem Soc Japan. 1980;53:502–507. [Google Scholar]
- 20.Some [(3H-indole)MLn] complexes are known for other metals. For examples of [Re] complexes, see Johnson TJ, Arif AM, Gladysz JA. Organometallics. 1994;13:3182–3193.; for [Ir] see Chen S, Noll BC, Peslherbe L, Rakowski DuBois M. Organometallics. 1997;16:1089–1092.
- 21.C3-H in 6a can be observed at early stages of kinetic runs (δ = 5.95 ppm, JPt-H = 87 Hz (500 MHz)), but N1-H was never found for coordinated indole.
-
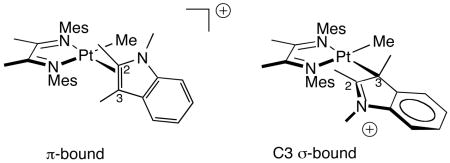
22.A reviewer proposed an alternative C3 σ-bound structure for 6a, 9, and 11, but we favor the π complexes as shown. The σ-bound structure is inconsistent with 1H and 13C chemical shifts for C3-H: δ C3-H = 5.4 and 6.0 for compounds 6a and 11, respectively, as identified by JPt-H and H/D exchange. This is more consistent with an sp2 carbon center. Moreover, no 13C signal is present in the 175–180 region of the spectrum of 11, as would be needed for C2 in the σ-bound structure. It is likely, however, that the platinum center is more tightly associated with the more electron rich C3 side of the olefin. This is a possible explanation for the low JPt-H for C2 in 11.
- 23.Reactions of N-protected indoles (Ac, Tf, Ts), and benzofuran afforded complex mixtures of C—H activation products. Pyrrole, carbazole, and 3-methylindole afforded complex mixtures of coordination complexes of the general form [(diimine)Pt(Me)(CnHnN-dn)]+. Hydrogenated homologs of indene and indole also react with 2a. Indane forms a mixture of C—H activation products, which includes an η3-benzyl complex analogous to that obtained from ethylbenzene; see notes 4cd. 1,2-Dihydroindole forms a stable N-bound coordination complex, which has 1H NMR (300 MHz, TFE-d3) δ: 7.21-7.14 (m, 3H), 7.07-7.06 (m, 4H), 6.89 (s, 1H), 3.47-3.30 (m, 2H), 2.87-2.75 (m, 1H), 2.58-2.47 (m, 1H), 2.33 (s, 3H), 2.30 (s, 6H), 2.17 (s, 3H), 2.17 (s, 3H), 1.93 (s, 3H), 1.80 (s, 3H), 1.75 (s, 3H), 0.53 (s, JPt-H = 76 Hz, 3H).
- 24.Rate constants for this experiment were determined by modeling [6a] as an intermediate in a scheme of consecutive reactions. For the reaction scheme 2a + indole (excess) − kbinding → 6a − kRAR → products, [6a]/[6a]o = (kbinding/(kbinding + kRAR)) (e−kbindingt − e−kRARt) See Laidler, K. J. Chemical Kinetics, 3rd ed.; Harper & Row: New York, 1987.
- 25.Brown HC, Johannesen RB. J Am Chem Soc. 1953;75:16–20. [Google Scholar]
- 26.Observed JPt-H = 22 Hz for this signal in a spectrum acquired at 300 MHz.
- 27.This signal (C3-H) is rapidly deuterated under the reaction conditions, but can be observed at the beginning of a kinetics run.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Spectral data for the crossover experiment between 9 and 1-methylindole, spectral and kinetic data for the conversion of 6a to 8a in the presence of excess acid, 2H NMR spectra for 4a and 4a-d2, and graphical 1H NMR spectra for 3a, 3a-d3, 4a, 4a-d2, 8a, 8b, 8c, 9, 10, 11, and 12 are available free of charge via the Internet at http://pubs.acs.org.