

Abstract
Designing RNAs that form specific secondary structures is enabling better understanding and control of living systems through RNA-guided silencing, genome editing and protein organization. Little is known, however, about which RNA secondary structures might be tractable for downstream sequence design, increasing the time and expense of design efforts due to inefficient secondary structure choices. Here, we present insights into specific structural features that increase the difficulty of finding sequences that fold into a target RNA secondary structure, summarizing the design efforts of tens of thousands of human participants and three automated algorithms (RNAInverse, INFO-RNA and RNA-SSD) in the Eterna massive open laboratory. Subsequent tests through three independent RNA design algorithms (NUPACK, DSS-Opt, MODENA) confirmed the hypothesized importance of several features in determining design difficulty, including sequence length, mean stem length, symmetry, and specific difficult-to-design motifs like zig-zags. Based on these results, we have compiled an Eterna100 benchmark of 100 secondary structure design challenges that span a large range in design difficulty to help test future efforts. Our in silico results suggest new routes for improving computational RNA design methods and for extending these insights to assessing “designability” of single RNA structures as well as of switches for in vitro and in vivo applications.
Introduction
RNA molecules play important roles in a variety of biological processes such as gene regulation, protein synthesis and viral infection. The folding of RNAs into specific secondary structures is necessary for performing these functions, and numerous methods have been developed to model this folding process [1,2]. Many emerging applications of RNA, including gene silencing, genome editing, and creation of complex nanostructures, rely on inverse folding, the design of RNAs that fold into specified secondary structures in their minimum free energy states [3–5]. Solving the inverse folding problem is an important step in moving towards the design of RNA molecules that carry out desired functions.
Several algorithms have been developed to computationally address the inverse folding problem. The pioneering tool RNAinverse used an adaptive walk, randomly sampling mutations and accepting those that improved the distance from the desired secondary structure [2]. Other algorithms took similar approaches, introducing additional features such as hierarchical decomposition of the target structure (RNA-SSD) [6], probabilistic sampling of sequences (INFO-RNA) [7], and multiple scoring functions (DSS-Opt) [8]. NUPACK combined some of these features and introduced the ensemble defect, a scoring function based on base pair probabilities [9], while MODENA introduced a genetic algorithm to optimize both structure stability and similarity to target [10]. Despite these advances, it remains difficult to distinguish, a priori, designable from intractable structures. Moreover, comparative benchmarks of design algorithms are not available, and it is unknown what features challenge which methods. Therefore, current design efforts can require multiple iterations to select a tractable secondary structure for a specific design problem with a specific design tool.
Assessing RNA designability is one of the main research areas of Eterna (previously EteRNA), a massive open laboratory which brings together tens of thousands of human participants (“players”) to tackle RNA design problem (http://eternagame.org/) [11]. Here, we report insights from Eterna players who made use of a puzzle solving interface, a puzzle creator tool, a puzzle selection interface, and a solver scripting tool (Figure 1) to create and analyze thousands of RNA secondary structures and their difficulty for sequence design. To help test which features are particularly challenging to design, Eterna integrated several automated solvers (RNAInverse, INFO-RNA and RNA-SSD) into the game (Figure 1C), as well as an EternaScript framework enabling simple player-designed algorithms (Figure 1D). These tools have allowed players to test numerous hypotheses about what makes puzzles difficult for these algorithms. While these studies have been described in many player-created documents (several compiled herein as Supporting Documents), they have not yet been presented in the formal scientific literature or subject to tests outside the Eterna platform. Thus, here, we confirm these hypotheses on what makes RNA secondary structure design problems difficult using further independent tests, based on three independent inverse folding algorithms (NUPACK, DSS-Opt, MODENA) run on a separate supercomputer. Aside from these independent tests, most of the writing herein is drawn directly or closely adapted from writing in the original player-created documents.
Figure 1. Tools used by Eterna community to assess difficulty of RNA secondary structure design puzzles.

[A] The puzzlemaker interface allows players to define a secondary structure string or insert bases or base pairs at specified positions and then deploy these puzzles to other players to solve. [B] The puzzle solver interface enables players to select nucleotides and paint them over the structure, which can switch between natural and target modes. [C] After puzzle design, players are able to see which bots are able to solve their puzzles. [D] The Eternascript interface allows for players to create and test their own puzzle solving algorithms.
We first describe several secondary structure features and specific structural idiosyncrasies that this internet-scale experiment has identified to be especially problematic for sequence design. Second, we describe the effects of symmetry on the design of RNAs, a less well-appreciated facet of difficult design challenges. Several of the discovered features arise frequently in natural RNA sequences as well as engineering challenges but, to our knowledge, have not been previously noted as difficult for design. Finally, we introduce the Eterna100 benchmark, which we propose as a standard set of structures to be used for challenging and evaluating the next generation of automated RNA design algorithms.
Results
Secondary structure features
Based on manual creation and curation of thousands of puzzles through the Eterna puzzle designing and solving interface (Figure 1), Eterna participants have identified several RNA secondary structure elements that result in the failure of state-of-the-art RNA design methods as well as player-designed algorithms. Most of these features can be simply rationalized: they introduce elements that have generally poor stability for any sequence and thus allow for the emergence of more stable alternative conformations. Beyond this simple rationalization, however, additional factors, such as the number of repetitions of each feature, also appear important in assessing design difficulty. To test these hypotheses, we evaluated six algorithms for their ability to solve various RNA design problems in silico. Henceforth, we define success as the design of a sequence whose minimum free energy structure matches the target structure, as computed by the energy function used by the algorithm.
First, Eterna players postulated that short stems are major contributors to the difficulty of an RNA design puzzle. As illustrated by secondary structures in Figure 2A, existing RNA design algorithms often fail to solve puzzles with many short stems and the difficulty increases as the number of stems increases. For example, Shortie 4 and Shortie 6 are essentially the same puzzle with different numbers of 2 base-pair stems. Algorithms such as NUPACK are able to solve the 2-stem Shortie 4 but not its 4-stem sibling Shortie 6 (colored squares in Figure 2A). The difficulty of designing short stems has been previously suggested in computational studies [7], but does not seem to be widely known. For example, recent work on RNA origami tiles involves 2 base-pair stems that act as 3D “dovetail” seams between parallel stacks of double helices [5] but may cause problems in larger origami constructions.
Figure 2. RNA design puzzles from Eterna demonstrate features that make design difficult.

Open or filled squares indicate failure or successful solutions, respectively, by existing RNA design algorithms – RNAinverse (red), INFO-RNA (yellow), RNA-SSD (green), NUPACK (cyan), DSS-Opt (blue), and MODENA (purple). [A] Stem length: Shortie 4, Shortie 6; [B] Adjacent Multiloops: Kyurem 5, Kyurem 7; [C] Loop next to a Multiloop; [D] Bulges: Just down to 1 bulge, 1,2,3 and 4 bulges; [E] Internal Loops: Mat – Lot 2-2 B, Crop Circle 2; [F] Zigzags: Hard Y; [G] Simple puzzles: This is ACTUALLY Small and Easy 6; [H] Quasispecies 2-2 Loop Challenge, Water Strider, The Fractal, Mutated Chicken Feet
There is a further reason for the difficulty of short-stem puzzles besides the generally low stability of these features, which, to our knowledge, has not been previously noted. Since the sequence space for 2 base-pair stems is small, there are very few stable sequence combinations for these stems. If such stems occur once or a few times in a structure, they can be stabilized with one of the known stabilizing sequence combinations. However, when many of these stems are present in a target secondary structure, subsequences will necessarily have to be repeated in order to ensure all stems are stable. This repetition of subsequences introduces opportunities for mispairing that must be “designed away” through careful positioning of subsequences into the structure. This issue is expected to increase the difficulty of design in large RNA structures that include series of nearby, repeated 2-bp stems (such as origami tiles in [5]) and may explain why natural RNAs do not seem to take advantage of repeated sub-tiles or stems. Further examples of this principle of repetition will be described below.
The problem of short stems is worsened when the stems are flanked by multiloops (junctions) or bulges, as in the Kyurem puzzles (Figure 2B). The design of multiloops often requires detailed optimization of the closing pairs of each stem, and when stems are difficult to maintain, this problem becomes exacerbated. There are extreme cases where two loops are joined by a stem with only one base pair, such as in the puzzle Loop next to a Multiloop (Figure 2C); these are especially difficult, as the same base pair must close more than one loop. Even though these arrangements occur with some frequency in natural RNAs and are permitted in most in silico secondary structure prediction models, most RNA design algorithms have difficulty solving them. This is distinct from the idea of short stems, because introducing a stronger base pair or permuting the stem pairs is often insufficient to stabilize the stem. These problems instead require the optimization of energies of neighboring loops and stems and are thus much more sensitive to the local sequence than other problems with short stems.
As an example, Figure 3A illustrates a near-miss sequence solution created by a player-written puzzle-solving script for Kyurem 7, while Figure 3B shows how a few changes to the design would have solved the puzzle. In Figure 3A, the short stem between the bulge and multiloop (region outlined in red circles) is not predicted to form in the minimum free energy solution. Strengthening the failing stem between the multiloop and bulge alone does not fix this near solution, but success is achieved by exchanging one closing pair on the multiloop from a GC to an AU, flipping another closing pair from a CG to a GC, and flipping a third AU pair in an adjacent stem to strengthen it (Figure 3B). These additional changes alter the energy of the potential misfolded states that occur relative to the target shape. These sorts of changes are routinely proposed by Eterna players to solve these puzzles, but are not found by current automatic algorithms
Figure 3. A case study for RNA design – Kyurem 7.

[A] A near-miss sequence design for Kyurem 7 (see Figure 2B) was designed by a player-created bot and misfolds only in one stem (red). [B] A successful solution with only afew base changes (green). [C] A slight variation in the Kyurem 7 target secondary structure has only minor changes in the lengths of the multiloops, but is much easier to solve due to the availability of low-energy designs for the expanded multiloops. Nucleotides are colored by base, with A in yellow, U in blue, G in red and C in green. Minimum free energy structures and loop energies (in kcal/mol) are based on the Turner 1999 parameters.
Bulges and internal loops within helices were also proposed to contribute to destabilization of RNA secondary structures and thereby to the general difficulty of designing sequences for these structures. Incremental introduction of additional bulges, as shown in Figure 2D, resulted in failure by more of the design algorithms as well as fewer successful player solutions (Table S1). These interruptions within a stem disrupt the favorable stacking interactions between base pairs, increasing (worsening) the free energy of the target RNA conformation. Last, as shown in Figure 2E, large internal loops, especially those bordered by short stems, present the problem of designing a low energy loop to compensate for the short stems and overall high energy of the structure. All algorithms fail at these problems.
Overall, secondary structural elements that are typically higher in predicted free energy change, such as multiloops with no unpaired bases, were generally viewed as being harder to design. For example, if the 0-0-0 multiloop (with 0 nucleotides separating the three stems) in Kyurem 7 is replaced by a 1-1-1 multiloop (with single nucleotides separating the three stems) and the 4-0 bulge is replaced by a 4-1 asymmetric loop, these slightly lengthened loops allow for sequence solutions with more favorable energy (compare energies, Figures 3B and C), and the overall structure becomes easier to solve.
The quantity and density of the previously described elements are important for determining puzzle difficulty. As a summary, players use a principle of least elements, which asserts that the fewer of the elements delineated in Figure 2, the more likely a design is to be feasible. This principle allows the design difficulty of a target secondary structure to be gauged by eye. Further examples of this principle are provided in the benchmark described below.
Structural idiosyncracies
Specific patterns of bulges were identified by Eterna players as particularly problematic regions for RNA design. The hard Y puzzle, shown in Figure 2F, has relatively long stems but contains two consecutive bulges opposite each other, a motif that players named the “zig-zag”. The presence of this pattern close to a small multiloop, in an otherwise straightforward puzzle, causes most existing algorithms to fail. Zig-zags appear uncommon in natural RNA structures, and we propose that their infrequency is related to the difficulty of designing them.
Players also noted that existing RNA design algorithms also fail on extremely simple problems of biological interest. This is ACTUALLY Small and Easy 6, shown in Figure 2G, consists of 400 unpaired nucleotides. While setting the entire sequence to the same nucleotide is sufficient to force complete unpairing and solve the problem, these algorithms overcomplicate the design problem by randomly initializing the sequence, causing them to fold incorrectly. These types of overly simple designs are an unexpected class of difficult problems for current algorithms. Nevertheless, automating strategies for solving them will be important for designing RNA nanostructures that need to present long single-stranded stretches to other molecular machines for translation, splicing or reverse transcription.
Symmetry
Players observed that striking visual symmetry in a target secondary structure is typically a hallmark of a difficult problem for sequence design, as demonstrated by the failure of existing algorithms to solve puzzles with high symmetry (Figure 2H).
The effect of symmetry on design puzzle difficulty appears mainly due to the presence of repeated elements. These elements may be similar or same-length stems, as described in the previous section. However, any other repeated structural motif can also lead to difficulty of symmetrical designs. The rationale is similar to the one described above for 2-bp stems: short, complex substructures, such as those in Mutated Chicken Feet (Figure 2H), can only be solved by a highly constrained space of subsequences. Repetition of these substructures can result in highly similar sequences for each of the repeated elements, increasing the probability of mispairing and decreasing the probability of a successful fold, requiring careful optimization. Supporting this ‘principle of repetition’ as the underlying factor and not symmetry per se, the difficulty of these problems appears agnostic to the type of visually apparent symmetry: the symmetry can be axial (e.g. Water Strider), rotational (e.g. Mutated Chicken Feet, The Fractal) or translational (e.g. Crop Circle 2), as depicted in Figure 2H.
Additionally, although the repetition of secondary structure features increases the difficulty of computing a correctly folding sequence, when repeating substructures are combined with the factors described above, the difficulty can further decrease or increase. On one hand, The Fractal demonstrates symmetry with a stable hairpin element; long stems make this solvable by five of six algorithms (Figure 2F). On other hand, Crop Circle 2 (Figure 2E), which contains a repeated element with short stems and large loops, is intractable for all existing secondary structure design algorithms tested.
Finally, many puzzles that are visually symmetric also include a multiloop, which is typically a high-energy element, as discussed previously, and can conspire with repeating substructures, large loops, or short stems to increase problem difficulty. In these multiloop-containing symmetric puzzles, a common misfold allows portions of symmetrically opposite structures to base-pair and replace a multiloop with a sequence of multiple simple stems and internal loops (Figure 4).
Figure 4. An example of a common near-miss fold.
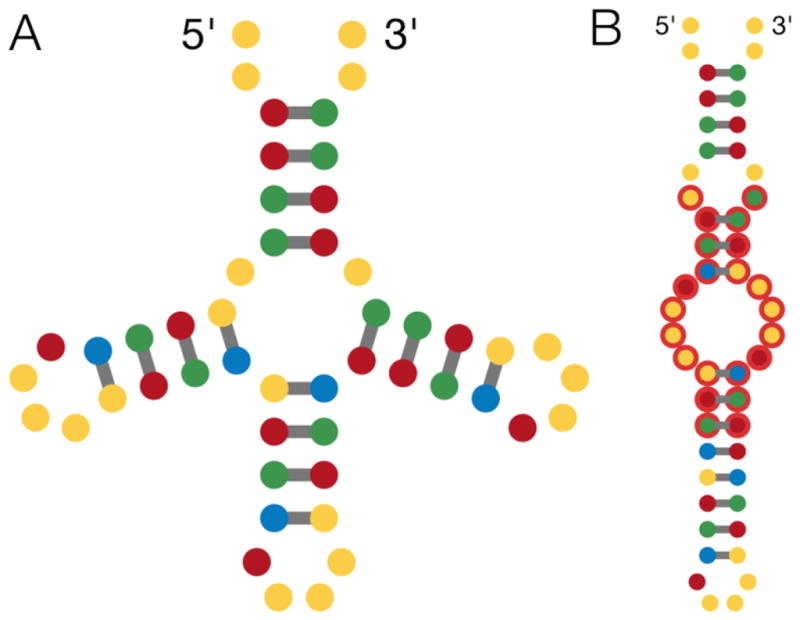
A symmetric, multiloop-containing puzzle created by players, in which designs for the target structure (A) frequently mispair to create the same misfolded structure (B).
In summary, visually apparent symmetry in a target secondary structure typically entails a number of features – repeated substructures, individually complex loops, and multiloops at the axes or points of symmetry – that herald difficulty for sequence design. This observation may explain why large natural structured RNAs, such as ribosomes and self-splicing introns, have fractal-like secondary structures but are still not symmetric despite the ease of creating such repetitious structures through processes analogous to gene duplication [12]. For natural RNAs with near symmetry, such as riboswitches with two tandem aptamers that bind glycine, the sequences of cognate helices in the repeated aptamers are distinct, as might be expected. In addition, however, the lengths of the cognate helices are not identical [13], even though exact secondary structure symmetry would be expected to stabilize these RNAs’ near-symmetric 3D folds when bound to their small molecule targets [14,15]. The slight breaking of symmetry at the secondary structure level is a design rule that does not appear to be widely appreciated outside Eterna. Recognition of this rule may aid RNA nanoengineering efforts seeking to build large-scale tiles, polyhedra, and tandem shRNAs and sgRNAs [3–5,16,17].
The Eterna100 benchmark
To test and help disseminate the principles described here, we have developed the Eterna100 benchmark for evaluating RNA design algorithms (Table S1). This set of 100 structures spans a wide range of lengths and difficulties, from short hairpins to complex 400-nucleotide designs. It includes all of the structures shown in Figure 2, which exemplify the features we have found to make design difficult, as well as additional structures that have different combinations of secondary structure features. The list also includes puzzles, like Misfolded Aptamer and This is ACTUALLY Small And Easy 6, whose difficulty for existing algorithms surprised us.
Figure 5 shows the performance of six current RNA design algorithms on these puzzles, highlighting the strengths and weaknesses of different methods. Five of the six algorithms seek sequences whose predicted minimum free energy structure is the target structure, the same success conditions as are used in Eterna in silico challenges. The exception, NUPACK, optimizes the ensemble defect of the sequence, a partition function based measure of the entire ensemble’s deviation from the desired structure, and has not yet been optimized for long RNA lengths; we nevertheless included it due to its wide use.
Figure 5. Performance of existing algorithms on the Eterna100 benchmark.

Six RNA design algorithms were evaluated using the Eterna100 benchmark. [A] The successes (green) and failures (red) are shown for each algorithm. The puzzles are ordered by the number of successful solvers on Eterna, from fewest to most, and the algorithms are labeled with the number of puzzles solved. [B] The amount of time required to reach a solution for puzzles of different lengths is shown for each algorithm. Lines show the median values over each bin of lengths.
Top Eterna players were able to solve all 100 puzzles. Overall, MODENA is unable to handle structures of fewer than 10 bases but outperforms all other algorithms on some of the longest designs, solving 54 of the 100 puzzles. INFO-RNA solves a similar number of puzzles (50) but runs 100 times faster. Despite these individual areas in which particular algorithms shine, most fare poorly on the harder puzzles. For example, symmetrical structures, including Mutated Chicken Feet, stump all of the algorithms but could be solved by players. We chose not to include EteRNABot, as it was created to design for in vitro rather than in silico performance.
Discussion
Player-led scientific writing
This article has presented scientific insights collated from extensive write-ups by citizen scientist “players” with no training in biochemistry or bioengineering prior to their involvement in Eterna, including two of the lead authors. Despite the unusual origin of these insights and writing, both the citizen and conventional scientist authors are comfortable presenting the results, in wording identical to or closely adapted from the original player write-ups, due to independent rigorous tests involving new algorithms and supercomputer resources unavailable to the players during their studies. Internet-scale hypothesis generation and subsequent independent tests devised by expert groups has served as a working template for scientific discovery [18–21], but the expert groups have typically written the resulting publications. Our work illustrates that players can take the lead in manuscript writing, and we suggest that this updated template may relieve bottlenecks in disseminating scientific results from internet-scale projects and better reflect the intellectual ownership of reported discoveries.
Design Lessons
Despite numerous algorithmic and experimental testing advances addressing the inverse RNA folding problem, a simple set of guidelines for which RNA secondary structures are more or less amenable to design has not been available. Citizen scientist participants of Eterna have now delineated specific structural features that increase the difficulty of the inverse RNA folding problem. These insights include relating average stem length with problem difficulty, a principle of least elements to reduce complexity, an avoidance of specific structural motifs such as the “zig-zag”, and the avoidance of repetitive elements and symmetrical structures. We have confirmed these insights herein through independent tests on six widely-used RNA design algorithms. We have also noted how biological RNA molecules appear to avoid difficult features such as exact symmetry. Recent RNA engineering efforts that seek to express RNAs in tandem arrays, to present unpaired RNA stretches available for recognition, or to create complex nanostructures, may have trouble scaling up secondary structure designs if their basic building blocks violate these principles.
Current state-of-the-art RNA design algorithms currently do not evaluate a priori the difficulty of designing a specific RNA secondary structure. This makes it difficult for algorithms to adapt and automatically adjust to puzzle features or to gracefully quit when a puzzle is overly difficult or impossible to solve in silico. Future algorithms might by improved by explicitly recognizing these features in RNA secondary structure design problems and applying strategies specific to each feature. For instance, one common Eterna player strategy is to modify the strength of RNA stacks by adjusting the amount of G-C or A-U base-pairs based on the average stem length. In addition, “boosts,” or sequences that allow for more energetically favorable loops, can be used to stabilize local secondary structure. A more detailed player strategy involves using a compiled knowledge of which sequences solve specific substructures, such as zig-zags, long stretches of unpaired residues, or single-pair stems between long loops, and placing them first. Algorithms challenged with symmetric/repetitive problems could request more computational time for those cases. Given the success of Eterna players on even the hardest problems (Figure 5), we are optimistic that integrating their strategies into algorithms will lead to rapid improvement in automated RNA secondary structure design.
Towards experiments
For the purposes of this paper, we focus on obtaining in silico solutions, or sequences that fold into the intended structure according to computational folding models. In vitro and in vivo tests [11] introduce additional factors that complicate the problem further. Distinct from this study, which assumes a specific energy model of RNA folding, in vitro design requires additional knowledge of which structure and sequence features are more likely to be sensitive to inaccuracies in current energy models of RNA folding. An example is the repetition of long subsequences in a design – even if a design is predicted by a given energy model to fold into a stable structure with a low probability of misfolding, such sequence repetition risks misfolding that might be exposed in in vitro tests [11,22].
The Eterna lab has already generated an abundance of chemical mapping data characterizing player designs, and the rapid improvement of player designs across multiple rounds of synthesis is evidence that players have developed a predictive intuition for how to design RNA molecules that will fold correctly. These experiments and analysis has already allowed for the identification of specific features that correlate with the success of player designs in the lab [11], strategies to surmount these challenges, and proposal of rules that expand the in silico principles for design difficulty, described herein, to the problem of predicting design difficulty for in vitro tests. Prospective wet-lab tests of these insights, expanding the in silico tests presented here, are possible with Eterna’s current massively parallel experimental pipeline, which tests 10,000 designs per monthly round and will provide the most useful framework for future RNA engineering efforts. Finally, multi-state riboswitch design puzzle creation and solving tools (see also [23–25]) are now available in Eterna, along with large-scale experimental tests. A similar study to the present one detailing features that increase the difficulty of riboswitches and providing a benchmark for switch puzzles is an important next investigation.
Materials & Methods
For players’ in-game tests, RNAinverse 1.8, INFO-RNA 2.0, and RNA-SSD were run on all Eterna puzzles on an Amazon EC2 instance. All puzzles were attempted 5 times by each algorithm with a total time limit of 2 hours. Eterna players conducted experiments using a puzzle maker interface and the automatic execution of these three design algorithms to determine the weaknesses of these design algorithms. Players also used the EternaScript framework to design their own JavaScript-based puzzle solvers. One group of players used iterative development (the ‘Random Mutation’ series of scripts) to develop a heuristic solver algorithm (see Supporting Documents). After each development cycle, the script was run against a benchmark set of puzzles and weaknesses were identified based on failures, allowing for further adaptations of the algorithm. In so doing, these players developed an intuition for what kinds of elements are hard to solve. Many of these original benchmark puzzles are part of the Eterna100, and the resulting insights were collated by players into Google documents (see Supporting Documents) and wiki pages available through the Eterna wiki (http://eternawiki.org/wiki/).
For the validation tests herein on the proposed Eterna100 benchmark, RNAinverse 2.1.9, INFO-RNA 2.1.2, NUPACK 3.0.5, RNA-SSD, DSS-Opt, and MODENA were selected as a representative set of existing RNA design algorithms. Each secondary structure was run through all six algorithms using default parameters, with five attempts and a time limit of 24 hours for each attempt. The computation was carried out on Intel Xeon Processors E5-2650. NUPACK uses its own custom fold function, based on the Turner 2004 parameter set [26], while the other five algorithms use the RNAfold function from ViennaRNA 2.1.9, based on the Turner 1999 parameters [27]. Eterna uses the fold function from ViennaRNA 1.8.5, which is also based on the Turner 1999 parameters. We chose to allow these differences in energetic parameters in order to produce an evaluation of each algorithm by its own favored rule set. Each solution was evaluated using the folding function used by the design algorithm to confirm whether or not the minimum free energy structure corresponded to the input structure.
Supplementary Material
Research Highlights.
We outline several secondary structure elements and structural idiosyncrasies that lead to difficult RNA design problems, and test a principle of least elements.
We describe the contributions of symmetry to design difficulty and its interplay with the presence and repetition of difficult secondary structure elements.
We introduce the Eterna100 benchmark for evaluating RNA design methods and evaluate six existing algorithms using this benchmark.
This is the first paper based on dominant writing contributions – and co-lead authorship – by non-expert citizen scientists recruited through a video game.
Acknowledgments
We thank Jeehyung Lee for his work on developing the Eterna platform. This work was funded through a Burroughs-Wellcome Foundation Career Award (to R.D.) and National Institutes of Health Grant R01 R01GM100953 (to R.D. and A.T.). M.W. was supported by the NSF Graduate Research Fellowship under Grant No. DGE-114747, and M.L. was supported by the Samsung Scholarship. Eterna100 benchmark calculations were performed on the Stanford BioX3 cluster.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Xia T, SantaLucia J, Burkard ME, Kierzek R, Schroeder SJ, Jiao X, et al. Thermodynamic parameters for an expanded nearest-neighbor model for formation of RNA duplexes with Watson-Crick base pairs. Biochemistry. 1998;37:14719–35. doi: 10.1021/bi9809425. [DOI] [PubMed] [Google Scholar]
- 2.Hofacker IL, Fontana W, Stadler PF, Bonhoeffer LS, Tacker M, Schuster P. Fast folding and comparison of RNA secondary structures. Monatshefte Für Chemie Chem Mon. 1994;125:167–88. doi: 10.1007/BF00818163. [DOI] [Google Scholar]
- 3.Patzel V, Rutz S, Dietrich I, Köberle C, Scheffold A, Kaufmann SHE. Design of siRNAs producing unstructured guide-RNAs results in improved RNA interference efficiency. Nat Biotechnol. 2005;23:1440–4. doi: 10.1038/nbt1151. [DOI] [PubMed] [Google Scholar]
- 4.Jinek M, Jiang F, Taylor DW, Sternberg SH, Kaya E, Ma E, et al. Structures of Cas9 endonucleases reveal RNA-mediated conformational activation. Science. 2014;343:1247997. doi: 10.1126/science.1247997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Geary C, Rothemund PWK, Andersen ES. RNA nanostructures. A single-stranded architecture for cotranscriptional folding of RNA nanostructures. Science. 2014;345:799–804. doi: 10.1126/science.1253920. [DOI] [PubMed] [Google Scholar]
- 6.Andronescu M, Fejes AP, Hutter F, Hoos HH, Condon A. A new algorithm for RNA secondary structure design. J Mol Biol. 2004;336:607–24. doi: 10.1016/j.jmb.2003.12.041. [DOI] [PubMed] [Google Scholar]
- 7.Busch A, Backofen R. INFO-RNA--a fast approach to inverse RNA folding. Bioinformatics. 2006;22:1823–31. doi: 10.1093/bioinformatics/btl194. [DOI] [PubMed] [Google Scholar]
- 8.Matthies MC, Bienert S, Torda AE. Dynamics in Sequence Space for RNA Secondary Structure Design. J Chem Theory Comput. 2012;8:3663–70. doi: 10.1021/ct300267j. [DOI] [PubMed] [Google Scholar]
- 9.Zadeh JN, Steenberg CD, Bois JS, Wolfe BR, Pierce MB, Khan AR, et al. NUPACK: Analysis and design of nucleic acid systems. J Comput Chem. 2011;32:170–3. doi: 10.1002/jcc.21596. [DOI] [PubMed] [Google Scholar]
- 10.Taneda A. MODENA: a multi-objective RNA inverse folding. Adv Appl Bioinform Chem. 2011;4:1–12. doi: 10.2147/aabc.s14335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee J, Kladwang W, Lee M, Cantu D, Azizyan M, Kim H, et al. RNA design rules from a massive open laboratory. Proc Natl Acad Sci U S A. 2014;111:2122–7. doi: 10.1073/pnas.1313039111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tokuriki N, Tawfik DS. Protein dynamism and evolvability. Science. 2009;324:203–7. doi: 10.1126/science.1169375. [DOI] [PubMed] [Google Scholar]
- 13.Mandal M, Lee M, Barrick JE, Weinberg Z, Emilsson GM, Ruzzo WL, et al. A glycine-dependent riboswitch that uses cooperative binding to control gene expression. Science. 2004;306:275–9. doi: 10.1126/science.1100829. [DOI] [PubMed] [Google Scholar]
- 14.Huang L, Serganov A, Patel DJ. Structural insights into ligand recognition by a sensing domain of the cooperative glycine riboswitch. Mol Cell. 2010;40:774–86. doi: 10.1016/j.molcel.2010.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Butler EB, Xiong Y, Wang J, Strobel SA. Structural basis of cooperative ligand binding by the glycine riboswitch. Chem Biol. 2011;18:293–8. doi: 10.1016/j.chembiol.2011.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Severcan I, Geary C, Chworos A, Voss N, Jacovetty E, Jaeger L. A polyhedron made of tRNAs. Nat Chem. 2010;2:772–9. doi: 10.1038/nchem.733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wendell D, Jing P, Geng J, Subramaniam V, Lee TJ, Montemagno C, et al. Translocation of double-stranded DNA through membrane-adapted phi29 motor protein nanopores. Nat Nanotechnol. 2009;4:765–72. doi: 10.1038/nnano.2009.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lintott CJ, Schawinski K, Slosar A, Land K, Bamford S, Thomas D, et al. Galaxy Zoo: morphologies derived from visual inspection of galaxies from the Sloan Digital Sky Survey ★. Mon Not R Astron Soc. 2008;389:1179–89. doi: 10.1111/j.1365-2966.2008.13689.x. [DOI] [Google Scholar]
- 19.Lee J, Kladwang W, Lee M, Cantu D, Azizyan M, Kim H, et al. RNA design rules from a massive open laboratory. Proc Natl Acad Sci U S A. 2014;111:2122–7. doi: 10.1073/pnas.1313039111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Treuille A, Das R. Scientific rigor through videogames. Trends Biochem Sci. 2014;39:507–9. doi: 10.1016/j.tibs.2014.08.005. [DOI] [PubMed] [Google Scholar]
- 21.Cooper S, Khatib F, Treuille A, Barbero J, Lee J, Beenen M, et al. Predicting protein structures with a multiplayer online game. Nature. 2010;466:756–60. doi: 10.1038/nature09304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Seeman NC, Zhang Y, Du SM, Chen J. Construction of DNA Polyhedra and Knots Through Symmetric Minimization. In: Siegel JS, editor. Supramol Stereochem. Dordrecht: Springer Netherlands; 1995. pp. 27–32. [DOI] [Google Scholar]
- 23.Lyngsø RB, Anderson JWJ, Sizikova E, Badugu A, Hyland T, Hein J. Frnakenstein: multiple target inverse RNA folding. BMC Bioinformatics. 2012;13:260. doi: 10.1186/1471-2105-13-260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Höner Zu Siederdissen C, Hammer S, Abfalter I, Hofacker IL, Flamm C, Stadler PF. Computational design of RNAs with complex energy landscapes. Biopolymers. 2013;99:1124–36. doi: 10.1002/bip.22337. [DOI] [PubMed] [Google Scholar]
- 25.Shu W, Liu M, Chen H, Bo X, Wang S. ARDesigner: a web-based system for allosteric RNA design. J Biotechnol. 2010;150:466–73. doi: 10.1016/j.jbiotec.2010.10.067. [DOI] [PubMed] [Google Scholar]
- 26.Mathews DH, Disney MD, Childs JL, Schroeder SJ, Zuker M, Turner DH. Incorporating chemical modification constraints into a dynamic programming algorithm for prediction of RNA secondary structure. Proc Natl Acad Sci U S A. 2004;101:7287–92. doi: 10.1073/pnas.0401799101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mathews DH, Sabina J, Zuker M, Turner DH. Expanded sequence dependence of thermodynamic parameters improves prediction of RNA secondary structure. J Mol Biol. 1999;288:911–40. doi: 10.1006/jmbi.1999.2700. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.