

Abstract
Dioxin is a ubiquitous environmental pollutant that induces toxicity when bound to the aryl hydrocarbon receptor (AhR). Significant differences in susceptibility of mouse strains to dioxin toxicity are largely accounted for by the dissociation constant of binding to dioxins of AhR subtypes encoded by different alleles. We showed that cyclooxygenase-2 (COX-2) and microsomal prostaglandin E synthase-1 (mPGES-1), components of a prostanoid synthesis pathway, play essential roles in the onset of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) induced hydronephrosis of neonatal mice. Although C57BL/6J and BALB/cA mice harbor AhR receptors highly responsive to TCDD, they were found by chance to differ significantly in the incidence of TCDD-induced hydronephrosis. Therefore, the goal of the present study was to determine the molecular basis of this difference in susceptibility to TCDD toxicity. For this purpose, we administered C57BL/6J and BALB/cA dams’ TCDD at an oral dose of 15 or 80 μg/kg on postnatal day (PND) 1 to expose pups to TCDD via lactation, and the pups’ kidneys were collected on PND 7. The incidence of hydronephrosis in C57BL/6J pups (64%) was greater than in BALB/cA pups (0%, p < 0.05), despite similarly increased levels of COX-2 mRNA. The incidence of hydronephrosis in these mouse strains paralleled the levels of renal mPGES-1 mRNA and early growth response 1 (Egr-1) that modulates mPGES-1 gene expression, as well as PGE2 concentrations in urine. Although these mouse strains possess AhR alleles tightly bound to TCDD, their difference in incidence and severity of hydronephrosis can be explained, in part, by differences in the expression of mPGES-1 and Egr-1.
Keywords: dioxin, hydronephrosis, prostaglandin, mouse strain difference
Dioxins and related compounds are comprised of polychlorinated dibenzo-p-dioxins (PCDDs), polychlorinated dibenzofurans (PCDFs), and polychlorinated biphenyls (PCBs). Of 419 congeners, 29 congeners (7 PCDDs, 10 PCDFs, and 12 PCBs) are classified as dioxin-like chemicals (van den Berg et al., 1998, 2006), which ubiquitously persist in the environment and biosphere. Therefore, they accumulate in various food items via biomagnification (Uemura, 2012). Among the 29 congeners, 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) is the most potent and is used as a prototype in toxicology research (Schecter and Gasiewicz, 2003). The deleterious effects of TCDD include carcinogenicity, reproductive and endocrine toxicity, developmental toxicity, and immunotoxicity (WHO, 2002). The toxicity of TCDD as well as other dioxin congeners is mediated by the aryl hydrocarbon receptor (AhR). The essentiality of AhR in the manifestation of dioxin toxicity is proved using AhR-null mice (Mimura et al., 1997; Peters et al., 1999). A widely accepted concept to explain dioxin toxicity maintains that the ligand-AhR (e.g., TCDD-AhR) complex that is associated with its transcription partner AhR nuclear translocator (Arnt) can be bound to the AhR-responsive element (also named xenobiotic responsive element or dioxin responsive element) present in the promoter regions of certain genes. This interaction upregulates the transcription of genes such as those encoding cytochrome P4501A1, AhR repressor (AhRR), and UDP-glucuronosyltransferase (Beischlag et al., 2008; Fujisawa-Sehara et al., 1987; Hoffman et al., 1991; Puga et al., 2009). The influence of AhR in mediating dioxin toxicity depends on its binding affinity for dioxin-like chemicals, which is determined by the structure of its ligand-binding domain (Poland and Glover, 1980; Poland et al., 1994).
AhR is encoded by four different alleles in inbred mice as follows: AhRb-1, AhRb-2, AhRb-3, and AhRd (Okey et al., 1989; Poland and Glover, 1980). The dissociation constants for TCDD for AhRs encoded by these AhR alleles differ. The mouse strains having different alleles manifest different dose response relationships for TCDD toxicity. For example, DBA/2 mice that harbor AhRd/d are much less responsive than C57BL/6 and BALB/c mice that have AhRb-1/b-1 and AhRb-2/b-2 alleles, respectively (Ema et al., 1994; Poland et al., 1994). The amino acid sequences of AhRs expressed by C57BL/6 and BALB/c mice are identical except for a single amino acid residue change in the second 50-amino acid sequence repeat (PAS boxes); their dissociation constants for TCDD are 6 and 10 pM (Poland et al., 1994), respectively, in contrast to 37 pM in DBA/2 mice. A unique feature of dioxin toxicity is the extremely large difference in susceptibility among animal species and strains that correlates with the dissociation constants of dioxin-like chemicals for AhR (Moriguchi et al., 2003). For example, the lethal dose (LD) 50 values of TCDD for guinea pigs and hamsters are approximately 0.6 μg/kg (McConnell et al., 1978; Schwetz et al., 1973) and 1160–5050 μg/kg (Henck et al., 1981; Olson et al., 1980), respectively. The LD50 values of rats, mice, monkeys, and others vary within this range. The correlation between LD50 and the dissociation constants is illustrated by findings that the LD50 values of TCDD of male Hartley guinea pigs and New Zealand albino rabbits are 0.6 μg/kg and 115 μg/kg, respectively; and their corresponding dissociation constants of AhR for TCDD are 0.32 nM and 7.82 nM, respectively (Denison and Wilkinson, 1985; Schwetz et al., 1973). Among mouse strains, LD50 and dissociation constant values of DBA/2 mice are 536 μg/kg and 37 pM, respectively, and those of C57BL/6 mice are 114 μg/kg and 9−10 pM, respectively (Ema et al., 1994; Poland et al., 1994; Shen et al., 1991).
In contrast to these findings, little is known about the role of AhR in determining susceptibility to specific diseases. To fill this gap in our knowledge, we chose to study hydronephrosis, a hallmark of TCDD toxicity, which is induced by lactational exposure to TCDD in rodents (Couture-Haws et al., 1991). Using this lactational exposure model, our previous studies discovered that cyclooxygenase (COX)-2 and microsomal prostaglandin E synthase-1 (mPGES-1) play essential roles in TCDD-induced hydronephrosis in C57BL/6J pups (Nishimura et al., 2008; Yoshioka et al., 2012a). In our previous study (Yoshioka et al., 2012a), we unexpectedly found that the incidence of TCDD-induced hydronephrosis in BALB/cA pups seemed to be lower than that of C57BL/6J pups, both of which express AhR subtypes with similar affinities for TCDD (Poland et al., 1994). Here, we confirm that BALB/cA pups do not develop TCDD-induced hydronephrosis, and show that the expression of early growth response 1 (Egr-1) that regulates mPGES-1 gene expression may determine the difference in susceptibility of C57BL/6J and BALB/cA to TCDD-induced neonatal hydronephrosis.
MATERIALS AND METHODS
Animals and treatments
TCDD (purity, >99.1%) purchased from AccuStandard (New Haven, MA) was diluted in corn oil containing 1% n-nonane (Wako Pure Chemicals, Osaka, Japan). Pregnant C57BL/6J and BALB/cA mice were purchased from CLEA Japan (Tokyo, Japan). Mice were housed at 23°C and 50% humidity with a 12-h light/12-h dark cycle. Laboratory rodent chow (Labo MR Stock; Nosan, Yokohama, Japan) and distilled water were freely available. The Animal Care and Use Committee of the Graduate School of Medicine of the University of Tokyo approved the experimental protocols.
Parturition was checked twice daily, and the day of birth was designated postnatal day 0 (PND 0). Dams were orally administered TCDD (15 or 80 μg/kg, 15 ml/kg body weight) or an equivalent volume of control on PND 1 to expose pups lactationally to TCDD. Pups were euthanized by inhalation of diethyl ether (Wako Pure Chemicals, Osaka, Japan) on PND 7 to collect urine from the bladder and kidney tissue. Dams were randomly assigned to a TCDD (N = 4–5) or control group (N = 4).
Histological analysis of kidneys
The right kidneys from male pups were fixed in 10% neutral-buffered formalin solution, cryoprotected in 10% sucrose for 5 h and then in 20% sucrose overnight, embedded in OCT compound (Sakura Finetek Japan, Tokyo, Japan), snap-frozen in liquid nitrogen, and then cut into 5 μm thick sections. The tissue sections were stained with hematoxylin and eosin. Severity scores of hydronephrosis ranging from 0 (undetectable) to +4 (most severe) were used (Bryant et al., 2001). The incidence of hydronephrosis was defined as a severity score ≥2 (Bryant et al., 2001; Yoshioka et al., 2012a).
Quantitative real-time reverse transcriptase-PCR (qRT-PCR)
The left kidneys of pups were minced in TRIzol (Invitrogen, Carlsbad, CA), and total RNA was isolated from the aqueous phase of the suspension using a NucleoSpin RNA II kit (Macherey-Nagel, Duren, Germany). Synthesis of cDNA was performed using an oligo-dT20 primer and SuperScript III (Invitrogen).
Gene expression levels were quantitated using a LightCycler System (Roche Molecular Biochemicals, Indianapolis, IN) and a premix containing SYBR Green (Thunderbird; Toyobo, Osaka, Japan). Specific primers were designed using Primer3 software (Rozen and Skaletsky, 2000) and their sequences are described in previous reports (Yoshioka et al., 2012a,b), except for COX-2 (sense 5′-TGTGAACAATCAAACAAAATGATG-3′ and antisense 5′-GCGTAA ATTCCAACAGCCTAAGT-3′). Cross-contamination between samples or nonspecific amplification was assessed using water instead of DNA templates. Melting curve analyses of the products were conducted for every PCR reaction to verify specificity. The mRNA expression levels were calculated using the delta Ct method and normalized with Ct for cyclophilin B.
PGE2 assay
Urine was collected using 29-gauge syringe needles from the bladder of pups on PND 7. An enzyme immunoassay kit was used to measure PGE2 concentrations according to the manufacturer's instructions (Cayman Chemicals, Ann Arbor, MI).
Urine osmolality assay
Urine osmolality was measured using a freezing point depression method with a Fiske 210 Micro-Sample Osmometer (Advanced Instruments, Norwood, MA) according to the manufacturer's instructions.
Chromatin immunoprecipitation assay
Chromatin immunoprecipitation (ChIP) assay was performed using a SimpleChIP Plus kit (Cell Signaling Technology, Danvers, MA) according to the manufacturer's instruction. Kidneys were harvested from a selected female pup of each dam, because there were no gender differences in the onset of hydronephrosis and gene profiles (Yoshioka et al., 2012a, 2014). After chromatin digestion, a rabbit polyclonal antibody against Egr-1 (Santa Cruz Biotechnology, Santa Cruz, CA; Cat No sc-110) was used for chromatin immunoprecipitation. Semi-quantitative PCR to detect the mPGES-1 promoter was conducted using the primers 5′-CACCTCCATTCCTCTGCTTC-3′ (sense) and 5′-CGGGCCAAGTCCTGAGTAG-3′ (antisense), and LA Taq Hot StartVersion (Takara Bio, Otsu, Japan). PCR products obtained after 25, 30, 32, 35, 37, 38, and 40 cycles were separated using electrophoresis through 2% agarose gels and analyzed for the band density, and 35-cycle was optimal for the quantification. Quantitative PCR to amplify the COX-2 promoter was performed using a LightCycler System and a premix containing SYBR Green (Thunderbird; Toyobo) with the primers 5′-CCCGGAGGGTAGTTCCATGAAAGACTTCAAC-3′ (sense) and 5′- GGTGGAGCTGGCAGGATGCAGTCCTG-3′ (antisense). The signal relative to input was calculated using a formula from the manufacture's protocol as follows:
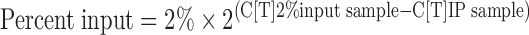 |
where C[T] = threshold cycle.
Statistical analysis
To minimize litter effects, the data for individual pups with the same variables such as sex, strain, and TCDD dose were averaged for each litter and then averaged among litters. The data represent the mean ± standard error of the mean (SEM) for the number of litters. Differences in mean values between groups were analyzed using one-way ANOVA followed by pairwise comparisons using the Bonferroni post hoc test. p values <0.05 were defined as significant.
RESULTS
Incidence and Severity of TCDD-Induced Neonatal Hydronephrosis of C57BL/6J and BALB/cA Pups
Spontaneous hydronephrosis was not observed in control C57BL/6J or BALB/cA pups on PND 7. On PND 7, the administration of 15 μg TCDD/kg to C57BL/6J dams induced hydronephrosis in 64% of their pups. The incidence and severity of hydronephrosis of C57BL/6J pups observed in this study is consistent with those observed in previous studies (Couture-Haws et al., 1991; Nishimura et al., 2008; Yoshioka et al., 2012a). In contrast, no hydronephrosis was observed in neonates of BALB/cA dams given either 15 μg TCDD/kg or 80 μg TCDD/kg (Table 1 and Fig. 1).
TABLE 1. Incidence and Severity of Hydronephrosis in C57BL/6J and BALB/cA Pups.
| Severityb | ||||||
|---|---|---|---|---|---|---|
| Dose (μg/kg) | Na[pups(dams)] | 0 | 1 | ≥2 | Incidencec (%) | |
| C57BL/6J | 0 | 13 (4) | 11 | 2 | 0 | 0 |
| 15 | 11 (5) | 0 | 4 | 7 | 64 | |
| BALB/cA | 0 | 12 (4) | 11 | 1 | 0 | 0 |
| 15 | 11 (4) | 9 | 2 | 0 | 0 | |
| 80 | 8 (4) | 6 | 2 | 0 | 0 | |
aNumber of pups and dams in each group.
bNumber of pups of each severity score.
cNormalized incidence of hydronephrosis (see the Materials and Methods section).
FIG. 1.
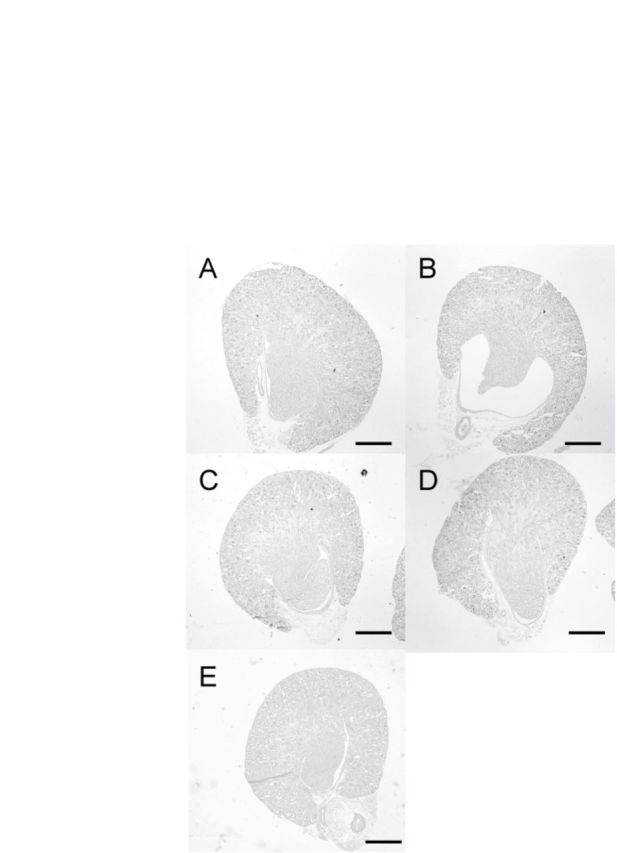
Histology of a representative kidney of a 7-day-old C57BL/6J and BALB/cA pup lactationally exposed to TCDD. TCDD (15 or 80μg/kg) or control was administered to dams on PND 1, and pups were exposed to TCDD via lactation. Hematoxylin and eosin staining of the kidneys of C57BL/6J pups treated with control (A) or 15 μg TCDD/kg (B), BALB/cA pups treated with control (C) or 15 μg TCDD/kg (D), or 80 μg TCDD/kg (E). Scale bars indicate 500 μm.
Expression of AhR-Target Genes in Response to TCDD
We next determined the effect of lactational exposure to TCDD on the transcription of AhR target genes CYP1A1 and AhRR (Mimura et al., 1999; Mimura and Fujii-Kuriyama, 2003) in the kidneys of neonatal C57BL/6J and BALB/cA mice. The basal levels of these mRNAs were significantly low and similar between the two mouse strains (Figs. 2A and B). Pups of both mouse strains exposed to TCDD significantly increased the expression of these two genes in the kidney tissues compared with controls (Figs. 2A and B). It has been reported that C57BL/6J and BALB/cA mice harbor AhR that binds TCDD with high affinity and that maternal exposure to AhR agonist, 3-methylcholanthrene, induces CYP1A1, a representative AhR target gene, at almost same levels in C57BL/6 and BALB/c fetal lungs (Poland et al., 1994; Xu et al., 2005). Likewise, CYP1A1 and AhRR transcript levels induced by TCDD in BALB/cA pups were comparative with those in C57BL/6J pups when BALB/cA dams were administered TCDD at a dose of 80 μg/kg compared with C57BL/6J dams given TCDD at a dose of 15 μg/kg (Figs. 2A and B). Thus, a remarkable difference in the incidence of TCDD-induced hydronephrosis between these two mouse strains cannot be explained by the affinity and transactivation activity of AhR subtypes to TCDD, suggesting that there is another yet unidentified factor that may determine susceptibility to TCDD-induced neonatal hydronephrosis.
FIG. 2.

Expression of genes regulated by AhR. CYP1A1 (A) and AhRR (B) on PND 7 pups. See the legend to Figure 1 for a TCDD exposure protocol. RT-PCR analysis of mRNA levels normalized to that of Cyclophilin B (CypB).
Analysis of PGE2 Levels and TCDD-Induced Expression of Genes Encoding PGE2 Synthases
To identify a factor that determines susceptibility to TCDD-induced neonatal hydronephrosis, we administered TCDD to C57BL/6J and BALB/cA dams and compared the PGE2 synthetic pathway, including COX-2 and mPGES-1 enzymes in the pups, because this pathway is responsible for the onset of TCDD-induced neonatal hydronephrosis (Nishimura et al., 2008; Yoshioka et al., 2012a). The levels of PGE2 in the urine of C57BL/6J pups lactationally exposed to TCDD were significantly higher compared with those of control pups (Fig. 3A). In contrast, the levels of PGE2 in the urine of BALB/cA pups exposed at a dose of 80 μg TCDD/kg were nearly half compared with those in the C57BL/6J pups exposed at a dose of 15 μg TCDD/kg (Fig. 3A). Further, the urinary concentration of PGE2 in TCDD exposed C57BL/6 pups with hydronephrotic kidneys was significantly higher than that with nonhydronephrotic kidneys (Supplementary fig. S1).
FIG. 3.

Strain differences in urinary PGE2 concentration (A) and mRNA levels of COX-2 (B) and mPGES-1(C), mPGES-2 (D), and cPGES (E) on PND 7. See the legend to Figure 1 for a TCDD exposure protocol. The mRNA levels are normalized to that of cyclophilin B (CypB). Histograms show the mean ± SEM for N = 4–5 dams per group.
Because COX-2 metabolizes arachidonic acid to prostaglandin H2, which is converted to PGE2 by PGE2 synthases, we next studied the effect of TCDD on the levels of mRNA encoding COX-2, microsomal prostaglandin E2 synthase-1 (mPGES-1), mPGES-2, and cytosolic prostaglandin E2 synthase (cPGES) in the kidneys of mouse pups. The basal expression levels of COX-2 mRNA were similar between C57BL/6J and BALB/cA pups (Fig. 3B), and TCDD exposure increased the levels of COX-2 mRNA in C57BL/6J and BALB/cA pups (Fig. 3B). There was no significant difference in the COX-2 induction between C57BL/6J and BALB/cA pups at a dose of 15 μg TCDD/kg. Among the three kinds of PGE2 synthases, TCDD exposure induced a significant increase in the levels of mPGES-1 mRNA in C57BL/6J pups compared with the control pups, but it did not alter the levels in BALB/cA pups (Fig. 3C). No alterations in the levels of mRNAs of the other two PGE2 synthases, mPGES-2 (Fig. 3D) or cPGES (Fig. 3E), were found in C57BL/6J or BALB/cA upon lactational exposure to TCDD.
Search for a Novel Factor that Regulates mPGES-1 Gene Expression in TCDD-Exposed C57BL/6J and BALB/cA Pups
Because we detected a strain difference in the upregulation of the gene encoding mPGES-1 following TCDD exposure, we next searched whether cis-acting elements potentially regulated mPGES-1 gene expression in C57BL/6J and BALB/cA mice using the mouse SNP database of the Center for Genome Dynamics of the Jackson laboratory (Mouse Genome Informatics, 2014). The nucleotide sequences of the cis-acting elements present in the two mouse strains are identical. Thus, we searched for trans-acting factors that may influence the expression of the gene encoding mPGES-1. First, we determined the mRNA levels encoding interleukin-1β (IL-1β) and tumor necrosis factor (TNF)-α in the kidneys of PND 7 pups, because these cytokines induce COX-2 and mPGES-1 synthesis through distinct pathways (Rzymkiewicz et al., 1995; Srivastava et al., 1994; Subbaramaiah et al., 2004; Wang et al., 2002). TCDD administration to dams at 15 μg/kg significantly increased the levels of IL-1β mRNA (Fig. 4A) in the kidneys of C57BL/6J and BALB/cA pups compared with control pups. A similar result was also observed for TNF-α mRNA levels (Fig. 4B). No significant differences in the basal or induced levels of IL-1β (Fig. 4A) and TNF-α (Fig. 4B) mRNAs were observed between the two mouse strains.
FIG. 4.

Gene expression of IL-1β (A) and TNF-α (B) on PND 7. TCDD (15 μg/kg) or control was administered to dams on PND 1, and pups were exposed to TCDD via lactation. The mRNA levels were measured using RT-PCR and normalized to that of cyclophilin B (CypB).
We next determined the levels of expression of early growth response protein 1 (Egr-1) gene in the kidneys of the two mouse strains on PND 7, because Egr-1 is a major transcription factor that induces the expression of mPGES-1 (Naraba et al., 2002). The basal expression level of Egr-1 mRNA was similar between the two mouse strains, and TCDD exposure increased Egr-1 mRNA levels in the kidneys of C57BL/6J pups by a factor of two on PND 7 compared with the control pups. In contrast, TCDD exposure did not increase the level of Egr-1 mRNA in BALB/cA pups (Fig. 5A).
FIG. 5.
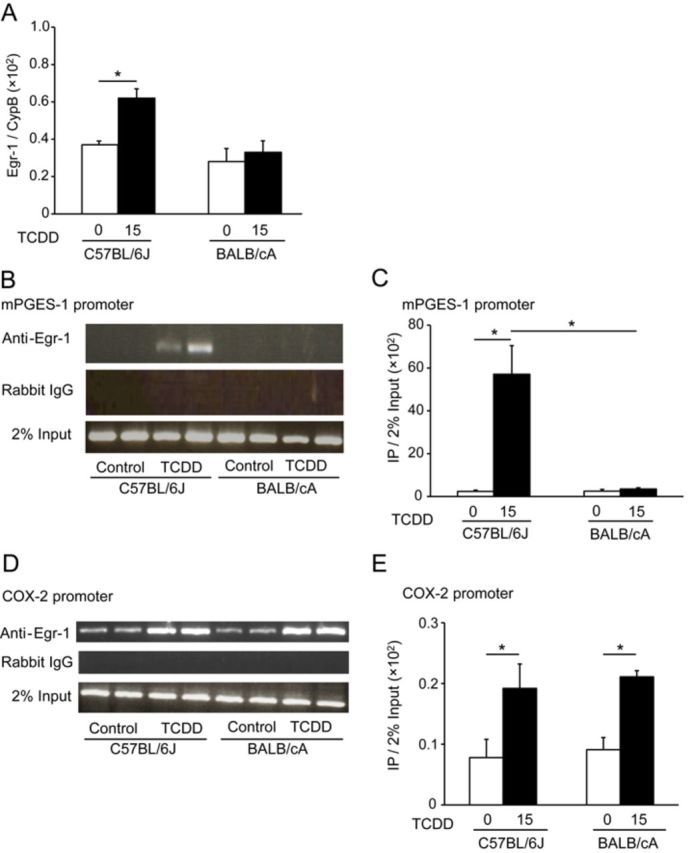
Egr-1 mRNA levels following TCDD exposure and Egr-1 binding to the mPGES-1 or COX-2 promoters in C57BL/6J and BALB/cA pups. See the legend to Figure 4 for a TCDD exposure protocol. The Egr-1 mRNA level was measured using RT-PCR and normalized to that of cyclophilin B (CypB). (A). The specific binding of Egr-1 to the mPGES-1 promoter was analyzed using a ChIP assay (B). Kidneys were collected from pups on PND 7 (N = 4). As a control, chromatin fragments were isolated before immunoprecipitation (2% input) using semi-quantitative PCR. Immunoprecipitation using rabbit IgG was performed in parallel. PCR products (10 μl) from different pups were loaded in each lane. The intensity of the bands was measured using ImageJ software (C). Binding of Egr-1 to the COX-2 promoter was analyzed using a ChIP assay. Kidneys were collected from pups on PND 7 (N = 4). Quantitative PCR was performed on chromatin fragments isolated before and after immunoprecipitation using an anti-Egr-1 antibody. Immunoprecipitation with normal rabbit serum was conducted to assess specificity. PCR products (5 μl) from different pups were analyzed (D). Signals relative to input were calculated using a formula included in the manufacturer's protocol. See the Materials and Methods section (E).
To determine whether the strain difference in Egr-1 mRNA levels led to the activation of mPGES-1 transcription, we performed ChIP assays of Egr-1 binding to the mPGES-1 promoter in vivo. Because the melting curve analyses of qPCR after ChIP assays produced ambiguous results possibly because of the complexity in the sequence of mPGES-1 promoter, we conducted semi-quantitative PCR (Fig. 5B). Binding of Egr-1 to the mPGES-1 promoter was detected only in kidney tissues of TCDD-exposed C57BL/6J pups, but not in those of control C57BL/6J pups (Fig. 5B). Under this assay condition, no bands were found in kidney specimens from TCDD-exposed BALB/cA pups and control BALB/cA pups (Fig. 5B). Furthermore, no bands were obtained using control rabbit IgG (Fig. 5B). The ChIP-PCR data are displayed quantitatively in Figure 5C.
Next, we investigated whether Egr-1 was bound to the COX-2 promoter in vivo, because such binding induces COX-2 gene expression in macrophages in vitro (Diaz-Munoz et al., 2010). Egr-1 was bound to the COX-2 promoter in the kidney of control and TCDD-exposed C57BL/6J pups, and the band intensity was increased by TCDD exposure (Fig. 5D). In the kidneys of control and TCDD-exposed BALB/cA pups, Egr-1 binding of the COX-2 promoter was similar to that of C57BL/6J pups (Fig. 5D). No visible bands were found in the use of control rabbit IgG (Fig. 5D). Quantitative PCR analysis revealed that the basal levels of Egr-1 bound to the COX-2 promoter were similar between C57BL/6J and BALB/cA pups, and that the amounts of Egr-1 bound to the COX-2 promoter were increased by factors of 2.5 and 2.3 in TCDD-induced C57BL/6J and BALB/cA pups, respectively, compared with controls (Fig. 5E).
Osmolality of Urine and Expression of Genes Encoding Water Channels and Electrolyte Transporters in the Kidneys of C57BL/6J and BALB/cA Pups
We analyzed the expression of genes encoding the water channel aquaporin 2 (AQP2) and the ROMK and NKCC2 electrolyte transporters, because our previous studies showed that the expression of these genes was decreased in hydronephrotic but not in nonhydronephrotic kidneys of TCDD-exposed C57BL/6J pups (Nishimura et al., 2008; Yoshioka et al., 2012a, 2014). In the present study, there was a tendency of decreased mRNA expression of AQP2 (Fig. 6A) and NKCC2 (Fig. 6B), while the decreased expression of ROMK (Fig. 6C) was statistically significant in the kidneys of TCDD-exposed C57BL/6J pups. A significant increase in AQP2 mRNA was found in TCDD-exposed BALB/cA pups (Fig. 6A). Because PGE2 influences AQP2 protein expression and trafficking (Li et al., 2009; Olesen and Fenton, 2013), we examined the correlation between urinary PGE2 concentration and AQP2 mRNA levels in TCDD-exposed C57BL/6J pups and BALB/cA pups. There were significant inverse correlations between PGE2 and AQP2 mRNA levels in both TCDD-exposed C57BL/6J pups and BALB/cA pups (R = −0.92 and −0.90, respectively) (Supplementary figs. S2A and B).
FIG. 6.

Strain difference in diuresis between C57BL/6J mice and BALB/cA mice on PND 7. The mRNA levels of AQP2 (A), NKCC2 (B), and ROMK (C) were measured using RT-PCR and normalized to that of cyclophilin B (CypB). Urine osmolality was measured using a freezing-point method (D). See the legend to Figure 4 for a TCDD exposure protocol.
AQP2, NKCC2, and ROMK affect the ability of kidneys to concentrate urine (Ares et al., 2011; Nielsen et al., 2002). Therefore, we determined urine osmolality because TCDD decreased the levels of ROMK mRNA in C57BL/6J pups and increased the levels of AQP2 mRNA in BALB/cA pups. Osmolality was significantly decreased in TCDD-exposed C57BL/6J pups, but not in BALB/cA pups, compared with the corresponding control pups (Fig. 6D).
DISCUSSION
A variety of dioxin toxicities, including cleft palate and hydronephrosis, is mediated by the cytosolic transcription factor AhR (Mimura et al., 1997; Peters et al., 1999; Schmidt et al., 1996), and AhR is thought to be a determinant of TCDD toxicity. In fact, a considerable difference in susceptibility to dioxin toxicity among animal species and strains is attributed to the binding affinity of TCDD to various AhR subtypes (Bank et al., 1992; Burbach et al., 1992). However, our present study revealed that TCDD-induced toxicity does not always depend on the binding affinity of TCDD. In the present study, the incidence and degree of hydronephrosis were greater in C57BL/6J pups than BALB/cA pups (Table 1) and were associated with induced levels of mPGES-1 mRNA in the kidney and those of PGE2 in urine (Figs. 3A and C) although AhR target genes, such as CYP1A1, AhRR, and COX-2, were induced in both strains (Figs. 2 and 3B). There are several studies of laboratory rodents which suggest that factors other than AhR affinity and AhR transcriptional activation influence TCDD toxicity. For example, TCDD exposure induces hepatic porphyria in SWR but not in DBA/2 mice, although they harbor an identical AhR allele (AhRd/d) (Smith et al., 1998). A quantitative trait loci (QTL) analysis suggested some modifier genes that are located on chromosome 11 and 14 in addition to AhR gene (chromosome 12) (Robinson et al., 2002). Keller and associates (Keller et al., 2007) investigated the effects of TCDD on molar development of C57BL/6J, BALB/cByJ, A/J, CBA/J, C3H/HeJ, and C57BL/10J mice, which harbor AhR alleles (AhRb-1/b-1 and AhRb-2/b-2) (Poland et al., 1994) that encode AhR with high binding affinity for TCDD. Despite similar dissociation constant values of their AhRs for TCDD, some of the mouse strains exhibit susceptibility to TCDD-induced disruption of mandibular development, whereas C57BL/6J and BALB/cByJ mice do not. Holtzman and Sprague Dawley rats harbor identical AhR alleles (Kawakami et al., 2006), but exhibit a large difference in the incidence of TCDD-induced fetal death because of abnormal vascular development under hypoxia in the placenta (Ishimura et al., 2002). Taken together, these studies using rodents suggest that genetic factors other than the AhR locus influence an organism's response to TCDD.
The mPGES-1 gene is activated by transcription factors NF-κB, Egr-1, hypoxia inducible factor (HIF), C/EBP, AP-1, and CACCC-binding factor that signal through distinct pathways (Diaz-Munoz et al., 2010; Maxwell, 2005; Naraba et al., 2002). To our knowledge, AhR response element (AhRE) was not present in mPGES-1 promoter. Moreover, there is no strain difference in transcriptional activity of AhR in CYP1A1 and AhRR gene expression. These findings suggest that the strain difference in the induction of mPGES-1 may be due to the factors other than canonical AhR/ARNT pathway. To explain the strain difference by a non-canonical pathway, the following speculations could be plausible. First, AhR/NF-κB may be involved in a mechanism of the strain difference because TCDD-activated AhR has crosstalk with NF-κB. For instance, NF-κB subunit RelA that is induced by inflammation regulates the expression of AhR (Vogel et al., 2014), and ligand-activated AhR binds RelA (Jensen et al., 2003; Tian et al., 1999) and RelB (Vogel et al., 2007). There is the possibility that such AhR/NF-κB crosstalk modulates TCDD-induced mPGES-1 gene expression. Second, AhR/Egr-1 crosstalk may be able to explain the strain difference because the complex of AhR and Egr-1 was reported to bind GC box, Egr-1 binding site in human endothelial cells under a high glucose concentration (Dabir et al., 2008), which may suggest that activated AhR may play a key role in Egr-1 recruitment in the neonatal kidney. Thus, yet-unidentified crosstalk between AhR and Egr-1 may be considered as a cause of strain difference in Egr-1 binding to mPGES-1 promoter. Third, the amount of protein of Egr-1, a main transcriptional regulator in the induction of mPGES-1, is increased in response to TCDD by the enhancement of stability of its mRNA in human lung epithelial cells (Martinez et al., 2004). Although the mechanism of TCDD-induced Egr-1 mRNA stability is not clear, a possible difference in the stability of Egr-1 mRNA between C57BL/6J and BALB/cA may explain the strain difference in mPGES-1 expression. Taken together, whether and how Egr-1 is involved in the difference in TCDD-induced hydronephrosis between mouse strains warrants future studies.
We present a scheme that depicts the possible roles of proteins that can play important roles in the regulation of the transcription of the mPGES-1 and Egr-1 genes (Fig. 7). Coordinated upregulation of COX-2 and mPGES-1 transcription occurs in mouse macrophages treated with lipopolysaccharide (Diaz-Munoz et al., 2010) and in certain tissues treated with proinflammatory molecules such as IL-1β and TNF-α (Murakami et al., 2000). Transcription of the COX-2 and mPGES-1 genes is induced by common signaling pathways that are mediated by NF-κB and Egr-1 (Diaz-Munoz et al., 2010; Dixon et al., 2013). In contrast, there are several reports showing apparently uncoordinated expression of the genes encoding COX-2 and mPGES-1. For example, dimethylcelecoxib, a non-COX-2 inhibiting derivative of celecoxib, inhibits PGE2 synthesis by inhibiting mPGES-1 expression. This inhibition was regulated by enhancing the binding of the NF-κB/histone deacetylase 1 (HDAC1) complex to the Egr-1 promoter, which decreases the amount of Egr-1 protein (Deckmann et al., 2012). Our present findings show that TCDD induced the expression of COX-2 and mPGES-1 mRNAs simultaneously in C57BL/6J pups. However, TCDD induced COX-2 mRNA only and failed to induce mPGES-1 mRNA in BALB/cA pups. The latter is likely due to a reduction in TCDD-induced Egr-1 gene expression, indicating the non-coordinated gene expression of COX2 and mPGES-1 in BALB/cA pups. The extent to which these transcription factors contribute to the strain-specific regulation of mPGES-1 and Egr-1 gene expression is unknown. How TCDD exposure regulates the complex signaling pathway that induces mPGES-1 during kidney development warrants future study.
FIG. 7.

Transcription factors that mediate the gene expression of mPGES-1 and Egr-1. The table below the scheme summarizes the gene expression data for Cyp1A1, AhRR, Egr-1, mPGES-1, and urinary PGE2 in TCDD-exposed C57BL/6J and BALB/cA pups compared with their corresponding controls. + indicates a complex of each trans-regulatory element. The dashed line is a proposed pathway. Up, upregulated; NC, not changed. (1) (Covert et al., 2005), (2) (Grimmer et al., 2007), (3) (Deckmann et al., 2012), and (4) (Jensen et al., 2003).
In TCDD-exposed pups with hydronephrotic kidneys, the levels of electrolyte transporters NKCC2 or ROMK are decreased (Nishimura et al., 2008; Yoshioka et al., 2012a, 2014). The results of our present findings demonstrate that the level of ROMK mRNA decreased similarly in TCDD-exposed C57BL/6J but not in TCDD-exposed BALB/cA pups. It therefore remains to be determined whether TCDD-induced alterations of the levels of these electrolyte transporters cause hydronephrosis or is a consequence of this condition. Furthermore, the levels of AQP2 mRNA represent another remarkable difference between the two mouse strains (Fig. 6A). PGE2 decreases AQP2 induction and trafficking from intracellular stores to the cellular membrane, which controls permeability of water in the collecting duct (Jia et al., 2012; Olesen and Fenton, 2013). In the present study, there was an inverse correlation between PGE2 and AQP2 mRNA levels in TCDD-exposed C57BL/6J pups and BALB/cA pups (Supplementary fig. S2). Thus, Egr-1-dependent regulation of mPGES-1 induction may alter the level of PGE2, which eventually determines the permeability of water in the collecting duct via AQP2 in TCDD-exposed C57BL/6J pups.
We reveal here a distinct difference in the onset of hydronephrosis between C57BL/6J pups and BALB/cA pups that have AhRs highly responsive to TCDD. We attribute this difference, in part, to less responsiveness of not only the induction of mPGES-1 transcription and subsequent PGE2 synthesis, but also Egr-1, which is a modulator of mPGES-1 gene expression. AhR is required to ensure the manifestation of dioxin toxicity by multiple signal transduction networks (Denison et al., 2011), but the present study presents a new example suggesting that the difference in dioxin susceptibility in animal strains that express AhRs with similar affinity for TCDD is regulated by other factors than AhRs.
SUPPLEMENTARY DATA
Supplementary data are available online at http://toxsci.oxfordjournals.org/.
FUNDING
Research grant from the Ministry of the Environment, Japan (to CT, in part).
Supplementary Material
Acknowledgments
We are grateful to Ms Yoshie Okuhara for her assistance in the preparation of illustrations.
Footnotes
Equally contributed to this work.
REFERENCES
- Ares G. R., Caceres P. S., Ortiz P. A. Molecular regulation of NKCC2 in the thick ascending limb. Am. J. Physiol. Renal Physiol. 2011;301:F1143–F1159. doi: 10.1152/ajprenal.00396.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bank P. A., Yao E. F., Phelps C. L., Harper P. A., Denison M. S. Species-specific binding of transformed Ah receptor to a dioxin responsive transcriptional enhancer. Eur. J. Pharmacol. 1992;228:85–94. doi: 10.1016/0926-6917(92)90016-6. [DOI] [PubMed] [Google Scholar]
- Beischlag T. V., Luis Morales J., Hollingshead B. D., Perdew G. H. The aryl hydrocarbon receptor complex and the control of gene expression. Crit. Rev. Eukaryot. Gene Expr. 2008;18:207–250. doi: 10.1615/critreveukargeneexpr.v18.i3.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant P. L., Schmid J. E., Fenton S. E., Buckalew A. R., Abbott B. D. Teratogenicity of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in mice lacking the expression of EGF and/or TGF-alpha. Toxicol. Sci. 2001;62:103–114. doi: 10.1093/toxsci/62.1.103. [DOI] [PubMed] [Google Scholar]
- Burbach K. M., Poland A., Bradfield C. A. Cloning of the Ah-receptor cDNA reveals a distinctive ligand-activated transcription factor. Proc. Natl. Acad. Sci. U. S. A. 1992;89:8185–8189. doi: 10.1073/pnas.89.17.8185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couture-Haws L., Harris M. W., Lockhart A. C., Birnbaum L. S. Evaluation of the persistence of hydronephrosis induced in mice following in utero and/or lactational exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicol. Appl. Pharmacol. 1991;107:402–412. doi: 10.1016/0041-008x(91)90304-w. [DOI] [PubMed] [Google Scholar]
- Covert M. W., Leung T. H., Gaston J. E., Baltimore D. Achieving stability of lipopolysaccharide-induced NF-kappaB activation. Science. 2005;309:1854–1857. doi: 10.1126/science.1112304. [DOI] [PubMed] [Google Scholar]
- Dabir P., Marinic T. E., Krukovets I., Stenina O. I. Aryl hydrocarbon receptor is activated by glucose and regulates the thrombospondin-1 gene promoter in endothelial cells. Circ. Res. 2008;102:1558–1565. doi: 10.1161/CIRCRESAHA.108.176990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deckmann K., Rorsch F., Geisslinger G., Grosch S. Dimethylcelecoxib induces an inhibitory complex consisting of HDAC1/NF-kappaB(p65)RelA leading to transcriptional downregulation of mPGES-1 and EGR1. Cell. Signal. 2012;24:460–467. doi: 10.1016/j.cellsig.2011.09.025. [DOI] [PubMed] [Google Scholar]
- Denison M. S., Soshilov A. A., He G., DeGroot D. E., Zhao B. Exactly the same but different: Promiscuity and diversity in the molecular mechanisms of action of the aryl hydrocarbon (dioxin) receptor. Toxicol. Sci. 2011;124:1–22. doi: 10.1093/toxsci/kfr218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denison M. S., Wilkinson C. F. Identification of the Ah receptor in selected mammalian species and induction of aryl hydrocarbon hydroxylase. Eur. J. Biochem. 1985;147:429–435. doi: 10.1111/j.1432-1033.1985.tb08767.x. [DOI] [PubMed] [Google Scholar]
- Diaz-Munoz M. D., Osma-Garcia I. C., Cacheiro-Llaguno C., Fresno M., Iniguez M. A. Coordinated up-regulation of cyclooxygenase-2 and microsomal prostaglandin E synthase 1 transcription by nuclear factor kappa B and early growth response-1 in macrophages. Cell. Signal. 2010;22:1427–1436. doi: 10.1016/j.cellsig.2010.05.011. [DOI] [PubMed] [Google Scholar]
- Dixon D. A., Blanco F. F., Bruno A., Patrignani P. Mechanistic aspects of COX-2 expression in colorectal neoplasia. Recent Results Cancer Res. 2013;191:7–37. doi: 10.1007/978-3-642-30331-9_2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ema M., Ohe N., Suzuki M., Mimura J., Sogawa K., Ikawa S., Fujii-Kuriyama Y. Dioxin binding activities of polymorphic forms of mouse and human arylhydrocarbon receptors. J. Biol. Chem. 1994;269:27337–27343. [PubMed] [Google Scholar]
- Fujisawa-Sehara A., Sogawa K., Yamane M., Fujii-Kuriyama Y. Characterization of xenobiotic responsive elements upstream from the drug-metabolizing cytochrome P-450c gene: A similarity to glucocorticoid regulatory elements. Nucleic Acids Res. 1987;15:4179–4191. doi: 10.1093/nar/15.10.4179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimmer C., Pfander D., Swoboda B., Aigner T., Mueller L., Hennig F. F., Gelse K. Hypoxia-inducible factor 1alpha is involved in the prostaglandin metabolism of osteoarthritic cartilage through up-regulation of microsomal prostaglandin E synthase 1 in articular chondrocytes. Arthritis Rheum. 2007;56:4084–4094. doi: 10.1002/art.23136. [DOI] [PubMed] [Google Scholar]
- Henck J. M., New M. A., Kociba R. J., Rao K. S. 2,3,7,8-tetrachlorodibenzo-p-dioxin: Acute oral toxicity in hamsters. Toxicol. Appl. Pharmacol. 1981;59:405–407. doi: 10.1016/0041-008x(81)90212-x. [DOI] [PubMed] [Google Scholar]
- Hoffman E. C., Reyes H., Chu F. F., Sander F., Conley L. H., Brooks B. A., Hankinson O. Cloning of a factor required for activity of the Ah (dioxin) receptor. Science. 1991;252:954–958. doi: 10.1126/science.1852076. [DOI] [PubMed] [Google Scholar]
- Ishimura R., Ohsako S., Kawakami T., Sakaue M., Aoki Y., Tohyama C. Altered protein profile and possible hypoxia in the placenta of 2,3,7,8-tetrachlorodibenzo-p-dioxin-exposed rats. Toxicol. Appl. Pharmacol. 2002;185:197–206. doi: 10.1006/taap.2002.9539. [DOI] [PubMed] [Google Scholar]
- Jensen B. A., Leeman R. J., Schlezinger J. J., Sherr D. H. Aryl hydrocarbon receptor (AhR) agonists suppress interleukin-6 expression by bone marrow stromal cells: An immunotoxicology study. Environ. Health. 2003;2:16. doi: 10.1186/1476-069X-2-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia Z., Liu G., Downton M., Dong Z., Zhang A., Yang T. mPGES-1 deletion potentiates urine concentrating capability after water deprivation. Am. J. Physiol. Renal Physiol. 2012;302:F1005–F1012. doi: 10.1152/ajprenal.00508.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawakami T., Ishimura R., Nohara K., Takeda K., Tohyama C., Ohsako S. Differential susceptibilities of Holtzman and Sprague-Dawley rats to fetal death and placental dysfunction induced by 2,3,7,8-teterachlorodibenzo-p-dioxin (TCDD) despite the identical primary structure of the aryl hydrocarbon receptor. Toxicol. Appl. Pharmacol. 2006;212:224–236. doi: 10.1016/j.taap.2005.08.007. [DOI] [PubMed] [Google Scholar]
- Keller J. M., Huet-Hudson Y. M., Leamy L. J. Qualitative effects of dioxin on molars vary among inbred mouse strains. Arch. Oral Biol. 2007;52:450–454. doi: 10.1016/j.archoralbio.2006.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J. H., Chou C. L., Li B., Gavrilova O., Eisner C., Schnermann J., Anderson S. A., Deng C. X., Knepper M. A., Wess J. A selective EP4 PGE2 receptor agonist alleviates disease in a new mouse model of X-linked nephrogenic diabetes insipidus. J. Clin. Invest. 2009;119:3115–3126. doi: 10.1172/JCI39680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez J.M., Baek S.J., Mays D.M., Tithof P.K., Eling T.E., Walker N.J. EGR1 is a novel target for AhR agonists in human lung epithelial cells. Toxicol. Sci. 2004;82:429–435. doi: 10.1093/toxsci/kfh272. [DOI] [PubMed] [Google Scholar]
- Maxwell P. H. The HIF pathway in cancer. Semin. Cell Dev. Biol. 2005;16:523–530. doi: 10.1016/j.semcdb.2005.03.001. [DOI] [PubMed] [Google Scholar]
- McConnell E. E., Moore J. A., Haseman J. K., Harris M. W. The comparative toxicity of chlorinated dibenzo-p-dioxins in mice and guinea pigs. Toxicol. Appl. Pharmacol. 1978;44:335–356. doi: 10.1016/0041-008x(78)90195-3. [DOI] [PubMed] [Google Scholar]
- Mimura J., Ema M., Sogawa K., Fujii-Kuriyama Y. Identification of a novel mechanism of regulation of Ah (dioxin) receptor function. Genes Dev. 1999;13:20–25. doi: 10.1101/gad.13.1.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mimura J., Fujii-Kuriyama Y. Functional role of AhR in the expression of toxic effects by TCDD. Biochim. Biophys. Acta. 2003;1619:263–268. doi: 10.1016/s0304-4165(02)00485-3. [DOI] [PubMed] [Google Scholar]
- Mimura J., Yamashita K., Nakamura K., Morita M., Takagi T. N., Nakao K., Ema M., Sogawa K., Yasuda M., Katsuki M., et al. Loss of teratogenic response to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in mice lacking the Ah (dioxin) receptor. Genes Cells. 1997;2:645–654. doi: 10.1046/j.1365-2443.1997.1490345.x. [DOI] [PubMed] [Google Scholar]
- Moriguchi T., Motohashi H., Hosoya T., Nakajima O., Takahashi S., Ohsako S., Aoki Y., Nishimura N., Tohyama C., Fujii-Kuriyama Y. Distinct response to dioxin in an arylhydrocarbon receptor (AHR)-humanized mouse. Proc. Natl. Acad. Sci. U.S.A. 2003;100:5652–5657. doi: 10.1073/pnas.1037886100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouse Genome Informatics, T. J. L. Mouse Genome Database (MGD) at the Mouse Genome Informatics website, The Jackson Laboratory, Bar Harbor, Maine. 2014. Available at: http://informatics.jax.org. Accessed January 13, 2014. [Google Scholar]
- Murakami M., Naraba H., Tanioka T., Semmyo N., Nakatani Y., Kojima F., Ikeda T., Fueki M., Ueno A., Oh S., et al. Regulation of prostaglandin E2 biosynthesis by inducible membrane-associated prostaglandin E2 synthase that acts in concert with cyclooxygenase-2. J. Biol. Chem. 2000;275:32783–32792. doi: 10.1074/jbc.M003505200. [DOI] [PubMed] [Google Scholar]
- Naraba H., Yokoyama C., Tago N., Murakami M., Kudo I., Fueki M., Oh-Ishi S., Tanabe T. Transcriptional regulation of the membrane-associated prostaglandin E2 synthase gene. Essential role of the transcription factor Egr-1. J. Biol. Chem. 2002;277:28601–28608. doi: 10.1074/jbc.M203618200. [DOI] [PubMed] [Google Scholar]
- Nielsen S., Frokiaer J., Marples D., Kwon T. H., Agre P., Knepper M. A. Aquaporins in the kidney: From molecules to medicine. Physiol. Rev. 2002;82:205–244. doi: 10.1152/physrev.00024.2001. [DOI] [PubMed] [Google Scholar]
- Nishimura N., Matsumura F., Vogel C. F., Nishimura H., Yonemoto J., Yoshioka W., Tohyama C. Critical role of cyclooxygenase-2 activation in pathogenesis of hydronephrosis caused by lactational exposure of mice to dioxin. Toxicol. Appl. Pharmacol. 2008;231:374–383. doi: 10.1016/j.taap.2008.05.012. [DOI] [PubMed] [Google Scholar]
- Okey A. B., Vella L. M., Harper P. A. Detection and characterization of a low affinity form of cytosolic Ah receptor in livers of mice nonresponsive to induction of cytochrome P1-450 by 3-methylcholanthrene. Mol. Pharmacol. 1989;35:823–830. [PubMed] [Google Scholar]
- Olesen E. T., Fenton R. A. Is there a role for PGE2 in urinary concentration? J. Am. Soc. Nephrol. 2013;24:169–178. doi: 10.1681/ASN.2012020217. [DOI] [PubMed] [Google Scholar]
- Olesen E. T., Rutzler M. R., Moeller H. B., Praetorius H. A., Fenton R. A. Vasopressin-independent targeting of aquaporin-2 by selective E-prostanoid receptor agonists alleviates nephrogenic diabetes insipidus. Proc. Natl. Acad. Sci. U.S.A. 2011;108:12949–12954. doi: 10.1073/pnas.1104691108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson J. R., Gasiewicz T. A., Neal R. A. Tissue distribution, excretion, and metabolism of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in the Golden Syrian hamster. Toxicol. Appl. Pharmacol. 1980;56:78–85. doi: 10.1016/0041-008x(80)90132-5. [DOI] [PubMed] [Google Scholar]
- Peters J. M., Narotsky M. G., Elizondo G., Fernandez-Salguero P. M., Gonzalez F. J., Abbott B. D. Amelioration of TCDD-induced teratogenesis in aryl hydrocarbon receptor (AhR)-null mice. Toxicol. Sci. 1999;47:86–92. doi: 10.1093/toxsci/47.1.86. [DOI] [PubMed] [Google Scholar]
- Poland A., Glover E. 2,3,7,8,-Tetrachlorodibenzo-p-dioxin: segregation of toxocity with the Ah locus. Mol. Pharmacol. 1980;17:86–94. [PubMed] [Google Scholar]
- Poland A., Palen D., Glover E. Analysis of the four alleles of the murine aryl hydrocarbon receptor. Mol. Pharmacol. 1994;46:915–921. [PubMed] [Google Scholar]
- Puga A., Ma C., Marlowe J. L. The aryl hydrocarbon receptor cross-talks with multiple signal transduction pathways. Biochem. Pharmacol. 2009;77:713–722. doi: 10.1016/j.bcp.2008.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson S. W., Clothier B., Akhtar R. A., Yang A. L., Latour I., Van Ijperen C., Festing M. F., Smith A. G. Non-ahr gene susceptibility Loci for porphyria and liver injury induced by the interaction of ‘dioxin’ with iron overload in mice. Mol. Pharmacol. 2002;61:674–681. doi: 10.1124/mol.61.3.674. [DOI] [PubMed] [Google Scholar]
- Rozen S., Skaletsky H. Primer3 on the WWW for general users and for biologist programmers. Methods Mol. Biol. 2000;132:365–386. doi: 10.1385/1-59259-192-2:365. [DOI] [PubMed] [Google Scholar]
- Rzymkiewicz D. M., DuMaine J., Morrison A. R. IL-1 beta regulates rat mesangial cyclooxygenase II gene expression by tyrosine phosphorylation. Kidney Int. 1995;47:1354–1363. doi: 10.1038/ki.1995.191. [DOI] [PubMed] [Google Scholar]
- Schecter A., Gasiewicz T. A. Dioxins and Health. 2nd ed. New York, NY: Wiley-Interscience; 2003. [Google Scholar]
- Schmidt J. V., Su G. H., Reddy J. K., Simon M. C., Bradfield C. A. Characterization of a murine Ahr null allele: Involvement of the Ah receptor in hepatic growth and development. Proc. Natl. Acad. Sci. U.S.A. 1996;93:6731–6736. doi: 10.1073/pnas.93.13.6731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwetz B. A., Norris J. M., Sparschu G. L., Rowe U. K., Gehring P. J., Emerson J. L., Gerbig C. G. Toxicology of chlorinated dibenzo-p-dioxins. Environ. Health Perspect. 1973;5:87–99. doi: 10.1289/ehp.730587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen E. S., Gutman S. I., Olson J. R. Comparison of 2,3,7,8-tetrachlorodibenzo-p-dioxin-mediated hepatotoxicity in C57BL/6J and DBA/2J mice. J. Toxicol. Environ. Health. 1991;32:367–381. doi: 10.1080/15287399109531491. [DOI] [PubMed] [Google Scholar]
- Smith A. G., Clothier B., Robinson S., Scullion M. J., Carthew P., Edwards R., Luo J., Lim C. K., Toledano M. Interaction between iron metabolism and 2,3,7,8-tetrachlorodibenzo-p-dioxin in mice with variants of the Ahr gene: A hepatic oxidative mechanism. Mol. Pharmacol. 1998;53:52–61. doi: 10.1124/mol.53.1.52. [DOI] [PubMed] [Google Scholar]
- Srivastava S. K., Tetsuka T., Daphna-Iken D., Morrison A. R. IL-1 beta stabilizes COX II mRNA in renal mesangial cells: Role of 3′-untranslated region. Am. J. Physiol. 1994;267:F504–F508. doi: 10.1152/ajprenal.1994.267.3.F504. [DOI] [PubMed] [Google Scholar]
- Subbaramaiah K., Yoshimatsu K., Scherl E., Das K. M., Glazier K. D., Golijanin D., Soslow R. A., Tanabe T., Naraba H., Dannenberg A. J. Microsomal prostaglandin E synthase-1 is overexpressed in inflammatory bowel disease. Evidence for involvement of the transcription factor Egr-1. J. Biol. Chem. 2004;279:12647–12658. doi: 10.1074/jbc.M312972200. [DOI] [PubMed] [Google Scholar]
- Tian Y., Ke S., Denison M. S., Rabson A. B., Gallo M. A. Ah receptor and NF-kappaB interactions, a potential mechanism for dioxin toxicity. J. Biol. Chem. 1999;274:510–515. doi: 10.1074/jbc.274.1.510. [DOI] [PubMed] [Google Scholar]
- Uemura H. Associations of exposure to dioxins and polychlorinated biphenyls with diabetes: Based on epidemiological findings. Nihon Eiseigaku Zasshi. 2012;67:363–374. doi: 10.1265/jjh.67.363. [DOI] [PubMed] [Google Scholar]
- van den Berg M., Birnbaum L., Bosveld A. T., Brunstrom B., Cook P., Feeley M., Giesy J. P., Hanberg A., Hasegawa R., Kennedy S. W., et al. Toxic equivalency factors (TEFs) for PCBs, PCDDs, PCDFs for humans and wildlife. Environ. Health Perspect. 1998;106:775–792. doi: 10.1289/ehp.98106775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Berg M., Birnbaum L. S., Denison M., De Vito M., Farland W., Feeley M., Fiedler H., Hakansson H., Hanberg A., Haws L., et al. The 2005 World Health Organization reevaluation of human and Mammalian toxic equivalency factors for dioxins and dioxin-like compounds. Toxicol. Sci. 2006;93:223–241. doi: 10.1093/toxsci/kfl055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel C. F., Khan E. M., Leung P. S., Gershwin M. E., Chang W. L., Wu D., Haarmann-Stemmann T., Hoffmann A., Denison M. S. Cross-talk between aryl hydrocarbon receptor and the inflammatory response: A role for nuclear factor-kappaB. J. Biol. Chem. 2014;289:1866–1875. doi: 10.1074/jbc.M113.505578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel C. F., Sciullo E., Li W., Wong P., Lazennec G., Matsumura F. RelB, a new partner of aryl hydrocarbon receptor-mediated transcription. Mol. Endocrinol. 2007;21:2941–2955. doi: 10.1210/me.2007-0211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D., Pedraza P. L., Abdullah H. I., McGiff J. C., Ferreri N. R. Calcium-sensing receptor-mediated TNF production in medullary thick ascending limb cells. Am. J. Physiol. Renal Physiol. 2002;283:F963–F970. doi: 10.1152/ajprenal.00108.2002. [DOI] [PubMed] [Google Scholar]
- WHO. Evaluation of certain food additives. Fifty-ninth report of the Joint FAO/WHO Expert Committee on Food Additives. World Health Organ. Tech. Rep. Ser. 2002 i-viii, 1–153, back cover. [PubMed] [Google Scholar]
- Xu M., Nelson G. B., Moore J. E., McCoy T. P., Dai J., Manderville R. A., Ross J. A., Miller M. S. Induction of Cyp1a1 and Cyp1b1 and formation of DNA adducts in C57BL/6, Balb/c, and F1 mice following in utero exposure to 3-methylcholanthrene. Toxicol. Appl. Pharmacol. 2005;209:28–38. doi: 10.1016/j.taap.2005.03.012. [DOI] [PubMed] [Google Scholar]
- Yoshioka W., Aida-Yasuoka K., Fujisawa N., Kawaguchi T., Ohsako S., Hara S., Uematsu S., Akira S., Tohyama C. Critical role of microsomal prostaglandin E synthase-1 in the hydronephrosis caused by lactational exposure to dioxin in mice. Toxicol. Sci. 2012a;127:547–554. doi: 10.1093/toxsci/kfs115. [DOI] [PubMed] [Google Scholar]
- Yoshioka W., Endo N., Kurashige A., Haijima A., Endo T., Shibata T., Nishiyama R., Kakeyama M., Tohyama C. Fluorescence laser microdissection reveals a distinct pattern of gene activation in the mouse hippocampal region. Sci. Rep. 2012b;2:783. doi: 10.1038/srep00783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshioka W., Kawaguchi T., Fujisawa N., Aida-Yasuoka K., Shimizu T., Matsumura F., Tohyama C. Predominant role of cytosolic phospholipase A2alpha in dioxin-induced neonatal hydronephrosis in mice. Sci. Rep. 2014;4:4042. doi: 10.1038/srep04042. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.