

Abstract
Methamphetamine (METH) is an extremely addictive stimulant drug that is widely used with high potential of abuse. Previous studies have shown that METH exposure damages the nervous system, especially dopaminergic neurons. However, the exact molecular mechanisms of METH-induced neurotoxicity remain unclear. We hypothesized that caspase-11 is involved in METH-induced neuronal apoptosis. We tested our hypothesis by examining the change of caspase-11 protein expression in dopaminergic neurons (PC12 and SH-SY5Y) and in the midbrain of rats exposed to METH with Western blotting. We also determined the effects of blocking caspase-11 expression with wedelolactone (a specific inhibitor of caspase-11) or siRNA on METH-induced apoptosis in PC12 cells and SH-SY5Y cells using Annexin V and TUNEL staining. Furthermore, we observed the protein expression changes of the apoptotic markers, cleaved caspase-3 and cleaved poly(ADP-ribose) polymerase 1 (PARP), after silencing the caspase-11 expression in rat midbrain by injecting LV-shcasp11 lentivirus using a stereotaxic positioning system. Results showed that METH exposure increased caspase-11 expression both in vitro and in vivo, with the effects in vitro being dose- and time-dependent. Inhibition of caspase-11 expression with either wedelolactone or siRNAs reduced the number of METH-induced apoptotic cells. In addition, blocking caspase-11 expression inhibited METH-induced activation of caspase-3 and PARP in vitro and in vivo, suggesting that caspase-11/caspase-3 signal pathway is involved in METH-induced neurotoxicity. These results indicate that caspase-11 plays an essential role in METH-induced neuronal apoptosis and may be a potential gene target for therapeutics in METH-caused neurotoxicity.
Keywords: methamphetamine, caspase-11, neurotoxicity, apoptosis, dopamine
Methamphetamine (METH) is a strong addictive psychoactive drug with high potential of abuse (classified as Schedule II drugs). It is commonly used illicitly and has become more widespread than heroin and cocaine (Carvalho et al., 2012; Kayagaki et al., 2011). The abuse of METH causes a variety of psychological outcomes, such as stimulant, euphoric, and hallucinogenic effects (Darke et al., 2008).
Multiple studies, including ours, have shown that METH exposure results in adverse outcomes on the dopaminergic system, including disruption of dopamine (DA) homeostasis (Chen et al., 2013; Kita et al., 2009), reduced tyrosine hydroxylase (TH) expression (Sasaki et al., 1987), and loss of TH-positive neurons in the substantia nigra pars compacta, along with destruction of dopaminergic terminals in the striatum (Ares-Santos et al., 2014; Li et al., 2012). Our recent study showed that up-regulation of protein tyrosine nitration through DDAH/ADMA/NOS pathway may contribute to METH-induced neuron apoptosis (Zhang et al., 2013). In addition, we also demonstrated that METH-induced dopaminergic toxicity may be mediated via IGFBP5/caspase-3 signal pathway (Qiao et al., 2014). However, the roles of other caspases in METH-induced neurotoxicity remain to be elucidated.
Caspases are a family of cysteine proteases that play essential roles in apoptosis (Thornberry and Lazebnik, 1998). A variety of apoptotic stimuli (e.g., lipopolysaccharides [LPS], stress, and ischemia) can activate initiator caspases (caspase-2, caspase-8, and caspase-9) to cleave pro-forms of effector caspases (e.g., caspase-3, caspase-6, and caspase-7) to degrade other protein substrates within the cells, thereby triggering the apoptotic pathway. Numerous studies suggest that caspases act as inducible killer protein in neuronal apoptosis. For example, induction and activation of caspase-2, -3, -7, -8, -10, or -11 were observed in neuronal apoptotic cell death that was induced by DNA damage, nitric oxide, cerebral ischemia, or chemical reagents, such as 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (Furuya et al., 2004; Harrison et al., 2001; Kang et al., 2000; Li et al., 2007; Rikhof et al., 2003). Among these caspases, caspase-11 can be prominently increased by a variety of apoptotic signals and has been shown to play an essential role in apoptosis of neurons, oligodendrocytes, astrocytes, and microglia (Fradejas et al., 2010; Hisahara et al., 2001; Kang et al., 2000; Lee et al., 2001; Shibata et al., 2000).
Caspase-11 is a murine caspase that shares the highest homology with human caspase-4 (60% identity), which plays diverse roles in apoptosis, inflammation, and cell migration. Caspase-11 is undetectable in healthy mice and highly inducible by apoptotic signals or by stress. Previous research found that caspase-11 is induced by stimulation with LPS (Kayagaki et al., 2011; Shin and Brodsky, 2013) and mediates the activation of caspase-1 by physical interaction with caspase-1 (Wang et al., 1998). Recent studies have found that the function of caspase-11 is similar to that of caspase-9 (Yamamuro et al., 2011). Specifically, caspase-11 can promote the processing of effector caspases, such as caspase-3 and act as the apical caspase in the cascade that activates downstream executioner caspases under pathological states (Kang et al., 2000). Thus, caspase-11 is crucial in both inflammation and apoptosis when under certain pathological states (Kang et al., 2000). Therefore, we hypothesized that capsase-11 would mediate METH-induced neuronal apoptosis and blockade of caspase-11 expression could partially protect against METH-induced dopaminergic neuron apoptosis.
The present study aimed to investigate the role of caspase-11 in neuronal apoptosis induced by METH. To this end, we examined the expression of caspase-11 and its active form (p30) in catecholaminergic PC12 cells, dopaminergic SH-SY5Y cells, and rats exposed to vehicle or METH. We also observed the effect on the protection against METH-induced neuronal death via blockade of caspase-11 expression by a specific inhibitors or synthetic siRNAs targeting caspase-11 in vitro. In addition, we checked the METH-induced activation level of caspase-3 and its substrate, PARP, after blockade of caspase-11 expression in vitro and in vivo. We demonstrated that caspase-11 is an upstream caspase, responsible for the activation of caspase-3 after METH exposure. This study provides a potential target for gene therapy for METH abuse.
MATERIALS AND METHODS
Animal protocol
Healthy adult male Sprague Dawley (SD) rats (180–220 g, 6–8 weeks old) were purchased from Laboratory Animal Center of Southern Medical University and were singly housed in tub cages on a 12-h light–12 h dark schedule with food and water available ad libitum. Animals were habituated to the animal facilities for 7 days before use. Rats were divided randomly into 2 groups (n = 4 each group). METH ( > 99% purity; National Institutes for Food and Drug Control, Guangzhou, China) was dissolved in saline. A cooling bath was used to counteract the hyperthermia produced by METH. Rats received intraperitoneally (i.p.) injections of saline or METH (8 injections, 15 mg/kg/injection, at 12 h intervals). This exposure paradigm was selected based on our and other previous studies (Cadet et al., 2003; Krasnova and Cadet, 2009; Li et al., 2008; Qiao et al., 2014); it is relevant to human exposure because the measured concentrations of METH in the blood and brain of rats at 1 h after the last injection (Supplementary Table 1) were in the range of reported blood concentrations (0.6–5 µg/ml [about 4–30µM]) in METH abusers (Winek et al., 2001). All animals survived during and after METH exposure. Rats were sacrificed 24 h after the last injection. Brain samples were rapidly removed and the midbrains were dissected on an ice cold glass plate, rapidly frozen and stored at −86°C until analysis. All animal procedure was conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Cell culture
Differentiated PC12 cells, a rat adrenal medulla pheochromocytoma cell line and SH-SY5Y cells, a human dopaminergic neuroblastoma cell line were obtained from the Cell Bank of Shanghai Institute for Biological Science, Chinese Academy of Science and cultured in high glucose DMEM medium containing 10% fetal bovine serum, 50 units/ml penicillin G, and 50 mg/ml streptomycin sulfate. Cells were grown in a CO2 incubator at 37°C, with 5% CO2 and 95% filtered air. The cells were passaged every 2–3 days.
Western blot analysis
PC12 cells, SH-SY5Y cells, midbrain samples from rats exposed to vehicle or METH were lysed in RIPA buffer at 4°C for 30 min. The samples were then separated by SDS-polyacrylamide gel electrophoresis and electroblotted onto polyvinylidenedifluoride membranes. The membranes were incubated overnight at 4°C or at 25°C for 2 h with primary antibodies at the following dilutions: rabbit polyclonal anti-caspase-11 (1:1000, BioVision), rabbit polyclonal anti-caspase-3 (1:1000, Cell Signaling Technology), or rabbit polyclonal anti-PARP (1:1000, Cell Signaling Technology). Next, membranes were incubated with corresponding HRP-conjugated (horse-radish peroxidase) secondary antibodies at 25°C for 1 h. The blot membrane was developed with Chemiluminescence ECL Plus-Western Blotting detection reagents. Proteins of interest were quantified (pixel density) with the Gel-Pro analyzer software and then normalized to correspondent β-actin prior to statistical analyses.
Annexin V apoptosis staining
PC12 cells and SH-SY5Y cells were seeded on 6-well plates at a density of 1.2 × 106/well. Cells were then exposed to wedelolactone (30µM, Santa Cruz Biotechnology, Inc) for 2 h prior to METH (3.0 mM for PC12 cells and 2.0 mM for SH-SY5Y cells) exposure for 24 h. These concentrations were selected based on our and other studies (Fernandes et al., 2014; Irie et al., 2011; Nara et al., 2010; Qiao et al., 2014; Wu et al., 2014) and based on the LC25 of METH in PC12 cells and SH-SY5Y cells (Supplementary Table 2). Cells were centrifuged to remove the medium, washed twice with 4°C PBS, and stained with Annexin V-FITC and propidiumiodide (Keygen, Nanjing, China) according to the Annexin V apoptosis detection kit instructions. The percentage of apoptotic cells was quantified with flow cytometry (FACSCalibur, BD Biosciences, San Jose, California).
TUNEL staining
PC12 cells and SH-SY5Y cells (5 × 105/well) were seeded on cover slips for 24 h. Cells were then exposed to wedelolactone (30µM) for 2 h followed by METH (3.0 mM for PC12 cells and 2.0 mM for SH-SY5Y cells) treatment for 24 h. Visualization of DNA fragmentation was performed using the fluorometric TUNEL system for apoptotic cells (Roche Applied Science) according to the manufacturer’s instructions. PC12 cells and SH-SY5Y cells were fixed in 4% paraformaldehyde in fresh PBS (pH 7.4) at room temperature for 15 min, incubated with fluorescein-conjugated TdT enzyme at 37°C for 1 h in the dark, and then mounted with 4′,6′-diamidino-2-phenylindole (DAPI) for nuclear counter staining. Cross-sections were imaged (×20 and ×40 objectives) using a fluorescence microscope (ECLIPSE 80i, Nikon, Tokyo, Japan). Both TUNEL- and DAPI-positive cells were counted. Data are reported as TUNEL index, which was calculated by counting the total number of TUNEL-positive cells.
Hoechst staining test
PC12 cells and SH-SY5Y cells were seeded on 12-well plates at a density of 5 × 105/well, and then were incubated in a standard condition for 24 h, followed by wedelolactone (30µM) exposure for 2 h and another 24 h treatment of METH (3.0 mM for PC12 cells and 2.0 mM for SH-SY5Y cells). At the end of METH exposure, cells were washed once with PBS and stained for 15 min in the dark at room temperature with 5 µg/ml Hoechst33342 (Invitrogen, Carlsbad, California). Nuclear fragmentation was visualized using a fluorescence microscope (ECLIPSE 80i, Nikon, Tokyo, Japan).
Immunofluorescence
For immunolabeling, all incubation solutions were prepared using PBS supplemented with 10% normal goat serum and 0.05% Triton X-100. The antibodies used were caspase-11 (mouse, 1:100, Santa Cruz) and fluorescein (FITC)-conjugated rabbit anti-mouse IgG (1:50, DingGuo, China) and were used together with DAPI nuclear labeling. Incubation periods with blocking buffer, primary antibody, and secondary antibody were 30 min at room temperature, overnight at 4°C, and 1 h at room temperature, respectively. Microphotographs were taken using a fluorescence microscopy (A1+/A1R+, Nikon, Tokyo, Japan). All digital images were processed using the same settings to improve the contrast.
siRNA and transfection
Three pieces of siRNA sequences that targeting caspase-11 were designed by Shanghai GenePharma Co Ltd, as shown below: siRNA No. 1 (Rat, 5′-UUCAAAUGAUUGUUGCACCTT-3′), siRNA No. 2 (Rat, 5′-UCAUUUCUGAUUCCAUGCCTT-3′), and siRNA No. 3 (Rat, 5′-UAUAAUAGCACAUCUGGAGTT-3′). siRNAs were dissolved in DEPC water at a concentration of 20μM. Appropriate volumes of Lipofectamine 2000 were added based on the instructions of Lipofectamine 2000 Transfection Reagent from Invitrogen. The siRNA for caspase-11 and control siRNA were mixed with Lipofectamine 2000 separately, vortexed for 20 s and then incubated at room temperature for 30 min prior to use.
Virus production and injection
The shRNA synthesis and stereotaxic injection protocol were based on a previous study (Lu et al., 2014). Briefly, the shRNA sequence targeting caspase-11 was designed as the following: GGTGCAACAATCATTTGAA. It was cloned into pGC-LV vector. pGC-LV-shcasp11, pHelper 1.0, and pHelper 2.0 were cotransfected into HEK293FT cells, and LV-shcasp11 was harvested with 109 transducing units per milliliter, and LV-GFP was used as control virus. Adult male SD rats (weight from 200 to 220 g) were divided randomly into 4 groups: LV-GFP group, LV-shcasp11 group, LV-GFP combined with METH group, and LV-shcasp11 combined with METH group. All surgical procedures were conducted under anesthesia following peritoneal injection with 2% pentobarbital. Anesthetized rats were fixed in a stereotaxic frame and a cut was made on the skin overlying the skull. Injections were made at the following stereotaxic coordinates: 5.04 mm rostral to bregma; 2.2 mm lateral to the midline (left or right side); 8.6 mm ventral to the dura, with tooth bar set at zero. A 10µl Hamilton syringe was used for microinjections and a total of 4μl of LV-shcasp-11 lentivirus or LV-GFP lentivirus was injected at a rate of 0.25 ml/min to the midbrain. After completion of the injection in the midbrain, the cannula was kept in situ for 5 min before being slowly withdrawn from the brain. Four days later, rats received i.p. injections of saline or METH (8 injections, 15 mg/kg/injection, at 12 h intervals) and sacrificed 24 h after the last injection. The brains were rapidly removed and the midbrain samples were dissected on an ice cold glass plate, rapidly frozen and stored at −86°C until use.
Statistical analysis
Data are expressed as mean ± standard error (SE) of at least 3 independent replicates. Statistical analysis was performed using parametric test or nonparametric test, as appropriate, with the scientific statistic software SPSS version 13. The parametric test includes 1-way ANOVA or independent-samples t-test and the post hoc test was done by LSD method when we use 1-way ANOVA. Also, the nonparametric test contains Mann Whitney U in 2-independent sample test or Kruskal-Wallis H in K independent samples test and the post hoc test was done by Bonferroni method when we use Kruskal-Wallis H. The value of P < 0.05 was considered statistically significant.
RESULTS
METH-Induced Caspase-11 Protein Expression in Neuronal Cells
To assess the role of caspase-11 in the METH-induced toxicity, we treated PC12 cells with different concentrations (0, 1, 2, 2.5, 3, or 3.5 mM) of METH for different duration (0, 2, 4, 8, 16, or 24 h) and then Western blot analysis was performed to detect caspase-11 expression. The results revealed that caspase-11 protein expression was dose-dependently increased (Figs. 1A and 1B). At 3.0 mM for 24 h, caspase-11 protein expression was 5.6-fold higher in the METH-treated PC12 cells than in the control. After exposure to 3.0 mM METH for 0, 2, 4, 8, 16, or 24 h, the results showed that caspase-11 protein expression was time-dependently increased (Figs. 1D and 1E). Specifically, at 2 h, caspase-11 protein expression was 2.5-fold higher in the METH-treated PC12 cells than in the control, and this effect became the greatest at 16 h. In particular, a processed form of caspase-11 (p30) was detected and exhibited a similar dose- and time-dependent increase as unprocessed caspase-11 (Figs. 1A, 1C, 1D, and 1 F), indicating activation of caspase-11 by METH exposure. Similar effects were observed in SH-SY5Y cells, that is, METH (2.0 mM) exposure for 24 h markedly induced the protein expression of both unprocessed caspase-11 and cleaved-caspase-11 (p30) (Figs. 1G and 1H). Furthermore, a rat model treated with METH (8 injections, 15 mg/kg/injection, at 12 h intervals) was used to ascertain whether METH induces caspase-11 expression in vivo. Western blot results showed that unprocessed caspase-11 and cleaved caspase-11 expression was up-regulated; cleaved capase-3 and cleaved PARP, both of which are the apoptotic markers, were also increased in the midbrain tissues of METH-treated rats (Figs. 2A and 2B). Immunofluorescence staining results showed that METH treatment increased the caspase-11 expression in rat midbrain tissues (Figs. 2C). These results suggest that METH exposure induces caspase-11 protein expression and its activation both in vivo and in vitro.
FIG. 1.

METH exposure increased unprocessed caspase-11 (Pro-casp11) and cleaved caspase-11 (Cleaved casp11) protein expression in PC12 cells and SH-SY5Y cells. A–C, PC12 cells were exposed to METH at concentrations ranging from 1.0 to 3.5 mM for 24 h. Western blot (A) and quantitative analyses (B, C) were performed to determine unprocessed caspase-11 and cleaved caspase-11 protein expression in cells receiving different treatments. D–F, PC12 cells were stimulated with 3.0 mM METH for indicated time (0, 2, 4, 8, 16, and 24 h), protein samples were collected. Western blot (D) and quantitative analyses (E, F) were performed to determine unprocessed caspase-11 and cleaved caspase-11 protein expression. G–H, SH-SY5Y cells were exposed to METH (2.0 mM) for 24 h. The protein expression of unprocessed caspase-11 and cleaved caspase-11 was determined with Western blot (G) and quantitative analyses (H). Fold induction relative to cells treated with vehicle is shown.*P < 0.05 versus vehicle-treated cells. Data in Figures 1B and 1C were analyzed by 1-way ANOVA followed by LSD post hoc analyses; data in Figures 1E and 1F were analyzed by Kruskal-Wallis H in K independent samples test and the post hoc test was Bonferroni; data in Figure 1H were analyzed by Mann Whitney U in 2-independent samples test.
FIG. 2.

Unprocessed caspase-11 (pro-casp11) and cleaved caspase-11 (cleaved casp11) expression was up-regulated while apoptosis markers were increased in the midbrain of METH-exposed male SD rats. Male SD rats were divided randomly into control and experimental groups (n = 4). Animals were injected i.p. with saline or METH (15 mg/kg × 8 injections, at 12 h interval). The midbrain tissues were harvested at 24 h after the last dosing. Western blot (A) and quantitative analyses (B) were performed to determine unprocessed caspase-11, cleaved caspase-11, cleaved caspase3 (cleaved-casp3), and cleaved-PARP protein expression. Fold induction relative to control group is shown. *P < 0.05 versus control group (1 sample t-test). Immunolabeling and confocal imaging analysis (C) showed elevated caspase-11 expression in the midbrain of METH-exposed male SD rats compared with controls.
Importance of Caspase-11 Induction in Neuronal Apoptosis Induced by METH
To examine whether caspase-11 is involved in METH-induced neurotoxicity, wedelolactone, a specific caspase-11 inhibitor (Kobori et al., 2004), was used to block caspase-11 expression. As expected, wedelolactone exposure inhibited caspase-11 expression and its activation induced by METH in PC12 cells (Figs. 3A–C) and in SH-SY5Y cells (Figs. 3F–H). To explore whether blockade of caspase-11 expression reduces METH-induced neuronal apoptosis in vitro, flow cytometry analysis was performed. In PC12 cells, as shown in Figures 3D and 3E, 30µM wedelolactone treatment alone induced apoptosis in 1.62 ± 0.06% of PC12 cells. There was no significant difference between untreated group and wedelolactone-treated group which indicated that wedelolactone had no effect on PC12 cells apoptosis. Treatment with 3.0 mM METH alone induced apoptosis in 17.9 ± 0.11% of PC12 cells, while treatment of 3.0 mM METH combined with 30µM wedelolactone induced apoptosis in 5.1 ± 0.29% of PC12 cells. Thus, wedelolactone significantly reduced the percentage of apoptotic cells caused by METH exposure (Fig. 3E). Similar results were obtained from SH-SY5Y cells, as shown in Figures 3I and 3J, 30µM wedelolactone treatment alone induced apoptosis in 0.8 ± 0.1% of SH-SY5Y cells. There was no significantly different between untreated group and wedelolactone-treated group, indicating that wedelolactone had no effect on SH-SY5Y cells apoptosis. METH (2.0 mM) treatment alone induced apoptosis in 17.6 ± 0.6% of SH-SY5Y cells, while treatment of 2.0 mM METH combined with 30µM wedelolactone induced apoptosis in 7.6 ± 0.36% of SH-SY5Y cells. Like in PC12 cells, wedelolactone also significantly reduced the percentage of apoptotic cells caused by METH exposure (Fig. 3J).
FIG. 3.
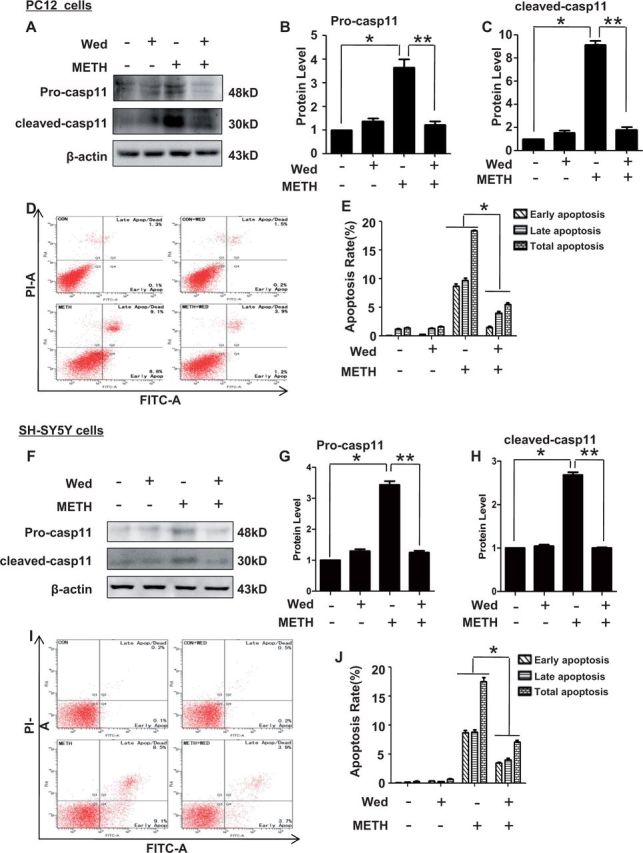
Blockade of caspase-11 expression with wedelolactone (Wed) attenuated METH-induced neuronal apoptosis. Effects of Wed on METH-induced neuronal apoptosis were evaluated with flow cytometry. PC12 cells and SH-SY5Y cells were exposed to Wed (30 µM) for 2 h prior to METH (3.0 mM in PC12 cells and 2.0 mM in SH-SY5Y cells) treatment as indicated. Western blot (A and F) and quantitative analyses (B–C and G–H) were performed to determine unprocessed caspase-11 and cleaved caspase-11 protein expression. Flow cytometry (D–E and I–J) was performed to assess the effects of Wed on apoptosis induced by METH. All the experiments were repeated 3 times. The data are presented as means ± SE from 3 experiments. *P < 0.01 versus non-METH-treated group, **P < 0.01 versus non-wedelolactone-treated group. Data were analyzed with Kruskal-Wallis H in K independent samples test followed by Bonferroni post hoc analyses.
To further evaluate the effect of caspase-11 on METH-caused neurotoxicity, we investigated whether caspase-11 expression blockade affects apoptosis induced by METH in PC12 cells using TUNEL and Hoechst staining. Both TUNEL and Hoechst staining were used to detect DNA damage. In PC12 cells, the TUNEL staining results (Fig. 4A) showed that the number of TUNEL-positive PC12 cells was increased by >9-fold in METH-treated group compared with control group (14.2 ± 0.3% vs 1.6 ± 0.26%, n = 5, P < 0.01, Fig. 4C). Wedelolactone treatment decreased the number of TUNEL-positive METH-exposed PC12 cells by>2.5-fold (14.2 ± 0.3% vs 4.98 ± 0.25%, n = 5, P < 0.01). Similarly, the Hoechst staining results (Fig. 4B) showed that the number of Hoechst 33342-positive cells was increased by >11-fold in METH-treated cells compared with the control cells (21.58 ± 0.42% vs 1.82 ± 0.14%, P < 0.01, Fig. 4D), and this effect was attenuated by >3-fold with wedelolactone treatment (21.58 ± 0.42% vs 6.89 ± 0.12%, n = 5, P < 0.01). In SH-SY5Y cells, the TUNEL staining results (Fig. 4E) showed that the number of TUNEL-positive SH-SY5Y cells was increased by about 12-fold in METH-treated group compared with control group (24.58 ± .67% vs 1.92 ± 0.14%, n = 5, P < 0.01, Fig. 4G). Wedelolactone treatment decreased the number of TUNEL-positive METH-exposed SH-SY5Y cells by >2-fold (24.58 ± 0.67% vs 10.89 ± 0.22%, n = 5, P < 0.01). Similarly, the Hoechst staining results (Fig. 4F) showed that the number of Hoechst 33342-positive cells was increased by >10-fold in METH-treated cells compared with the control cells (20.48 ± 0.67% vs 1.89 ± 0.09%, P < 0.01, Fig. 4H), and this effect was attenuated by >3-fold with wedelolactone treatment (20.48 ± 0.67% vs 7.56 ± 0.35%, n = 5, P < 0.01). These results suggest that blockade of caspase-11 expression reduces METH-induced apoptosis in PC12 cells and SH-SY5Y cells, indicating that caspase-11 is involved in METH-induced neuronal apoptosis in vitro.
FIG. 4.
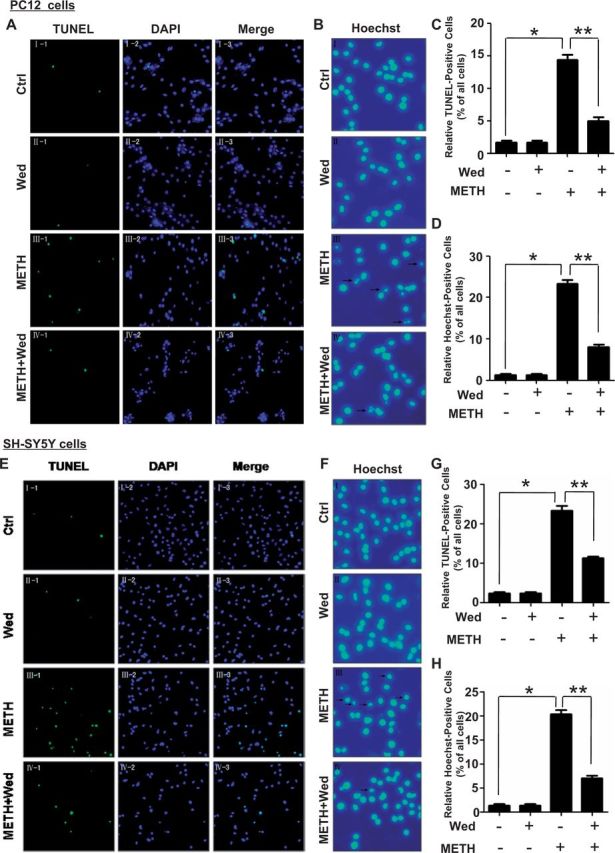
Caspase-11 is necessary for METH-induced apoptosis in PC12 cells and SH-SY5Y cells. Effects of Wed on METH-induced neuronal apoptosis were evaluated using TUNEL (A, C for PC12 cells and E, G for SH-SY5Y cells) and Hoechst staining (B, D for PC12 cells and F, H for SH-SY5Y cells). A and E, PC12 cells and SH-SY5Y cells were exposed to Wed (30 µM) for 2 h before METH treatment (3.0 mM in PC12 cells and 2.0 mM in SH-SY5Y cells). Nuclei were counterstained with DAPI (blue). Apoptotic cells were stained with TUNEL (green). C and G, Quantitative analysis of the percentage of apoptotic cells using a standard cell counting method with the TUNEL assay. B and F, Apoptotic positive cells were detected by their fragmented nuclei that exhibited blue (Hoechst 33342) pykno-fluorescence. D and H, Quantitative analysis of the percentage of apoptotic cells using a standard cell counting method. Representative calculations from 3 independent experiments are shown. The number of positive cells is presented as mean ± SE of 3 experiments. *P < 0.01 versus no METH treated, **P < 0.01 versus no wedelolactone treated. Data were analyzed with Kruskal-Wallis H in K independent samples test followed by Bonferroni post hoc analyses.
Silencing of Caspase-11 Protected PC12 Cells from METH-Induced Apoptosis
To confirm that blockade of caspase-11 expression protects against METH-induced apoptosis, we used synthetic siRNAs targeting caspase-11 to silence caspase-11 expression and then examined their effects on METH-caused apoptosis in PC12 cells. Firstly, we investigated the knockdown efficacy of each siRNA sequence targeting caspase-11 at the protein level. We transfected the siRNA Nos. 1–3 (100 nM) or scrambled siRNA (control siRNA, 100 nM) into PC12 cells for 48 h followed by METH exposure for 24 h. Western blot analysis showed that METH exposure induced caspase-11 protein expression in control siRNA group; this effect was significantly attenuated by coexposure to any of the 3 siRNA sequences (Figs. 5A and 5B). Notably, these siRNA sequences not only attenuated the unprocessed caspase-11 expression, but also inhibited the expression of its activated form p30. To explore whether silencing of caspase-11 expression by synthetic siRNA sequences also reduces METH-induced apoptosis in vitro, a TUNEL assay was performed to detect DNA damage in PC12 cells. The number of TUNEL-positive cells was increased by more than 15-fold in PC12 cells transfected with control siRNA exposed to METH compared with vehicle treatment (16.9 ± 0.57% vs 1.13 ± 0.25%, Figs. 5C and 5D). The number of TUNEL-positive cells was decreased by >3-fold in METH-treated PC12 cells transfected with siRNA Nos. 1–3 compared with control siRNA group (16.9 ± 0.57% vs 4.66 ± 0.49%, 4.33 ± 0.68% and 4.12 ± 0.23% for siRNA No. 1, siRNA No. 2, and siRNA No. 3, respectively, n = 5, P < 0.05, Fig. 5D). These results suggest that silencing of caspase-11 expression can reduce apoptosis induced by METH in PC12 cells.
FIG. 5.

Effects of caspase-11 siRNA on METH-caused apoptosis in PC12 cells. A–B, Synthetic caspase-11 siRNAs effectively suppressed endogenous unprocessed caspase-11 and cleaved caspase-11 expression. PC12 cells were transfected with siRNAs targeting caspase-11 or control siRNA for 48 h followed by METH (3.0 mM) treatment for 24 h. Protein samples were collected. Western blot (A) and quantitative analyses (B) were performed to evaluate the efficiency of caspase-11 knockdown. C, Effects of suppressing the caspase-11 gene in 3.0 mM METH-treated PC12 cells assessed by TUNEL assays. PC12 cells were pretreated with caspase-11 siRNAs or control siRNA. Nuclei were counterstained with DAPI (blue). Apoptotic cells were stained with TUNEL (green). D, Quantitative analysis of the percentage of apoptotic cells using a standard cell counting method with the TUNEL assay. All the results were repeated by 3 independent experiments. The data are presented as means ± SE from 3 experiments (n = 3). *P < 0.05 versus control siRNA group. Data were analyzed with Kruskal-Wallis H in K independent samples test followed by Bonferroni post hoc analyses.
Blockade of Caspase-11 Expression Can Reduce Caspase-3 and PARP Activation in vitro
Previous studies have shown that caspase-11 plays a critical role in the activation of caspase-3 under certain pathological conditions (Kang et al., 2000) and METH-induced neuronal apoptosis is accompanied with caspase-3 and PARP activation (Jayanthi et al., 2004; Qiao et al., 2014). We sought to determine whether caspase-11 mediates neuronal apoptosis via caspase-3 pathway. Therefore, we examined the activation level of caspase-3 and PARP after inhibition of caspase-11 expression by wedelolactone in PC12 cells and SH-SY5Y cells treated with or without METH. As shown in Figures 6A and 6B, Western blot analyses showed that cleaved caspase-3 expression was increased up to >6-fold in METH-treated PC12 cells compared with vehicle-treated cells, this effect was attenuated by coexposure to wedelolactone. These data indicate that caspase-11 mediates neuronal apoptosis caused by METH, in part, via activation of caspase-3. Similar results were observed for cleaved PARP. As shown in Figures 6A and 6C, the cleaved PARP was increased up to about 3.5-fold in PC12 cells treated with METH compared with control cells, and the increased cleavage of PARP was attenuated by blocking caspase-11 expression with wedelolactone. Meanwhile, we determined the effect of blockade of caspase-11 on caspase-3 and PARP activation in SH-SY5Y cells. As shown in Figures 6D and 6E, Western blot analyses showed that cleaved caspase-3 expression was increased up to >3.5-fold in METH-treated SH-SY5Y cells compared with vehicle-treated cells, this effect was antagonized by coexposure to wedelolactone. Similar results were observed for cleaved PARP. As shown in Figures 6D and 6F, the cleaved PARP was increased up to about 3.5-fold in SH-SY5Y cells treated with METH compared with control cells, and the increased cleavage of PARP was neutralized by blocking caspase-11expression with wedelolactone. These results further demonstrated that the caspase-3 pathway is involved in caspase-11-mediated METH-induced neuronal apoptosis.
FIG. 6.

Blockade of caspase-11 expression reduced caspase-3 and PARP activation caused by METH in PC12 cells and SH-SY5Y cells. PC12 cells and SH-SY5Y cells were exposed to Wed (30 µM) for 2 h followed by treatment with or without METH (3.0 mM in PC12 cells and 2.0 mM in SH-SY5Y cells) for 24 h. A and D, Cells were lysed and lysates were subjected to Western blot using antibodies against cleaved caspase-3 or PARP. B, C and E, F, Quantitative analyses were performed to determine cleaved caspase-3 and cleaved PARP protein expression. *P < 0.01 versus non-METH-treated group, **P < 0.01 versus non-wedelolactone-treated group. Data were analyzed with Kruskal-Wallis H in K independent samples test followed by Bonferroni post hoc analyses.
Silencing of Caspase-11 Expression Reduced Caspase-3 and PARP Activation In Vivo
To further testify the roles of caspase-11 in METH-caused neurotoxicity in vivo, LV-GFP and LV-shcasp11 lentivirus were injected separately to the rat midbrain using a standard stereotaxic positioning system to silence caspase-11 expression in the midbrain region and then rats were treated with METH or vehicle. As shown in Supplementary Figure 1, fluorescence analysis showed lentivirus had been successfully infected into midbrains. And then we checked the protein level of unprocessed caspase-11, cleaved-caspase-11, cleaved-caspase-3, and cleaved-PARP in each group. LV-shcasp11 lentivirus effectively silenced the caspase-11 expression and its activation in the midbrain of METH-treated rats (Figs. 7A–C). The expression level of cleaved caspase-3 and cleaved-PARP was decreased following caspase-11 expression knockdown (Figs. 7A, 7D, and 7 E). These in vivo results were consistent with those in vitro as mentioned before. These results further demonstrated that caspase-3 pathway is involved in caspase-11-mediated METH-induced neuronal apoptosis.
FIG. 7.

Silencing of caspase-11 expression reduced caspase-3 and PARP activation in the midbrain of METH-exposed male SD rats. LV-GFP and LV-shcaspase11 (LV-shcasp11) lentivirus were injected separately to the rat midbrain using a standard stereotaxic positioning system (n = 3/group). After injecting with lentivirus for 4 days, animals were injected i.p. with saline or METH (15 mg/kg × 8 injections, at 12 h interval). The midbrain tissues were harvested at 24 h after the last dosing. Western blot (A) and quantitative analyses (B–E) were performed to determine unprocessed caspase-11, cleaved caspase-11, cleaved caspase3 (cleaved-casp3), and cleaved-PARP protein expression. *P < 0.01 compared with saline-treated group, **P < 0.01 compared with LV-GFP-treated group. Data were analyzed with Kruskal-Wallis H in K independent samples test followed by Bonferroni post hoc analyses.
DISCUSSION
Caspases are subdivided into 2 classes: apoptotic caspases or inflammatory caspases (Hotchkiss and Nicholson, 2006).Caspase-11, however, has dual roles in both apoptosis and inflammation (Kang et al., 2000). The formation of the inflammasome by caspase-11, caspase-1, apoptosis-associated speck-like protein containing a CARD, and cytosolic NOD-like receptor is important in the caspase-11-mediated inflammatory response. Additionally, activation of caspase-3 by caspase-11 is a key step in caspase-11-mediated apoptosis (Hotchkiss and Nicholson, 2006; Kang et al., 2000). In the present study, we for the first time report that caspase-11 expression is increased after METH exposure in vivo and in vitro. We also demonstrate that caspase-11 plays an essential role in dopaminergic cell death caused by METH and inhibition of caspase-11 expression by caspase-11-specific inhibitor wedelolactone or synthetic siRNA sequences can attenuate METH-exposed dopaminergic cell death in vitro. In addition, we injected lentivirus into the rat midbrain to inhibit the caspase-11 expression induced by METH, and found that the expression of cleaved capase-3 and cleaved PARP in the rat midbrain was decreased following the caspase-11 expression silencing. These findings indicate that caspase-11 plays a crucial role in METH-exposed neurotoxicity.
The expression of caspase-11 is the most stringently regulated among all the caspases identified so far. Unlike most caspases that can be detected in healthy, unstimulated cells, caspase-11 is undetectable in healthy cells, but highly inducible by apoptotic signals or by stress. In the present study, we did not detect the expression of caspase-11 in untreated PC12 cells or SH-SY5Y cells, but it was detectable in PC12 cells and SH-SY5Y cells after METH treatment. Moreover, the induction of caspase-11 expression by METH occurred in a dose- and time-dependent manner. Intriguingly, a cleaved caspase-11, p30, was detected in vivo and in vitro, which indicates that METH not only inducescaspase-11 expression but also activates itself. There are 2 possible mechanisms of caspase-11 activation, namely auto-activation model and scaffold mediated activation. The auto-activation model was proposed by Rathinam et al. (2012), who suggested that induction of caspase-11 expression is both necessary and sufficient for its own activation, and indeed when expresses at significant levels, procaspase-11 does undergo auto-processing (Kang et al., 2000; Rathinam et al., 2012). Another possible mechanisms of caspase-11 activation, scaffold-mediated activation model, were proposed by Broz et al. (2012), who revealed a requirement for type-I-IFN signaling in caspase-11 activation that is consistent with a model in which an interferon inducible activator mediates caspase-11 activation in response to intracellular Salmonella. In this study, we detected that caspase-11 was activated in neuronal cells after METH exposure. However, among above-mentioned 2 mechanisms, which one is responsible for caspase-11 activation after METH treatment is still unknown. Additional experiments are needed to provide a definitive conclusion.
Our results show that wedelolactone attenuates up-regulation of METH-induced caspase-11 expression in PC12 and SH-SY5Y cells. Previous studies have demonstrated that wedelolactone could block caspase-11 expression by inhibition of NF-KB activation via mediating phosphorylation and degradation of IKBα (Kobori et al., 2004). Hence, wedelolactone may attenuate METH-caused caspase-11 induction via inhibition of NF-KB activation. This explanation is consistent with the finding that METH induces inflammation directly in neurons by activating NF-KB and other cytokines (Liu et al., 2012; Permpoonputtana and Govitrapong, 2013; Shah et al., 2012).
Caspase-11 has been demonstrated to play important roles in apoptosis through caspase-3 pathway. We examined the activation of caspase-3 after blockade of caspase-11 expression. The results showed that blockade of caspase-11 expression can inhibit caspase-3 activation by METH in vitro and in vivo. These results indicate that caspase-11 mediates neuronal apoptosis caused by METH via caspase-3 pathway. Although our data demonstrated that caspase-11 can directly activate caspase-3, we do not exclude the possibility that cytokines may also be involved in the regulation of METH-induced cell death. Previous studies have demonstrated that exposure to METH increases the levels of the inflammatory cytokines IL-8, IL-1β, and NF-KB (Liu et al., 2012). Hence, it is possible that these cytokines may be involved in caspase-11-mediated caspase-3 activation during METH exposure, which requires additional studies to verify. In addition, in the in vivo portion of this study, we conducted all analyses using the rat midbrain, which contains substantia nigra that is rich of dopaminergic neurons and some other nuclei. Further studies collecting specific regions of the basal ganglia such as substantia nigra and striatum will help to identify the specific target region(s) of METH.
In conclusion, the present study demonstrates that caspase-11 is increased after METH treatment in vivo and in vitro, and blockade of caspase-11 expression significantly protects neuronal cells against METH-induced toxicity in vivo and in vitro. Caspase-11 mediates neuronal apoptosis through activation of caspase-3 pathway. However, further studies are needed to elucidate the inflammatory effect of caspase-11 on neuron death induced by METH.
SUPPLEMENTARY DATA
Supplementary data are available online at http://toxsci.oxfordjournals.org/.
ACKNOWLEDGMENT
The authors declare that there are no conflicts of interest.
FUNDING
Natural Science Foundation of China (81273345 to H.W.; 81370227 to W.-B.X.; and 81172907 to D.Q.).
REFERENCES
- Ares-Santos S., Granado N., Espadas I., Martinez-Murillo R., Moratalla R. (2014). Methamphetamine causes degeneration of dopamine cell bodies and terminals of the nigrostriatal pathway evidenced by silver staining. Neuropsychopharmacology 39, 1066–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broz P., Ruby T., Belhocine K., Bouley D. M., Kayagaki N., Dixit V. M., Monack D. M. (2012). Caspase-11 increases susceptibility to Salmonella infection in the absence of caspase-1. Nature 490, 288–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadet J. L., Jayanthi S., Deng X. (2003). Speed kills: cellular and molecular bases of methamphetamine-induced nerve terminal degeneration and neuronal apoptosis. FASEB J. 17, 1775–1788. [DOI] [PubMed] [Google Scholar]
- Carvalho M., Carmo H., Costa V. M., Capela J. P., Pontes H., Remiao F., Carvalho F., Bastos Mde L. (2012). Toxicity of amphetamines: an update. Arch. Toxicol. 86, 1167–1231. [DOI] [PubMed] [Google Scholar]
- Chen L., Huang E., Wang H., Qiu P., Liu C. (2013). RNA interference targeting alpha-synuclein attenuates methamphetamine-induced neurotoxicity in SH-SY5Y cells. Brain Res. 1521, 59–67. [DOI] [PubMed] [Google Scholar]
- Darke S., Kaye S., McKetin R., Duflou J. (2008). Major physical and psychological harms of methamphetamine use. Drug Alcohol Rev. 27, 253–262. [DOI] [PubMed] [Google Scholar]
- Fernandes S., Salta S., Bravo J., Silva A. P., Summavielle T. (2014). Acetyl-L-carnitine prevents methamphetamine-induced structural damage on endothelial cells via ILK-related MMP-9 activity. Mol. Neurobiol . doi: 10.1007/s12035-014-8973-5. [DOI] [PubMed] [Google Scholar]
- Fradejas N., Pastor M. D., Burgos M., Beyaert R., Tranque P., Calvo S. (2010). Caspase-11 mediates ischemia-induced astrocyte death: involvement of endoplasmic reticulum stress and C/EBP homologous protein. J. Neurosci. Res. 88, 1094–1105. [DOI] [PubMed] [Google Scholar]
- Furuya T., Hayakawa H., Yamada M., Yoshimi K., Hisahara S., Miura M., Mizuno Y., Mochizuki H. (2004). Caspase-11 mediates inflammatory dopaminergic cell death in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson’s disease. J. Neurosci. 24, 1865–1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison D. C., Davis R. P., Bond B. C., Campbell C. A., James M. F., Parsons A. A., Philpott K. L. (2001). Caspase mRNA expression in a rat model of focal cerebral ischemia. Brain Res. Mol. Brain Res. 89, 133–146. [DOI] [PubMed] [Google Scholar]
- Hisahara S., Yuan J., Momoi T., Okano H., Miura M. (2001). Caspase-11 mediates oligodendrocyte cell death and pathogenesis of autoimmune-mediated demyelination. J. Exp. Med. 193, 111–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotchkiss R. S., Nicholson D. W. (2006). Apoptosis and caspases regulate death and inflammation in sepsis. Nature Rev. 6, 813–822. [DOI] [PubMed] [Google Scholar]
- Irie Y., Saeki M., Tanaka H., Kanemura Y., Otake S., Ozono Y., Nagai T., Kondo Y., Kudo K., Kamisaki Y., et al. (2011). Methamphetamine induces endoplasmic reticulum stress related gene CHOP/Gadd153/ddit3 in dopaminergic cells. Cell Tissue Res. 345, 231–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayanthi S., Deng X., Noailles P. A., Ladenheim B., Cadet J. L. (2004). Methamphetamine induces neuronal apoptosis via cross-talks between endoplasmic reticulum and mitochondria-dependent death cascades. FASEB J. 18, 238–251. [DOI] [PubMed] [Google Scholar]
- Kang S. J., Wang S., Hara H., Peterson E. P., Namura S., Amin-Hanjani S., Huang Z., Srinivasan A., Tomaselli K. J., Thornberry N. A., et al. (2000). Dual role of caspase-11 in mediating activation of caspase-1 and caspase-3 under pathological conditions. J. Cell Biol. 149, 613–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayagaki N., Warming S., Lamkanfi M., Vande Walle L., Louie S., Dong J., Newton K., Qu Y., Liu J., Heldens S., et al. (2011). Non-canonical inflammasome activation targets caspase-11. Nature 479, 117–121. [DOI] [PubMed] [Google Scholar]
- Kita T., Miyazaki I., Asanuma M., Takeshima M., Wagner G. C. (2009). Dopamine-induced behavioral changes and oxidative stress in methamphetamine-induced neurotoxicity. Int. Rev. Neurobiol. 88, 43–64. [DOI] [PubMed] [Google Scholar]
- Kobori M., Yang Z., Gong D., Heissmeyer V., Zhu H., Jung Y. K., Gakidis M. A., Rao A., Sekine T., Ikegami F., et al. (2004). Wedelolactone suppresses LPS-induced caspase-11 expression by directly inhibiting the IKK complex. Cell Death Differ. 11, 123–130. [DOI] [PubMed] [Google Scholar]
- Krasnova I., Cadet J. (2009). Methamphetamine toxicity and messengers of death. Brain Res. Rev. 60, 379–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J., Hur J., Lee P., Kim J. Y., Cho N., Kim S. Y., Kim H., Lee M. S., Suk K. (2001). Dual role of inflammatory stimuli in activation-induced cell death of mouse microglial cells. Initiation of two separate apoptotic pathways via induction of interferon regulatory factor-1 and caspase-11. J. Biol. Chem. 276, 32956–32965. [DOI] [PubMed] [Google Scholar]
- Li L., Zhang J., Jin B., Block E. R., Patel J. M. (2007). Nitric oxide upregulation of caspase-8 mRNA expression in lung endothelial cells: role of JAK2/STAT-1 signaling. Mol. Cell. Biochem. 305, 71–77. [DOI] [PubMed] [Google Scholar]
- Li X. F., Wang H. J., Qiu P. M., Luo H. (2008). Proteomic profiling of proteins associated with methamphetamine-induced neurotoxicity in different regions of rat brain. Neurochem. Int. 52, 256–264. [DOI] [PubMed] [Google Scholar]
- Li Y., Hu Z., Chen B., Bu Q., Lu W., Deng Y., Zhu R., Shao X., Hou J., Zhao J., et al. (2012). Taurine attenuates methamphetamine-induced autophagy and apoptosis in PC12 cells through mTOR signaling pathway. Toxicol. Lett. 215, 1–7. [DOI] [PubMed] [Google Scholar]
- Liu X., Silverstein P. S., Singh V., Shah A., Qureshi N., Kumar A. (2012). Methamphetamine increases LPS-mediated expression of IL-8, TNF-alpha and IL-1beta in human macrophages through common signaling pathways. PLoS One 7, e33822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y., Sun X. D., Hou F. Q., Bi L. L., Yin D. M., Liu F., Chen Y. J., Bean J. C., Jiao H. F., Liu X., et al. (2014). Maintenance of GABAergic activity by neuregulin 1-ErbB4 in amygdala for fear memory. Neuron 84, 835–846. [DOI] [PubMed] [Google Scholar]
- Nara A., Aki T., Funakoshi T., Uemura K. (2010). Methamphetamine induces macropinocytosis in differentiated SH-SY5Y human neuroblastoma cells. Brain Res. 1352, 1–10. [DOI] [PubMed] [Google Scholar]
- Permpoonputtana K., Govitrapong P. (2013). The anti-inflammatory effect of melatonin on methamphetamine-induced proinflammatory mediators in human neuroblastoma dopamine SH-SY5Y cell lines. Neurotox. Res. 23, 189–199. [DOI] [PubMed] [Google Scholar]
- Qiao D., Xu J., Le C., Huang E., Liu C., Qiu P., Lin Z., Xie W. B., Wang H. (2014). Insulin-like growth factor binding protein 5 (IGFBP5) mediates methamphetamine-induced dopaminergic neuron apoptosis. Toxicol. Lett. 230, 444–453. [DOI] [PubMed] [Google Scholar]
- Rathinam V. A., Vanaja S. K., Waggoner L., Sokolovska A., Becker C., Stuart L. M., Leong J. M., Fitzgerald K. A. (2012). TRIF licenses caspase-11-dependent NLRP3 inflammasome activation by gram-negative bacteria. Cell 150, 606–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rikhof B., Corn P. G., El-Deiry W. S. (2003). Caspase 10 levels are increased following DNA damage in a p53-dependent manner. Cancer Biol. Therapy 2, 707–712. [PubMed] [Google Scholar]
- Sasaki M., Hashizume K., Manaka S., Takakura K. (1987). [The influences of neurotransmitters on the traumatic unconsciousness, immediate convulsion and mortality in the experimental mice model]. No To Shinkei 39, 983–990. [PubMed] [Google Scholar]
- Shah A., Silverstein P. S., Singh D. P., Kumar A. (2012). Involvement of metabotropic glutamate receptor 5, AKT/PI3K signaling and NF-kappaB pathway in methamphetamine-mediated increase in IL-6 and IL-8 expression in astrocytes. J. Neuroinflammation 9, 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata M., Hisahara S., Hara H., Yamawaki T., Fukuuchi Y., Yuan J., Okano H., Miura M. (2000). Caspases determine the vulnerability of oligodendrocytes in the ischemic brain. J. Clin. Invest. 106, 643–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin S., Brodsky I. E. (2013). Caspase-11: the noncanonical guardian of cytosolic sanctity. Cell Host Microbe 13, 243–245. [DOI] [PubMed] [Google Scholar]
- Thornberry N. A., Lazebnik Y. (1998). Caspases: enemies within. Science 281, 1312–1316. [DOI] [PubMed] [Google Scholar]
- Wang S., Miura M., Jung Y. K., Zhu H., Li E., Yuan J. (1998). Murine caspase-11, an ICE-interacting protease, is essential for the activation of ICE. Cell 92, 501–509. [DOI] [PubMed] [Google Scholar]
- Winek C. L., Wahba W. W., Winek C. L., Jr, Balzer T. W. (2001). Drug and chemical blood-level data 2001. Forensic Sci. Int. 122, 107–123. [DOI] [PubMed] [Google Scholar]
- Wu X., Wang A., Chen L., Huang E., Xie W., Liu C., Huang W., Chen C., Qiu P., Wang H. (2014). S-nitrosylating protein disulphide isomerase mediates alpha-synuclein aggregation caused by methamphetamine exposure in PC12 cells. Toxicol. Lett. 230, 19–27. [DOI] [PubMed] [Google Scholar]
- Yamamuro A., Kishino T., Ohshima Y., Yoshioka Y., Kimura T., Kasai A., Maeda S. (2011). Caspase-4 directly activates caspase-9 in endoplasmic reticulum stress-induced apoptosis in SH-SY5Y cells. J. Pharmacol. Sci. 115, 239–243. [DOI] [PubMed] [Google Scholar]
- Zhang F., Chen L., Liu C., Qiu P., Wang A., Li L., Wang H. (2013). Up-regulation of protein tyrosine nitration in methamphetamine-induced neurotoxicity through DDAH/ADMA/NOS pathway. Neurochem. Int. 62, 1055–1064. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.