

Abstract
Levels of amyloid beta (Aβ) in the central nervous system are regulated by the balance between its synthesis and degradation. Neprilysin (NEP) is associated with Alzheimer’s disease (AD) by its ability to degrade Aβ. Some studies have involved the exposure to mercury (Hg) in AD pathogenesis; therefore, our aim was to investigate the effects on the anabolism and catabolism of Aβ in differentiated SH-SY5Y cells incubated with 1–20 μM of Hg. Exposure to 20 µM of Hg induced an increase in Aβ-42 secretion, but did not increase the expression of the amyloid precursor protein (APP). Hg incubation (10 and 20 µM) increased NEP protein levels; however, it did not change NEP mRNA levels nor the levels of the amyloid intracellular domain peptide, a protein fragment with transcriptional activity. Interestingly, Hg reduced NEP activity at 10 and 20 µM, and circular dichroism analysis using human recombinant NEP showed conformational changes after incubation with molar equivalents of Hg. This suggests that the Hg-induced inhibition of NEP activity may be mediated by a conformational change resulting in reduced Aβ-42 degradation. Finally, the comparative effects of lead (Pb, 50 μM) were evaluated. We found a significant increase in Aβ-42 levels and a dramatic increase in APP protein levels; however, no alteration in NEP levels was observed nor in the enzymatic activity of this metalloprotease, despite the fact that Pb slightly modified the rhNEP conformation. Overall, our data suggest that Hg and Pb increase Aβ levels by different mechanisms.
Keywords: neprilysin, mercury, lead, beta-amyloid peptide, Alzheimer’s disease, amyloid precursor protein, retinoic acid
Alzheimer’s disease (AD) is a neurodegenerative disorder characterized by the formation of neurofibrillary tangles and amyloid plaques in the brain. Neurofibrillary tangles are primarily formed by the hyperphosphorylation of a microtubule-associated protein called tau, and amyloid plaques that are mostly constituted by the amyloid beta (Aβ) peptide. The amyloid cascade hypothesis of AD suggests that Aβ production and accumulation are a key early step in the onset of the disease (Hardy and Higgins, 1992). The amyloid precursor protein (APP) is a type I transmembrane protein related to different physiological processes. Its induction has been related to pathological conditions, such as AD and Down’s syndrome (Armstrong, 1994). APP is enzymatically processed through 2 independent pathways: the amyloidogenic pathway, in which it is cleaved first by a beta (β) secretase (BACE1) followed by a gamma (γ) secretase complex to produce Aβ-40 and Aβ-42 fragments and other products such as soluble APPβ (sAPPβ) and the amyloid intracellular domain (AICD) (Thinakaran and Koo, 2008). On the other hand, when gamma (γ) and alpha (α) secretase cleavage takes place, soluble APPα and AICD but not Aβ are produced; this pathway is known as nonamyloidogenic (Thinakaran and Koo, 2008). The increased levels of Aβ found in sporadic AD could be caused by a reduction in its clearance rate. Iwata et al. (2005) demonstrated that Aβ is degraded by a group of peptidases, and it was proposed that the major peptidase involved in Aβ degradation, both in vivo and in vitro, is the zinc-dependent metalloprotease neprilysin (NEP).
NEP, also known as CD10 or CALLA, is a transmembrane protein of 750 amino acids involved in the cleavage of a broad group of peptides in different organs including the brain (Turner et al., 2001). Some studies have demonstrated that NEP mRNA is reduced in the brain of AD patients (Yasojima et al., 2001), as well as its immunoreactivity and enzymatic activity (Wang et al., 2005). Gene therapy studies in AD transgenic mice have examined the role of the overexpression of human NEP and found a reduction in amyloid plaques, reduced oxidation and inflammation, and improved spatial orientation in contrast with transgenic mice overexpressing only the control vector (El-Amouri et al., 2008).
Biogenic and toxic metals including copper (Cu), zinc (Zn), iron (Fe), aluminum (Al), cadmium (Cd), lead (Pb), and mercury (Hg) have been associated with AD (Duce and Bush, 2010; Mutter et al., 2010; Wu et al., 2008), and some of these metals have the ability to aggregate Aβ in vitro (Atwood et al., 1998). Despite that Hg does not induce aggregation of Aβ, it has been associated with cognitive alterations and the development of AD. Hock et al. (1998) found increased Hg levels in blood samples of AD patients, and similar results were reported in the temporal lobe of AD brains (Wenstrup et al., 1990). Also, memory loss was observed in patients diagnosed with chronic Hg toxicity (Wojcik et al., 2006), and an association between Hg exposure and the allele ApoE4, which is the principal genetic risk factor for sporadic AD has been suggested (Godfrey et al., 2003). In animal models, increased phosphorylation of tau in the cortex, but not in the hippocampus was reported in mice treated with MeHg (Fujimura et al., 2009). In vitro studies suggest increased Aβ-40 levels and the phosphorylation of tau in cells treated with inorganic Hg (Olivieri et al., 2000). MeHg (50–100 nM) also increases tau phosphorylation by mechanisms that include reactive oxygen species production (Petroni et al., 2012). Furthermore, disruption of the membrane structure and linear growth rates of neurites were observed in primary cultures of snail neurons exposed to inorganic Hg (Leong et al., 2001).
Hg exposure has been linked with some alterations observed in AD; however, the mechanism by which it produces this effect is not known. The objective of this study was to investigate the effect of inorganic Hg exposure on NEP, a degrading enzyme of Aβ, in differentiated SH-SY5Y cells. The comparative effect of Pb exposure was used in this study to assess the outcomes on Aβ, APP, and NEP.
MATERIALS AND METHODS
Cell differentiation
SH-SY5Y cells were routinely cultured in Dulbecco’s modified Eagle’s medium with high glucose (DMEM-HG) (GIBCO, Grand Island, New York) supplemented with inactivated 10% fetal bovine serum (FBS, GIBCO Grand Island, New York), 100 U/ml penicillin and 2 mM l-glutamine and maintained at 5% CO2 and 37°C. To induce cell differentiation, cells at 80% of confluence were treated with 10 μM retinoic acid (RA) (Sigma Aldrich, St Louis, Missouri) dissolved in DMEM-HG with 1% FBS for 7 days. Culture medium was removed every 3 days. Cell differentiation was confirmed by the formation of abundant and large processes and by the expression of specific neuronal markers (see below).
Cell viability assay
Aliquots of 200 000 cells were seeded per well in 12-well culture plates and differentiated with RA. Cells were exposed to 0, 1, 5, 10, 20, 50, or 100 μM Hg as HgCl2 (Sigma Aldrich, St Louis, Missouri) for 48 h. Then, 50 µg/ml of 3 -(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT) (Sigma Aldrich, St. Louis, Missouri) dissolved in DMEM-HG medium with 1% FBS were added and incubated at 37°C for 3 h. The medium was removed and formazan crystals were homogenized by adding 500 µl of dimethyl sulfoxide (DMSO) and shaken for 15 min. The absorbance at 590 nm was measured in a microtiter plate reader, and data were expressed as percent of cell viability.
Determination of Aβ-42
The production of Aβ-42 in differentiated cells was determined using a ‘sandwich’ ELISA Kit (Invitrogen, Carlsbad, California) according to the manufacturer’s instructions. Fifty micrograms of total protein extract from 1.5 × 106 differentiated cells incubated with Hg or Pb for 48 h was used for this determination. Data were expressed as pg/ml.
Immunodetection of APP and NEP
Total protein from 1.5 × 106 differentiated cells treated with 1–20 µM Hg or 50 µM Pb for 48 h were extracted with the radioimmunoprecipitation assay (RIPA) buffer containing 50 mM HEPES, pH 7.4, 150 mM NaCl, 1 mM EGTA (Sigma Aldrich, St. Louis, Missouri), 1 mM sodium orthovanadate, 100 mM NaF, 10 mM sodium pyrophosphate, 10% glycerol, 1% Triton X-100, 1% sodium deoxycholate, 1.5 mM MgCl2, 0.1% SDS, and 1 mM phenylmethylsulfonyl fluoride. Aliquots of 100 μg of total protein were separated in 10% SDS-PAGE gels and transferred to nitrocellulose membranes that were blocked with 6% nonfat dried milk in Tris buffer saline (TBS) for 2 h, and then incubated with primary antibodies anti-APP (Santa Cruz Biothecnology Inc., Dallas, Texas) at a 1:500 dilution, anti-NEP (Abcam Laboratories, Cambridge, UK) at a 1:850 dilution, and anti-actin (Santa Cruz Biothecnology Inc., Dallas, Texas) at a 1:4000 dilution overnight at 4°C. Membranes were washed 5 times with TBS/0.1% Tween 20, incubated with the secondary antibody (horseradish peroxidase-conjugated, rabbit anti-IgG mouse) for 1 h at room temperature, and finally washed 5 times with TBS/0.1% Tween 20. The intensity of bands was quantified by chemioluminiscence and analyzed by densitometry using the Image J software.
Transfection of siRNA NEP
Aliquots of 1.5 × 106 undifferentiated SH-SY5Y cells were seeded in 60 mm Petri dishes and allowed to proliferate for 2 days. Twenty microliters of Lipofectamine 2000 (Invitrogen Carlsbad, California) was dissolved in 500 μl of Optimem medium (Gibco, Grand Island, New York) and incubated for 20 min at room temperature, and then mixed with 40 μl of siRNA NEP (Santa Cruz Biotechnology Inc., Dallas, Texas) dissolved in 500 μl of Optimem culture medium. After this, Lipofectamine 2000 and siRNA NEP were added to cells, previously mixed with 1 ml of Optimem, and incubated for 4 h at 37°C. Then, the medium was replaced by DMEM-HG 10% FBS and incubated for 24 h. Finally, total proteins were extracted with RIPA buffer as previously described and used for NEP protein and activity determinations.
Enzymatic activity of NEP
The enzymatic activity of NEP was determined by a 2-step enzymatic reaction (Li and Hersh, 1995) with some modifications. This method is based on the use of a synthetic NEP substrate, Glutaryl-Ala-Ala-Phe-4-methoxy-2-naphthylamine (Sigma Aldrich, St Louis, Missouri) to produce Phe-4-methoxy-2-naphthylamine, which is used as a substrate by leucine aminopeptidase (Sigma Aldrich, St Louis, Missouri) to form the fluorescent product 4-methoxy-2-naphthylamine. Briefly, 100 µM of Glutaryl-Ala-Ala-Phe-4-methoxy-2-naphthylamine was added to 100 µg of total protein from differentiated cells treated with 10 or 20 µM of Hg or 50 µM of Pb and incubated at 37°C for 1 h in the dark to form Phe-4-methoxy-2-naphthylamine. Cells treated with 50 µM of phosphoramidon (a NEP inhibitor) (Sigma Aldrich, St Louis, Missouri) for 30 min were used as a control. The enzymatic reaction was carried out in 20 mM 2-(N-morpholino) ethanesulfonic acid (MES) (Sigma Aldrich, St Louis, Missouri) buffer at pH 6.5 in a total volume of 100 µl. To generate the fluorescent product, 3 µg of kidney leucine aminopeptidase (Sigma Aldrich, St Louis, Missouri) were added to the reaction mixture and allowed to react for 10 min, and 50 µM of phosphoramidon was added to stop the reaction. Finally, 100 μl of the reaction mixtures were loaded in triplicate in a 96-well plate and then the fluorescent 4-methoxy-2-naphthylamine was determined at an emission wavelength of 425 nm and an excitation wavelength of 340 nm. Data were reported as area under the curve of each treatment.
Immunocytochemistry
Fifty thousand cells were seeded in coverslips and differentiated as previously described. Cells were incubated with 10 or 20 µM of Hg or 50 µM of Pb for 48 h and then washed with PBS 1× and fixed with paraformaldehyde 4%. Fixed cells were washed 3 times with PBS 1×, permeabilized with PBS-Triton ×100 0.1%, and finally washed with PBS 1×. Then, cells were blocked with IgG 1%-albumin free for 1 h at room temperature, and afterwards incubated with specific primary antibodies anti-tubulin 1:100 (Covance Princenton, New York), anti-synapsin 1:100 (Santa Cruz Biotechnology Inc., Dallas, Texas), anti-AICD 1:25 (Covance, Princenton, New York) overnight at 4×C. Cells were washed thre times with PBS 1× and twice with PBS-Triton ×100 0.1% and incubated for 1 h at room temperature with the appropriate secondary antibody (TRITC anti-IgG mouse 1:80, FITC anti-IgG goat 1:80 and TRITC anti-IgG rabbit 1:80, respectively). Finally, cells were mounted with Vectashield and DAPI and immunofluorescence were observed. Images were analyzed with the Image Pro Plus software.
RNA extraction and quantification
Total RNA from 1.5 × 106 differentiated SH-SY5Y cells incubated with 1–20 µM of Hg or 50 µM of Pb was extracted with Trizol reagent (Invitrogen Carlsbad, California). One microgram of total RNA was used to synthesize the cDNA using MMLV Reverse Transcriptase (Promega Madison, Wisconsin), oligo(dT) and primers according to the manufacturer’s instructions. For q-PCR assays, an aliquot of 3 µl of cDNA was used as a template and primers for NEP and actin were added (for actin sense TGGCACCACACCTTCTACA and antisense TCACGCACGATTTCCC, for NEP sense ATGGGCAAGTCAGAAAGTCA and antisense TGCTTTCTGCACTGCTACTA) in the presence of oligonucleotides (12.5 mM), MgCl2 12.5 mM and DMSO 10% in a final volume of 50 μl. For q-PCR, 3 µl of cDNA was used as a template and Taqman probes (Applied Biosystems, California) and Master Mix in a final volume of 12.5 µl. The fluorescence intensity of NEP and hypoxanthine phosphoribosyltransferase (HPRT) (a control housekeeping gene) was determined using a 7500 real time PCR instrument (Applied Biosystems, California). Data were analyzed by the method and reported as the relative expression with respect to the control group.
Circular dichroism assay of hrNEP
To study the ability of Hg and Pb to interact with NEP and therefore to alter its conformational structure, human recombinant NEP (hrNEP) generated according to Dale et al. (2000) was used. Five micrograms of hrNEP was dissolved in PBS, pH 7 and the spectrum was recorded at 2 nm intervals from 218 to 300 nm in a CD spectrometer (JASCO J 815). Hg and Pb titrations were carried out by adding 2–20 molar equivalents from 25 µM salt solutions, pH 7. Values of Δξ at 222 nm were used to compare the effect of molar equivalents of Hg and Pb in the conformational structure of NEP. CD data were collected in duplicate experiments.
Statistical analyses
Data are expressed as the mean ± the standard error, and analyzed by 1-way ANOVA or Kruskal-Wallis tests followed by Dunnet’s or Tukey post hoc tests to determine differences among groups. A value of P ≤ .05 was considered statistically significant. GraphPad Prism 5 Software was used for all analyses.
RESULTS
Cell Differentiation and Metal Toxicity
SH-SY5Y cells were differentiated using RA (10 µM for 7 days), and changes in cell morphology such as large and abundant projections were observed by phase contrast microscopy, which were not observed in cells treated with DMSO (Fig. 1A). To confirm cell differentiation, we evaluated the expression of tubulin beta III, a marker of both mature and immature neurons. Cells treated with RA strongly expressed this marker in both the cell body and the processes, whereas cells treated with DMSO only showed a faint signal in the cell body. Synapsin, a marker of mature neurons, was also observed only in cells treated with RA (Fig. 1B). These data demonstrate that RA induced the differentiation of SH-SY5Y cells to a more mature neuronal.
FIG. 1.
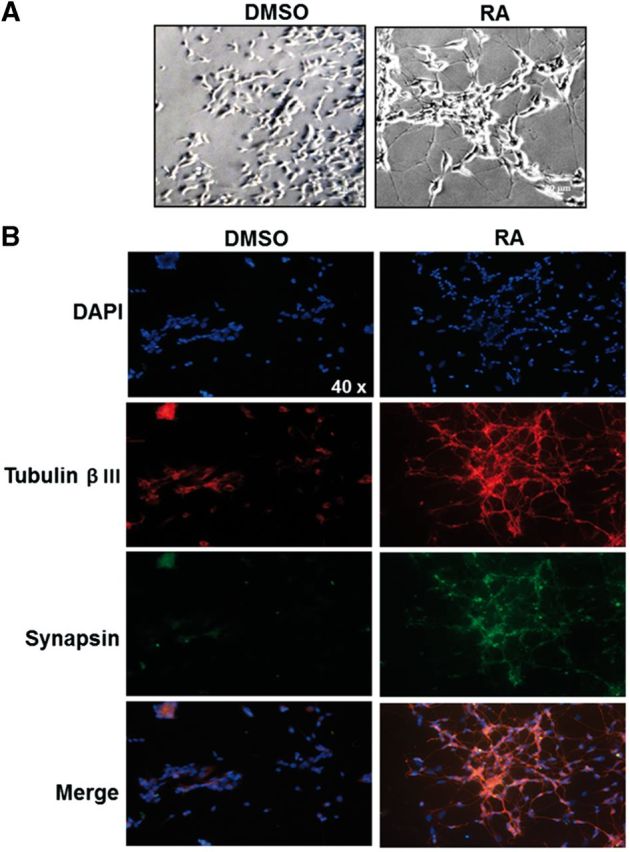
RA promotes differentiation of SH-SY5Y cell line. SH-SY5Y cells (50 000) were seeded on cover-slips for 24 h and then treated with 10 µM RA for 7 days. A, Cell morphology was evaluated by contrast phase microscopy; a representative image is presented. B, Representative immunofluorescence microscopy images showing changes in the expression of proteins of neuronal markers: tubulin βIII (TRITC, red color) and synapsin (FITC, green color). The blue staining indicates the nuclei (DAPI). RA = retinoic acid.
To determine nontoxic concentrations of Hg, we evaluated the viability of differentiated SH-SY5Y cells by the MTT assay. Cells were treated with 0, 1, 5, 10, 20, 50, or 100 µM of Hg for 48 h. We observed that cells tolerated up to 20 µM of Hg, and surprisingly, the toxicity of Hg did not show a dose-dependent response, since there were no significant effects at 1, 5, 10, or 20 µM, whereas an abrupt reduction in cell viability was observed with 50 and 100 µM (Fig. 2A). We confirmed that none of the concentrations below 50 µM of Hg were toxic to differentiated SH-SY5Y cells because no changes in p-AKT, a protein involved in cell survival, were observed (Fig. 2B). Therefore, concentrations ranging from 1 to 20 µM Hg were used for most of the experiments.
FIG. 2.

Cell viability in differentiated SH-SY5Y cells treated with HgCl2. Differentiated SH-SY5Y cells (200 000) were incubated with different concentrations of Hg (1, 5, 10, 20, 50, or 100 µM) for 48 h. A group without treatment was used as a negative control. A, MTT test was used to determine cell viability. B, Densitometric analysis of p-AKT protein levels. Total AKT was used as a loading control. The media of 3 independent experiments ± SE is presented. *P ≤ .05 compared with control cells according to 1-way ANOVA and Tukey’s post-hoc tests.
Effect of Hg on Aβ-42 and APP Levels
We found a statistically significant increase in the production of Aβ-42 when cells were exposed to 20 µM Hg. A similar effect was observed in cells treated with 50 µM Pb (Fig. 3A). To determine whether the increased production of Aβ-42 was due to an effect on its anabolism, we investigated APP protein levels by immunodetection. Treatment with Hg (1, 5, 10, or 20 µM) did not show a statistically significant increase in this protein, whereas APP was dramatically increased by the treatment with Pb (50 µM) (P < 0.05; Figs. 3B and C).
FIG. 3.

Effect of metals in Aβ-42 and APP levels. Differentiated SH-SY5Y cells (1.5 × 106) were incubated with 1, 5, 10, or 20 µM of Hg or 50 µM of Pb for 48 h. Cells treated with 50 µM Pb were used as a positive control. The group without treatment was used as a control. A, Aβ-42 levels in total protein extracts. B, A representative blot of Hg and Pb effects on APP levels. C, Densitometric analysis of APP protein levels from 3 independent experiments; mean ± SE. Actin was used as a loading control. *P ≤ .05 different with respect to control cells according to 1-way ANOVA and Dunnet’s multiple comparison post-hoc tests. RA = retinoic acid.
Effect of Metals on NEP Protein and mRNA Levels
We evaluated the effect of Hg on NEP protein levels. Unexpectedly, treatment of differentiated cells with 10 and 20 µM Hg for 48 h significantly increased NEP levels (Figs. 4A and B). In contrast, Pb treatment (50 µM) did not induce significant changes in NEP levels (Figs. 4A and B). Since this was an a non-expected result, we evaluated the protein levels of NEP in SH-SY5Y undifferentiated cells treated with 0.1, 1, and 5 µM of Hg and observed the same effect (Fig. 1A, Supplementary material). In contrast, mRNA levels of NEP evaluated by end point PCR (Fig. 4C) or q-PCR (Fig. 4D) did not increase in cells incubated with any of the metals when compared with control cells. This suggests that Hg does not alter the expression of the NEP gene.
FIG. 4.

Hg increases NEP protein but not mRNA levels in differentiated SH-SY5Y cells. Differentiated cells (1.5 × 106) were exposed to 1, 5, 10, or 20 µM of Hg or 50 µM Pb for 48 h. The negative control was the group without treatment while cells treated with 50 µM Pb were used as a positive control. One hundred micrograms of total protein was used for electrophoresis and WB analysis. A, A representative blot image. B, Densitometric analysis of 3 independent experiments; mean ± SE. Actin was used as a loading control. C, A representative image of the end-point PCR assay. Actin was used as a housekeeping gene. D, Relative values of 2ΔCt data from q-PCR assay of 3 independent experiments expressed as the mean ± SE. HPRT was used as housekeeping gene. *P ≤ .05 different with respect to control cells according to 1-way ANOVA and Dunnet’s multiple comparison post-hoc tests. NEP = neprilysin.
Effect of Metals on AICD Levels
We evaluated whether the treatment of differentiated SH-SY5Y cells with Hg or Pb for 48 h induced an increase in nuclear AICD, a product of APP cleavage, which has been implicated in the transcriptional activation of some genes, including NEP. Immunofluorescence analysis showed that AICD was preferentially found in the nucleus and treatments with 5 or 20 µM of Hg did not increase the AICD levels compared with control cells. Pb exposure did not show any change in the immunofluorescence signal compared with control cells (Fig. 5). This result is in agreement with the lack of a significant effect observed in NEP mRNA levels after the exposure to metals.
FIG. 5.

Hg does not affect nuclear levels of AICD. SH-SY5Y cells (1.5 × 106) were differentiated with RA for 7 days, and then treated with 0, 10, or 20 µM of Hg or 50 µM Pb for 48 h. Cells were fixed with 4% PFA and tested for AICD by immunofluorescence. A representative image is presented, the blue staining indicates nuclei labeled with DAPI and in green are nuclei positive to AICD peptide.
Effect of Metals on NEP Enzymatic Activity
Since the increase in NEP protein was unexpected, we evaluated whether this would be reflected in an increase in NEP enzymatic activity. We used only the higher concentrations of Hg (10 and 20 µM), and found that both concentrations clearly reduced the enzymatic activity of NEP, showing a stronger effect at 20 µM. Incubation with 50 µM phosphoramidon (an inhibitor of a limited number of metallopeptidases including NEP) dramatically reduced the activity of NEP, as it was expected (Figs. 6A and B). Interestingly, Pb (50 µM) did not reduce the NEP activity (Figs. 6A and B), suggesting a specific effect of Hg on this metallopeptidase.
FIG. 6.

Hg reduces the enzymatic activity of NEP. SH-SY5Y cells (1.5 × 106) were differentiated with RA for 7 days, and then treated with 10 or 20 µM of Hg or 50 µM Pb for 48 h. A, A kinetic evaluation of NEP activity. B, The area under the curve from each treatment is presented. Data represent the mean ± SE from 3 independent experiments. Phosphoramidon was used as a positive control of NEP activity inhibition. *P ≤ .05 different with respect to control cells according to 1-way ANOVA and Tukey’s post-hoc tests. NEP=neprilysin. Phospho = phosphoramidon.
Because NEP activity was evaluated using total protein extracts from cells treated with Hg or Pb, we investigated whether NEP was the principal enzyme responsible for the substrate degradation in the fluorometric assay. To accomplish this, we transfected a siRNA for NEP in SH-SY5Y undifferentiated cells (we were unable, under our experimental conditions to transfect differentiated SH-SY5Y cells). Immunodetection analysis showed that the siRNA successfully reduced NEP protein levels compared with a control siRNA and untreated cells (Fig. 7A). siRNA NEP also showed a statistically significant reduction in the enzyme activity of NEP compared with the control siRNA and the untreated groups; however, phosphoramidon treatment caused a further inhibition of this enzyme (Figs. 7B and C). These results suggest that the major enzymatic activity detected by the assay was that of NEP.
FIG. 7.

NEP is the principal protease responsible for the fluorogenic substrate degradation. Nondifferentiated SH-SY5Y cells were transfected with siRNA NEP or siRNA control for 24 h. A, Protein expression 24 h after the transfection. B, A kinetic evaluation of NEP activity. C, The area under the curve is presented. Phosphoramidon, an inhibitor of NEP and related metalloproteases, was used as a positive control for inhibition of the enzymatic activity. *P ≤ .05 different with respect to control cells according to 1-way ANOVA and Tukey’s post-hoc tests. NEP = neprilysin. Phospho = phosphoramidon.
Hg Interaction With NEP
Mercury is a heavy metal able to inhibit the enzymatic activity of a great number of proteins due to its ability to bind to their functional groups. To study if Hg-induced enzymatic inhibition of NEP could be due to changes in the conformational structure of the protein, we performed CD studies. rhNEP showed a characteristic spectrum of an α-helix, showing a characteristic negative band at about 222 nm (Figs. 8A and B). Since the pH alters the conformation of proteins, metal solutions were adjusted to pH 7, and then molar equivalents of both metals were added to hrNEP. The negative 222 nm band gradually increased upon addition of Hg (2–20 molar equivalents) (Fig. 8A), and the titration with 2–20 molar equivalents of Pb showed similar but smaller changes in the conformation of hrNEP (Fig. 8B). When comparing the effect of titrating the protein with Hg or Pb on the intensity of the band at 222 nm, Hg addition showed a greater negative slope compared with Pb ( − 0.0098 ± 0.0011 vs − 0.0069 ± 0.00081), however it was not significant (P = 0.095) (Fig. 8C). These data support our suggestion that Hg, but not Pb, inhibits the enzymatic activity of NEP probably by altering its conformation.
FIG. 8.

Hg changes the conformational structure of hrNEP. Recombinant hrNEP (0.5 µM in 50 µl of PBS pH 7) was used for titration with 2–20 molar equivalents of Hg or Pb dissolved in the same buffer. The spectrum was recorded every 2 nm in a CD spectrophotometer. Changes in the conformational structure of hrNEP induced by the addition of molar equivalents of Hg (A) or Pb (B) are presented. C, Effect of titration of molar equivalents of Hg or Pb at 222 nm in the hrNEP spectrum. The data were normalized to 1 when the rhNEP was free of any metal.
DISCUSSION
Environmental exposures to Hg include the intake of MeHg from seafood, inorganic Hg from activities such as mining and vapor of Hg from amalgam filling (Clarkson and Magos, 2006). It has been reported that MeHg can be slowly demethylated and sequestered in the brain of rodents and monkeys (Friberg and Mottet, 1989; Vahter et al., 1995), and also suggested by in vitro studies (Shapiro and Chan, 2008). Therefore, we believe that regardless of the chemical species of Hg that reaches the brain, it will eventually be converted to inorganic Hg.
Differentiated and undifferentiated SH-SY5Y cells are used to study neurodegenerative diseases (Cheung et al., 2009); however, differentiated cells showed characteristics more similar to mature neurons. Here, we observed a more neuron-like morphology and the expression of neuronal markers after RA treatment, as well as that differentiated cells showed higher APP levels (Fig. 1B, Supplementary material), as previously reported (Beckman and Iverfeldt, 1997). Others have shown increased levels of proteins involved in APP processing (Holback et al., 2008) or Aβ degradation, such as NEP and insulin-like degrading enzyme (IDE) (Culpan et al., 2011). In this study, differentiated SH-SY5Y cells showed a higher resistance to Hg toxicity than undifferentiated cells (data not shown), therefore we employed higher concentrations of Hg than those used in other studies performed in nondifferentiated cells (µM vs nM) (Monnet-Tschudi et al., 2006; Olivieri et al., 2000; Song and Choi, 2013).
We observed increased Aβ-42 levels in cells treated with 20 μM Hg or 50 µM Pb. It was previously reported that Pb increased Aβ-40 levels after in vitro and in vivo exposures (Huang et al., 2011; Wu et al., 2008). Regarding Hg, a study showed increased Aβ-40 levels in undifferentiated SH-SY5Y cells exposed to 180 nM inorganic Hg for 6 h (Olivieri et al., 2000), and recently, a significant increase in Aβ-40 levels was observed in PC12 cells exposed to 100 nM of inorganic Hg or MeHg for 48 h (Song and Choi, 2013). In addition, a study conducted in male rats exposed to MeHg (20–2000 µg/kg/day) found increased Aβ-42 levels in the hippocampus (Kim et al., 2014). Our data support the hypothesis that toxic metals such as Hg and Pb can be involved in the overproduction of Aβ. In contrast, a recent study showed that MeHg reduces APP proteolytic processing by inhibiting the activity of the γ secretase complex, reducing the levels of both Aβ-40 and Aβ-42 in a cell-free model and in Drosophila embryos (Alattia et al., 2011).
We did not observe increased APP protein levels in differentiated cells treated with Hg, while others reported an APP increase in aggregating brain cells, mainly in astrocytes of fetal rat telencephalon exposed to MeHg (0.1 μM/10 days) (Monnet-Tschudi et al., 2006) or PC12 cells treated with inorganic Hg or MeHg (100 nM) (Song and Choi, 2013). Interestingly, a recent study showed a significant reduction in the low-density lipoprotein receptor-related protein 1, a protein involved in the Aβ clearance in the brain capillary endothelium of male rats exposed to 20–2000 µg/kg/day/4 weeks of MeHg (Kim et al., 2014). This suggests that Hg-induced Aβ-42 is not necessarily mediated by increasing APP levels, and that other mechanisms may be involved. Regarding Pb exposure, we found that it increased APP levels, which agrees with a study done in brains of monkeys (Wu et al., 2008) treated with Pb early in life, or in differentiated SH-SY5Ycells incubated with 5–50 µM Pb (Huang et al., 2011). This increase in APP levels correlated with an increased Aβ secretion, elevated tau phosphorylation and alterations in cognitive functions (Liu et al., 2014).
On the other hand, we showed that differentiated SH-SY5Y cells incubated with 10 and 20 µM Hg increased NEP protein levels; similar results were observed with undifferentiated cells (Fig. 1A, Supplementary material). Some reports have shown reduced NEP levels in AD cases (Yasojima et al., 2001); however, one study showed increased levels of this protein in AD brain samples, which correlated with the severity of the disease (Miners et al., 2009). This can be due to a positive feedback regulation induced by Aβ, as it was reported by the intracranial administration of Aβ in mice (Mohajeri et al., 2002). Therefore, Hg-induced increased NEP levels could be secondary to an Aβ accumulation; however, we did not detect changes in NEP mRNA in cells treated with either Hg or Pb. This was confirmed by a lack of the induction of the transcriptional fragment, AICD, although it was recently reported that AICD does not bind to the promoter of NEP gene in SH-SY5Y cells as it does in other cells such as NB7 (Belyaev et al., 2009).
In addition, we showed that NEP-reduced activity induced by Hg does not appear to be mediated by the reduced expression of the protein, as it happens after the exposure to other metals such as Pb and Cd (Huang et al., 2011; Li et al., 2012). A recent study suggests that inorganic Hg and MeHg reduce NEP protein levels in PC12 cells (Song and Choi, 2013); however, Kim et al. (2014) did not find changes in NEP levels in male rats exposed to different doses of MeHg for 4 weeks. Thus the increased NEP levels observed in our work could be due to a longer half-life of the protein probably by a direct metal-protein interaction. Metal-dependent reduction of NEP activity observed in our work could be explained by Hg binding to cysteine residues, as NEP is known to be sensitive to inactivation by reaction of its thiol groups, which is suggested by our CD findings (see discussion below). Regarding Pb, we did not find changes in NEP protein level neither in its, in contrast with a previous study in which differentiated SH-SY5Y cells exposed to 5–50 µM of Pb showed a reduced expression and enzymatic activity of NEP (Huang et al., 2011). This discrepancy could be due to differences in the specificity of substrates used in the assays. Huang et al. employed N-dansyl-d-Ala-Gly-p-(nitro)-Phe-Gly, whereas we used Glutaryl-Ala-Ala-Phe-4-methoxy-2-naphthylamine; we confirmed by using a siRNA that NEP was the principal enzymatic activity measured with our fluorescent method.
CD analyses showed that Hg produces a stronger change in NEP compared to Pb. Thus, it is reasonable to suggest that Hg interacts with NEP, which could in turn contribute to the reduced activity of this enzyme, and probably to its longer half-life represented by the higher NEP levels observed in Hg-treated cells. The metal-ion substitution in the active site can change the substrate specificity and the activity of enzymes (Arima et al., 2009). By mass spectrometry and kinetic studies, Grasso et al. (2011) reported that Cu affected the IDE activity, an Aβ degrading enzyme probably by the interaction with cysteine residues and by substituting endogenous Zn. As NEP has some free cysteine residues that are important for its activity (Navarrete Santos et al., 2002), it is possible that Hg-induced NEP conformational changes and reduced activity may be due to the binding to cysteine residues. In line with this, Hg and Pb can reduce the activity of protein kinase C by interacting with cysteine residues (Rajanna et al., 1995). On the other hand, it was reported that Cd increases free Zn levels in brain mice, probably by displacing it from its native site in metalloenzymes (Li et al., 2012). In line with this, a computational study suggested that Hg can displace Zn from the active sites of enzymes and structural proteins, because it is a far better electron acceptor than Zn (Tai and Lim, 2006). However, further studies are needed to better evaluate Hg binding to NEP.
Finally, some studies demonstrated that NEP-decreased activity can be due to post-transductional alterations such as oxidation or adduct formation with products of lipid peroxidation (Shinall et al., 2005). As Hg is a pro-oxidant metal (Hussain et al., 1999), we cannot discard that Hg-reduced NEP activity may be mediated by a mechanism involving protein oxidation.
Taken together, our results show that Hg and Pb increase Aβ-42 levels, and probably act through different mechanisms. Hg seems to mainly impair the catabolism of Aβ, whereas Pb appears to target the anabolic pathway. Although Hg does not decrease NEP expression, it inhibits its activity, probably by its ability to affect NEP’s secondary structure. However, as previously discussed, we cannot discard that Hg inhibition of NEP activity might be mediated by protein oxidation. Further studies are necessary to investigate this mechanism. Our results support the hypothesis that the exposure to Hg can contribute to AD pathogenesis, despite the inconsistency of epidemiologic studies that have the genetic background as an important variable to consider.
SUPPLEMENTARY DATA
Supplementary data are available online at http://toxsci.oxfordjournals.org/.
ACKNOWLEDGMENTS
The authors want to thank Araceli Navarrete B.Sc. for her technical assistance with the q-PCR assays, Atenea Villegas B.Sc. for her technical support with CD experiments, and Elizabeth Bautista M.Sc. for her suggestions with the immunofluorescence images. M.C.C. was a recipient of a scholarship from Consejo Nacional de Ciencia y Tecnología (CONACYT-México). The authors declare no conflicts of interest.
FUNDING
This study was partially supported by CONACYT-México grant # 127357 given to J.S.
REFERENCES
- Alattia J. R., Kuraishi T., Dimitrov M., Chang I., Lemaitre B., Fraering P. C. (2011). Mercury is a direct and potent gamma-secretase inhibitor affecting Notch processing and development in Drosophila. FASEB J. 25, 2287–2295. [DOI] [PubMed] [Google Scholar]
- Arima J., Uesugi Y., Hatanaka T. (2009). Bacillus d-stereospecific metallo-amidohydrolase: active-site metal-ion substitution changes substrate specificity. Biochimie 91, 568–576. [DOI] [PubMed] [Google Scholar]
- Armstrong R. A. (1994). Differences in beta-amyloid (beta/A4) deposition in human patients with Down's syndrome and sporadic Alzheimer's disease. Neurosci. Lett. 169, 133–136. [DOI] [PubMed] [Google Scholar]
- Atwood C. S., Moir R. D., Huang X., Scarpa R. C., Bacarra N. M., Romano D. M., Hartshorn M. A., Tanzi R. E., Bush A. I. (1998). Dramatic aggregation of Alzheimer abeta by Cu(II) is induced by conditions representing physiological acidosis. J. Biol. Chem. 273, 12817–12826. [DOI] [PubMed] [Google Scholar]
- Beckman M., Iverfeldt K. (1997). Increased gene expression of beta-amyloid precursor protein and its homologues APLP1 and APLP2 in human neuroblastoma cells in response to retinoic acid. Neurosci. Lett. 221, 73–76. [DOI] [PubMed] [Google Scholar]
- Belyaev N. D., Nalivaeva N. N., Makova N. Z., Turner A. J. (2009). Neprilysin gene expression requires binding of the amyloid precursor protein intracellular domain to its promoter: implications for Alzheimer disease. EMBO Rep. 10, 94–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarkson T. W., Magos L. (2006). The toxicology of mercury and its chemical compounds. Crit. Rev. Toxicol. 36, 609–662. [DOI] [PubMed] [Google Scholar]
- Culpan D., Palmer J., Miners J. S., Love S., Kehoe P. G. (2011). The influence of tumour necrosis factor- alpha (TNF-alpha) on amyloid-beta (Abeta)-degrading enzymes in vitro. Int. J. Mol. Epidemiol. Genet. 2, 409–415. [PMC free article] [PubMed] [Google Scholar]
- Cheung Y. T., Lau W. K., Yu M. S., Lai C. S., Yeung S. C., So K. F., Chang R. C. (2009). Effects of all-trans-retinoic acid on human SH-SY5Y neuroblastoma as in vitro model in neurotoxicity research. Neurotoxicology 30, 127–135. [DOI] [PubMed] [Google Scholar]
- Dale G. E., D'Arcy B., Yuvaniyama C., Wipf B., Oefner C., D'Arcy A. (2000). Purification and crystallization of the extracellular domain of human neutral endopeptidase (neprilysin) expressed in Pichia pastoris. Acta Crystallogr. D Biol. Crystallogr. 56, 894–897. [DOI] [PubMed] [Google Scholar]
- Duce J. A., Bush A. I. (2010). Biological metals and Alzheimer's disease: implications for therapeutics and diagnostics. Prog. Neurobiol. 92, 1–18. [DOI] [PubMed] [Google Scholar]
- El-Amouri S. S., Zhu H., Yu J., Marr R., Verma I. M., Kindy M. S. (2008). Neprilysin: an enzyme candidate to slow the progression of Alzheimer's disease. Am. J. Pathol. 172, 1342–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friberg L., Mottet N. K. (1989). Accumulation of methylmercury and inorganic mercury in the brain. Biol. Trace Elem. Res. 21, 201–206. [DOI] [PubMed] [Google Scholar]
- Fujimura M., Usuki F., Sawada M., Takashima A. (2009). Methylmercury induces neuropathological changes with tau hyperphosphorylation mainly through the activation of the c-jun-N-terminal kinase pathway in the cerebral cortex, but not in the hippocampus of the mouse brain. Neurotoxicology 30, 1000–1007. [DOI] [PubMed] [Google Scholar]
- Godfrey M. E., Wojcik D. P., Krone C. A. (2003). Apolipoprotein E genotyping as a potential biomarker for mercury neurotoxicity. J. Alzheimers Dis. 5, 189–195. [DOI] [PubMed] [Google Scholar]
- Grasso G., Pietropaolo A., Spoto G., Pappalardo G., Tundo G. R., Ciaccio C., Coletta M., Rizzarelli E. (2011). Copper(I) and copper(II) inhibit Abeta peptides proteolysis by insulin-degrading enzyme differently: implications for metallostasis alteration in Alzheimer's disease. Chemistry 17, 2752–2762. [DOI] [PubMed] [Google Scholar]
- Hardy J. A., Higgins G. A. (1992). Alzheimer's disease: the amyloid cascade hypothesis. Science 256, 184–185. [DOI] [PubMed] [Google Scholar]
- Hock C., Drasch G., Golombowski S., Muller-Spahn F., Willershausen-Zonnchen B., Schwarz P., Hock U., Growdon J. H., Nitsch R. M. (1998). Increased blood mercury levels in patients with Alzheimer's disease. J. Neural Transm. 105, 59–68. [DOI] [PubMed] [Google Scholar]
- Holback S., Adlerz L., Gatsinzi T., Jacobsen K. T., Iverfeldt K. (2008). PI3-K- and PKC-dependent up-regulation of APP processing enzymes by retinoic acid. Biochem. Biophys. Res. Commun. 365, 298–303. [DOI] [PubMed] [Google Scholar]
- Huang H., Bihaqi S. W., Cui L., Zawia N. H. (2011). In vitro Pb exposure disturbs the balance between Abeta production and elimination: the role of AbetaPP and neprilysin. Neurotoxicology 32, 300–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussain S., Atkinson A., Thompson S. J., Khan A. T. (1999). Accumulation of mercury and its effect on antioxidant enzymes in brain, liver, and kidneys of mice. J. Environ. Sci. Health B 34, 645–660. [DOI] [PubMed] [Google Scholar]
- Iwata N., Higuchi M., Saido T. C. (2005). Metabolism of amyloid-beta peptide and Alzheimer’s disease. Pharmacol. Ther. 108, 129–148. [DOI] [PubMed] [Google Scholar]
- Kim D. K., Park J. D., Choi B. S. (2014). Mercury-induced amyloid-beta (Abeta) accumulation in the brain is mediated by disruption of Abeta transport. J. Toxicol. Sci. 39, 625–635. [DOI] [PubMed] [Google Scholar]
- Leong C. C., Syed N. I., Lorscheider F. L. (2001). Retrograde degeneration of neurite membrane structural integrity of nerve growth cones following in vitro exposure to mercury. Neuroreport 12, 733–737. [DOI] [PubMed] [Google Scholar]
- Li C., Hersh L. B. (1995). Neprilysin: assay methods, purification, and characterization. Methods Enzymol. 248, 253–263. [DOI] [PubMed] [Google Scholar]
- Li X., Lv Y., Yu S., Zhao H., Yao L. (2012). The effect of cadmium on Abeta levels in APP/PS1 transgenic mice. Exp. Ther. Med. 4, 125–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F., Xue Z., Li N., Huang H., Ying Y., Li J., Wang L., Li W. (2014). Effects of lead exposure on the expression of amyloid beta and phosphorylated tau proteins in the C57BL/6 mouse hippocampus at different life stages. J. Trace Elem. Med. Biol. 28, 227–232. [DOI] [PubMed] [Google Scholar]
- Miners J. S., Baig S., Tayler H., Kehoe P. G., Love S. (2009). Neprilysin and insulin-degrading enzyme levels are increased in Alzheimer disease in relation to disease severity. J. Neuropathol. Exp. Neurol. 68, 902–914. [DOI] [PubMed] [Google Scholar]
- Mohajeri M. H., Wollmer M. A., Nitsch R. M. (2002). Abeta 42-induced increase in neprilysin is associated with prevention of amyloid plaque formation in vivo. J. Biol. Chem. 277, 35460–35465. [DOI] [PubMed] [Google Scholar]
- Monnet-Tschudi F., Zurich M. G., Boschat C., Corbaz A., Honegger P. (2006). Involvement of environmental mercury and lead in the etiology of neurodegenerative diseases. Rev. Environ. Health 21, 105–117. [DOI] [PubMed] [Google Scholar]
- Mutter J., Curth A., Naumann J., Deth R., Walach H. (2010). Does inorganic mercury play a role in Alzheimer's disease? A systematic review and an integrated molecular mechanism. J. Alzheimers Dis. 22, 357–374. [DOI] [PubMed] [Google Scholar]
- Navarrete Santos A., Wulfanger J., Helbing G., Blosz T., Langner J., Riemann D. (2002). Two C-terminal cysteines are necessary for proper folding of the peptidase neprilysin/CD10. Biochem. Biophys. Res. Commun. 295, 423–427. [DOI] [PubMed] [Google Scholar]
- Olivieri G., Brack C., Muller-Spahn F., Stahelin H. B., Herrmann M., Renard P., Brockhaus M., Hock C. (2000). Mercury induces cell cytotoxicity and oxidative stress and increases beta-amyloid secretion and tau phosphorylation in SHSY5Y neuroblastoma cells. J. Neurochem. 74, 231–236. [DOI] [PubMed] [Google Scholar]
- Petroni D., Tsai J., Agrawal K., Mondal D., George W. (2012). Low-dose methylmercury-induced oxidative stress, cytotoxicity, and tau-hyperphosphorylation in human neuroblastoma (SH-SY5Y) cells. Environ. Toxicol. 27, 549–555. [DOI] [PubMed] [Google Scholar]
- Rajanna B., Chetty C. S., Rajanna S., Hall E., Fail S., Yallapragada P. R. (1995). Modulation of protein kinase C by heavy metals. Toxicol. Lett. 81, 197–203. [DOI] [PubMed] [Google Scholar]
- Shapiro A. M., Chan H. M. (2008). Characterization of demethylation of methylmercury in cultured astrocytes. Chemosphere 74, 112–118. [DOI] [PubMed] [Google Scholar]
- Shinall H., Song E. S., Hersh L. B. (2005). Susceptibility of amyloid beta peptide degrading enzymes to oxidative damage: a potential Alzheimer's disease spiral. Biochemistry 44, 15345–15350. [DOI] [PubMed] [Google Scholar]
- Song J. W., Choi B. S. (2013). Mercury induced the Accumulation of Amyloid Beta (Abeta) in PC12 Cells: the role of production and degradation of Abeta. Toxicol. Res. 29, 235–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai H. C., Lim C. (2006). Computational studies of the coordination stereochemistry, bonding, and metal selectivity of mercury. J. Phys. Chem. A 110, 452–462. [DOI] [PubMed] [Google Scholar]
- Thinakaran G., Koo E. H. (2008). Amyloid precursor protein trafficking, processing, and function. J. Biol. Chem. 283, 29615–29619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner A. J., Isaac R. E., Coates D. (2001). The neprilysin (NEP) family of zinc metalloendopeptidases: genomics and function. Bioessays 23, 261–269. [DOI] [PubMed] [Google Scholar]
- Vahter M. E., Mottet N. K., Friberg L. T., Lind S. B., Charleston J. S., Burbacher T. M. (1995). Demethylation of methyl mercury in different brain sites of Macaca fascicularis monkeys during long-term subclinical methyl mercury exposure. Toxicol. Appl. Pharmacol. 134, 273–284. [DOI] [PubMed] [Google Scholar]
- Wang D. S., Lipton R. B., Katz M. J., Davies P., Buschke H., Kuslansky G., Verghese J., Younkin S. G., Eckman C., Dickson D. W. (2005). Decreased neprilysin immunoreactivity in Alzheimer disease, but not in pathological aging. J. Neuropathol. Exp. Neurol. 64, 378–385. [DOI] [PubMed] [Google Scholar]
- Wenstrup D., Ehmann W. D., Markesbery W. R. (1990). Trace element imbalances in isolated subcellular fractions of Alzheimer’s disease brains. Brain Res. 533, 125–131. [DOI] [PubMed] [Google Scholar]
- Wojcik D. P., Godfrey M. E., Christie D., Haley B. E. (2006). Mercury toxicity presenting as chronic fatigue, memory impairment and depression: diagnosis, treatment, susceptibility, and outcomes in a New Zealand general practice setting (1994–2006). Neuro Endocrinol. Lett. 27, 415–423. [PubMed] [Google Scholar]
- Wu J., Basha M. R., Brock B., Cox D. P., Cardozo-Pelaez F., McPherson C. A., Harry J., Rice D. C., Maloney B., Chen D., et al. (2008). Alzheimer’s disease (AD)-like pathology in aged monkeys after infantile exposure to environmental metal lead (Pb): evidence for a developmental origin and environmental link for AD. J. Neurosci. 28, 3–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasojima K., Akiyama H., McGeer E. G., McGeer P. L. (2001). Reduced neprilysin in high plaque areas of Alzheimer brain: a possible relationship to deficient degradation of beta-amyloid peptide. Neurosci. Lett. 297, 97–100. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.