

Abstract
Knowledge of which genes and pathways are affected by inbreeding may help understanding the genetic basis of inbreeding depression, the potential for purging (selection against deleterious recessive alleles), and the transition from outcrossing to selfing. Arabidopsis lyrata is a predominantly self-incompatible perennial plant, closely related to the selfing model species A. thaliana. To examine how inbreeding affects gene expression, we compared the transcriptome of experimentally selfed and outcrossed A. lyrata originating from two Scandinavian populations that express similar inbreeding depression for fitness (∂ ≈ 0.80). The number of genes significantly differentially expressed between selfed and outcrossed individuals were 2.5 times higher in the Norwegian population (≈500 genes) than in the Swedish population (≈200 genes). In both populations, a majority of genes were upregulated on selfing (≈80%). Functional annotation analysis of the differentially expressed genes showed that selfed offspring were characterized by 1) upregulation of stress-related genes in both populations and 2) upregulation of photosynthesis-related genes in Sweden but downregulation in Norway. Moreover, we found that reproduction- and pollination-related genes were affected by inbreeding only in Norway. We conclude that inbreeding causes both general and population-specific effects. The observed common effects suggest that inbreeding generally upregulates rather than downregulates gene expression and affects genes associated with stress response and general metabolic activity. Population differences in the number of affected genes and in effects on the expression of photosynthesis-related genes show that the genetic basis of inbreeding depression can differ between populations with very similar levels of inbreeding depression.
Keywords: inbreeding, gene expression, stress-related genes, Arabidopsis lyrata, RNA-Seq
Introduction
The fitness consequences of inbreeding were investigated already by Darwin in order to understand why outcrossing is the prevalent mechanism of reproduction in nature. He found self-fertilization to be highly disadvantageous because the inbred progeny were often less viable and less fertile—a phenomenon known as inbreeding depression (Darwin 1876). Inbreeding depression results from exposure of recessive harmful alleles due to increased homozygosity at inbreeding, and may be the product of several mechanisms. According to the most generally accepted model, the dominance or partial dominance model, deleterious recessive mutations, which occur at low frequencies throughout the genome, will be detrimentally expressed as inbreeding increases homozygosity. In contrast, the overdominance model suggests that balancing selection favors heterozygous genotypes over homozygote ones, and that fitness is therefore reduced during inbreeding (Charlesworth D and Charlesworth B 1987). Other models assume that 1) closely linked genes with different deleterious recessive alleles in repulsion will result in lower fitness in homozygotes compared with heterozygotes in an apparently overdominant fashion (pseudo-overdominance) or 2) unlinked deleterious alleles can compensate each other in a positive epistatic manner in hybrids, causing heterosis (Charlesworth and Willis 2009). Thus, there are several possible mechanisms resulting in the expression of inbreeding depression on selfing.
However, the negative consequences of inbreeding can in some situations be more than balanced by the transmission advantage and the reproductive assurance that a selfing mutant may enjoy in an otherwise outcrossing population. Selfing allows the transmission of two parental copies of genes to the progeny through female function compared with only one copy per parent among outcrossing individuals. This is advantageous because a higher proportion of own genetic material will be transmitted (Fisher 1941), but also because locally adapted genotypes will not be broken up by recombination in each generation. Moreover, reproductive assurance may favor the evolution of selfing in environments with few suitable mates and where pollinators are scarce (Jarne and Charlesworth 1993). The direction of mating system evolution is thus expected to depend on the magnitude of inbreeding depression, but also on environmental context. Indeed, the magnitude of inbreeding depression is much stronger in regularly outcrossing species than in obligate selfers (Barrett and Charlesworth 1991; Husband and Schemske 1996; Charlesworth and Willis 2009; Winn et al. 2011).
Although the fitness consequences of inbreeding are relatively well studied in some systems (Husband and Schemske 1996; Keller and Waller 2002), little is known about the effects of inbreeding on gene expression (Kristensen et al. 2010; Hansson et al. 2014). Recent developments in “omic” technologies and systems biology allow new approaches for studying inbreeding-induced gene expression variation. For example, transcriptomics and proteomics allow visualization of gene expression and protein changes in response to inbreeding and may thus help unraveling pathways affected. Furthermore, single nucleotide polymorphism (SNP) arrays and quantitative trait locus mapping can assist in finding the deleterious alleles causing inbreeding depression. Transcriptomic studies of inbreeding effects in Drosophila melanogaster have found that inbreeding mainly affects response to stress and fundamental metabolic processes (Kristensen et al. 2010). One of the most common effects is increased expression of heat shock proteins, which are known to bind to unstable proteins and facilitate correct protein folding (Gething and Sambrook 1992). The induction of heat shock proteins is likely to be a general stress response that buffers the negative effects of expression of deleterious mutations causing protein misfolding (Ayroles et al. 2009). It is also well documented that inbreeding increases sensitivity toward environmental stress in inbred individuals (Bijlsma et al. 2000), suggesting that the upregulation of stress response genes is a general response to both genetic and environmental stress (Sorensen et al. 2003). Stress responses are associated with energetic costs, and inbred individuals in general show a lower metabolic efficiency and an upregulation of genes involved in metabolic processes (Kristensen et al. 2006; Ayroles et al. 2009). However, so far it is not known to what extent changes in gene expression reflect 1) direct effects of deleterious mutations in the corresponding genes and 2) indirect downstream responses to buffer the negative effects of stress caused by exposure of other deleterious alleles, respectively.
Several transcriptomics and genome-wide expression studies of inbreeding have been conducted using Drosophila, but there are only few corresponding studies of plants. Therefore, how inbreeding affects gene expression is still largely unknown. This is unfortunate because knowledge of which genes and pathways are affected by inbreeding is of importance for understanding central questions in evolutionary and conservation biology, including the genetic basis of inbreeding depression, the potential for purging (selection against deleterious recessive alleles), and the transition from outcrossing to selfing (Bijlsma et al. 2000; Charlesworth and Willis 2009; Kristensen et al. 2010). Purging and transition to selfing are for instance less likely when inbreeding depression is caused by many genes with weakly deleterious effects and when gene interaction is pronounced (Charlesworth et al. 1990, 1993; Ayroles et al. 2009).
In this study, we investigate the effects of inbreeding on gene expression in experimentally selfed and outcrossed Arabidopsis lyrata, a perennial herb in the family Brassicaceae and a close relative of the model organism A. thaliana (Kaul et al. 2000). Experimental plants originated from two self-incompatible, Scandinavian A. lyrata populations, which both express severe inbreeding depression for overall fitness measured across two growing seasons in a glasshouse (∂ ≈ 0.80), with similar timing and magnitude of inbreeding depression across life stages (Sletvold et al. 2013). The experimentally induced inbreeding level, that is, selfing (f = 0.5), will on average reduce heterozygosity by 50% relative to the outcrossed controls (f = 0). Therefore, any effect of the experiment is likely to be caused by increased homozygosity and expression of partly recessive alleles in the selfed individuals. We measure gene expression levels in the leaves of young plants (12 weeks old) because we want to test inbreeding effects at an early life stage. We apply transcriptome and pathway analyses and identify a large number of genes and biological functions that are sensitive to inbreeding. We also conduct gene interaction network analyses of the genes that were significantly affected by inbreeding. The results are discussed in light of the similar magnitude of inbreeding depression in the two study populations (Sletvold et al. 2013) and the fact that a mating-system transition from self-incompatibility to self-fertility has occurred in several Arabidopsis lineages, including in A. thaliana and some North American A. lyrata populations (Clauss and Koch 2006; Mable and Adam 2007).
Results
Mapping Statistics and Clustering of Samples
The number of reads and mapping statistics for the transcriptome assembly of each of the 24 samples are given in supplementary table S1, Supplementary Material online.
Clustering by Euclidean distance among individuals based on their count data revealed that the transcriptomes of the Swedish and Norwegian populations differed from each other and largely clustered separately, whereas selfed and outcrossed progeny within populations were more weakly separated, in particular in the Swedish population (fig. 1).
Fig. 1.

Clustering of individuals based on count data. The two Arabidopsis lyrata populations Sweden and Norway with the treatments selfed and outcrossed were clustered by Euclidean distance in DESeq. Individuals from Norway are shown in gray and individuals from Sweden in black, with plant ID indicating the identity of the maternal parent and whether the plant was a selfed (S) or outcrossed (W) progeny.
Differential Gene Expression between Selfed and Outcrossed Individuals
Differential gene expression between selfed and outcrossed individuals was analyzed for each population separately. In Norway, 507 genes were significantly differentially expressed between selfed and outcrossed progeny, whereas in Sweden only 195 genes were differentially expressed (fig. 2A and B). The Venn diagram in figure 2C shows the overlap between these comparisons. Sixty-two differentially expressed genes were common to both comparisons. Among those genes, a majority showed upregulation in selfed progeny, while quite a few show upregulation in one population (Sweden) and downregulation in the other (Norway; fig. 2D). A visual inspection showed that the significantly differentially expressed genes were distributed all over the genome in both populations (fig. 3).
Fig. 2.

Differential expression of selfed and outcrossed progeny and its overlap between the populations. Differential expression of genes was calculated using DESeq with an FDR and an adjusted P value of 0.05. (A) Scatter plot of log2 fold change versus mean of normalized counts for the comparison selfed versus outcrossed in Norway showing in red 507 significantly differentially expressed genes between the conditions. (B) Scatter plot for the comparison selfed versus outcrossed in Sweden, resulting in 195 significantly differentially expressed genes. The Venn diagrams represent the overlap of significantly differentially expressed genes between the two populations with focus on (C) the overlap of genes differentially expressed within Norway and within Sweden and (D) the overlap according to genes up- and downregulated in the selfed progeny of a given population.
Fig. 3.

Distribution of the differentially expressed genes in each population on the Arabidopsis lyrata genome. Genes that were significantly differentially expressed in Norway are shown in blue and those differentially expressed in Sweden are shown in orange. Their chromosomal locations were retrieved by BioMart and plotted in Circos. Numbers refer to the A. lyrata chromosome number.
The following analyses of functional annotation, gene ontology (GO) term enrichment, and pathways affected were first conducted on the 62 genes that were differentially expressed in both populations (fig. 2C), and second on the genes that were significantly differentially expressed in one population only (445 genes in the Norwegian population and 133 genes in the Swedish population; fig. 2C).
Annotation and GO Term Enrichment of Genes that Were Significantly Differentially Expressed in Both Populations
Functional annotation analyses allowed the assignment of domains and protein families to nearly all genes that were significantly affected by inbreeding in both the Norwegian and the Swedish population. Genes without an annotation included genes (or predicted genes) of unknown function, and some relatively unspecific genes, like transcription factors (supplementary table S2, Supplementary Material online). It is worth noting that some protein families and predicted domains had more than one gene assigned—like cytochrome P450 (5 genes), glycosyl hydrolases (4 genes), and chlorophyll A–B binding proteins (6 genes) to mention a few (supplementary table S2, Supplementary Material online). Furthermore, the effect of selfing on the expression profiles of many of the genes with multiple entries differed between the Norwegian and Swedish populations. Inbreeding was associated with overexpression of photosynthesis-related genes in Sweden, whereas the opposite was true in the Norwegian population (supplementary table S2, Supplementary Material online).
Annotation of GO terms and their enrichment showed that two categories of genes were particularly common among the differentially expressed genes (fig. 4). A large portion of the genes was associated with stress responses and there were also many photosynthesis-related genes. “Response to stimulus” was with 41.2% of assigned GO terms—the largest group of biological processes—and “plastid” was with 24% of assigned GO terms—the largest group of cell compartments within the category cellular component (fig. 4A). Enrichment of significant GO terms based on this annotation showed a similar outcome (fig. 4B).
Fig. 4.

GO annotation and enrichment analysis of the genes that were significantly differentially expressed in both populations. (A) Distribution of GO annotation for the candidates performed using BLAST2GO. Percentages indicate the percentage of candidate sequences assigned to each subcategory of either biological process or cellular component. (B) Enrichment map, built on GO annotation (Cytoscape plugins BiNGO and EnrichmentMap), providing insights into GO terms that are highly significant and their interactions.
Pathway Visualization of Genes that Were Differentially Expressed in Both Populations
The genes that were differentially expressed in both populations mapped to different pathways, but could be assigned to two major categories. These categories are “biotic stress” (fig. 5) and “chloroplast” (fig. 6). A closer examination of these categories showed that genes falling into the category of biotic stress were predominantly upregulated in selfed compared with outcrossed progeny in both populations (fig. 5A and B). Photosynthesis-related genes, on the other hand, were downregulated among selfed offspring in the Norwegian population (fig. 6A), but upregulated in the Swedish population (fig. 6B).
Fig. 5.
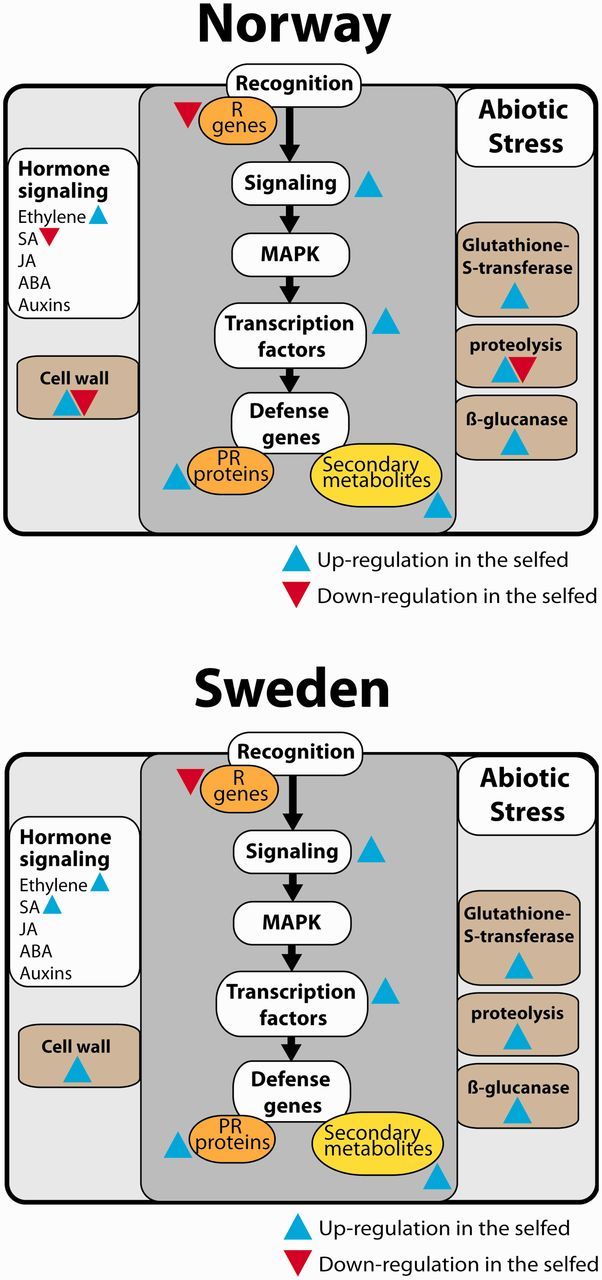
Results from biotic stress pathway analyses in the (A) Norwegian and (B) Swedish population, respectively. The analyses were based on genes that were significantly differentially expressed in both populations. Assignment of the genes into pathways was achieved by ortholog Markov clustering against Arabidopsis thaliana and mapping of orthologs into pathways. Blue signifies a positive log2 fold change in the selfed compared with the outcrossed (upregulation), whereas red signifies a negative log2 fold change in the selfed compared with the outcrossed (downregulation).
Fig. 6.
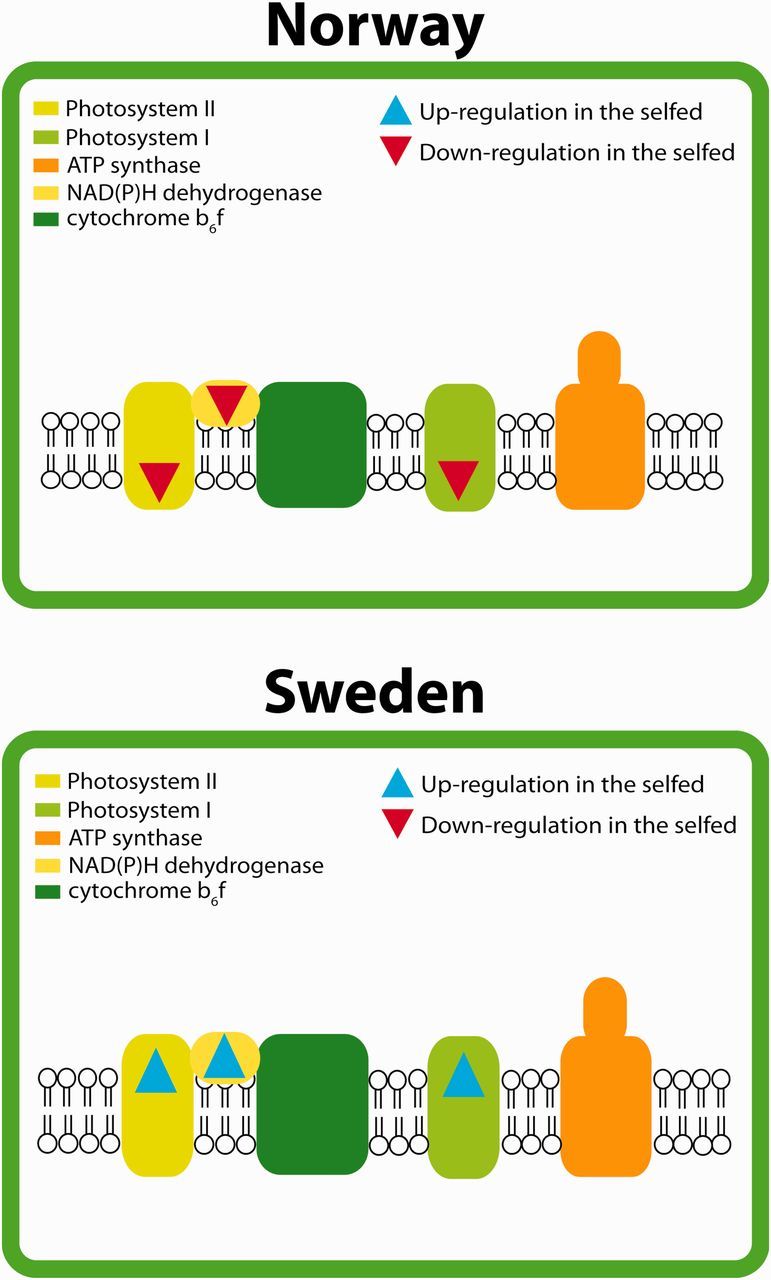
Results from chloroplast pathway analyses in the (A) Norwegian and (B) Swedish population, respectively. The analyses were based on genes that were significantly differentially expressed in both populations. Assignment of genes into pathways was achieved by ortholog Markov clustering against Arabidopsis thaliana and mapping of orthologs into pathways. Blue signifies a positive log2 fold change in the selfed compared with the outcrossed (upregulation), whereas red signifies a negative log2 fold change in the selfed compared with the outcrossed (downregulation).
Annotation, GO Term Enrichment, and Pathway Visualization of Genes that Were Differentially Expressed in One Population Only
We list the annotation to domains and protein families of the genes that were significantly differentially expressed between selfed and outcrossed progeny only in Norway in supplementary table S3, Supplementary Material online, and only in Sweden in supplementary table S4, Supplementary Material online.
GO annotation, enrichment, and pathway analyses showed that the genes that were differentially expressed in only one population to a large extent shared the patterns detected for genes being differentially expressed in both populations: A large proportion of genes was associated with biotic stress (upregulation after selfing in both populations) and photosynthesis (downregulation after selfing in Norway; upregulation in Sweden) (supplementary figs. S1–S3, Supplementary Material online). However, there were also some novel patterns detected, most notably that genes related to reproduction and pollination were affected by inbreeding, and only so in the Norwegian population (supplementary fig. S1, Supplementary Material online).
All significant GO terms, their significance levels, and associated differentially expressed genes are listed in supplementary table S5, Supplementary Material online, for the Norwegian population and in supplementary table S6, Supplementary Material online, for the Swedish population.
Gene Interaction Networks
Mapping of the differentially expressed A. lyrata genes of both populations to their A. thaliana orthologs resulted in 476 matches for the Norwegian population and 177 matches for the Swedish population, of which 224 and 104, respectively, were involved in interactions with other genes (supplementary figs. S4 and S5, Supplementary Material online). Among the genes that were significantly differentiated in both populations, 33 were involved in interactions (results not shown).
Discussion
This study has demonstrated that inbreeding affects the expression of a large number of genes in two self-incompatible populations of A. lyrata. Many of the identified genes are involved in gene interactions or are transcription factors and proteases likely to have very general effects, consistent with the earlier finding of strong reductions in several components of fitness following inbreeding (Sletvold et al. 2013). Moreover, the two populations differed substantially in expression patterns affected. The number of differentially expressed genes was approximately 2.5 times higher in the Norwegian compared with the Swedish population, and genes associated with photosynthetic activity were downregulated in selfed progeny in the Norwegian population but upregulated in the Swedish population. That the two populations differ at least partly in how inbreeding affects gene expression contrasts the situation on the level of reproduction: The two populations show almost identical inbreeding depression on several fitness components (Sletvold et al. 2013).
Population Differences in Expression Patterns
The comparison of selfed and outcrossed offspring identified 507 differentially expressed genes in the Norwegian population, compared with only 195 differentially expressed genes in the Swedish population. Furthermore, clustering by Euclidean distance revealed a separate grouping of individuals from Norway and Sweden, respectively, regardless of selfing. These population differences corroborate the pronounced differentiation observed between Scandinavian populations in a previous marker-based study (Gaudeul et al. 2007), and in traits such as trichome production (Løe et al. 2007) and tolerance to drought (Sletvold and Ågren 2012). Thus, differentiation throughout the genome, which is likely due to different demographic histories and lack of gene flow between these populations as well as differences in landscape structure and selection regimes (mountains in Norway vs. coastal habitat in Sweden), can potentially explain why these populations at least partly differ in the genetic basis of inbreeding depression.
Several factors may influence the number of inbreeding-sensitive genes detected in a population. First, short-term inbreeding effects are expected to be stronger in older and more genetically variable populations because such populations should have had time to accumulate more deleterious recessives alleles. However, SNP calling detected more SNPs in the Swedish than in the Norwegian population (Methods and Results given in supplementary material S2, fig. S6, and table S7, Supplementary Material online). Furthermore, a previous study observed a higher diversity at microsatellite loci in the Swedish population (Gaudeul et al. 2007). This suggests that the Swedish population should have had more time to accumulate recessive mutations and thus be more sensitive to inbreeding—a prediction opposite to what we observe. Second, the magnitude of within-population variance in gene expression should affect the number of significant differences detected. However, the variance in expression was higher in the Norwegian than in the Swedish population (supplementary table S8, Supplementary Material online), and this could thus not explain why fewer significant differences were detected in the latter. Finally, it may be that the population difference does not directly reflect a difference in the number of deleterious alleles exposed, but rather a difference in the number of “repair genes” induced as a secondary response (Kristensen et al. 2010). More stress response genes may be active at the 12-week stage in the Norwegian population, because of differences in seasonal phenology of growth and in the proportion of plants flowering in the first year. All plants were vegetative at the time of sampling and gene expression was measured in leaves, but 8 weeks later a higher proportion of plants had produced flowers in the Norwegian population than in the Swedish population (Sletvold et al. 2013). At the time of sampling, gene expression (in particular for genes associated with amino acid and sugar synthesis) differed between outcrossed progeny of the two populations (data not shown), which is consistent with a difference in the physiological states of plants.
Differences between Selfed and Outcrossed Offspring
In total, 62 of the significantly differentially expressed genes were common to both populations, whereas 445 genes were differentially expressed only in Norway and 133 only in Sweden. The functional annotation analyses identified two major underlying biological functions: Biotic stress response and photosynthesis. Moreover, among the genes that were significantly affected by inbreeding only in Norway, genes related to reproduction and pollination were represented.
The inbreeding effect on gene expression that we have detected in this study is likely to have been caused by a reduction in heterozygosity across the genome produced by the experimental treatment (selfing; inbreeding coefficient, f = 0.5). Indeed, the selfed progeny had much lower level of heterozygosity than had the outcrossed plants both 1) at the seven loci we used for parentage analyses (Hselfed = 29%, Houtbred = 52%, P < 0.001; data from 358 selfed and 490 outcrossed plants included in a larger experiment; Sletvold et al. 2013) and 2) at genome-wide SNPs estimated from the present RNA sequences (SNP calling, see supplementary material S2, Supplementary Material online; Norway: Hselfed = 15%, Houtcrossed = 28%, t-test: P < 0.0001; Sweden: Hselfed = 18%, Houtcrossed = 26%, t-test: P < 0.003). Another type of variation in heterozygosity occurs due to random segregation of a finite number of chromosome parts. This will make individuals with exactly the same inbreeding coefficients to differ somewhat in heterozygosity. However, such variation is much less pronounced than the variation in heterozygosity between outcrossed and selfed progeny (Chakraborty 1981; Hansson and Westerberg 2008; Hill and Weir 2011; Schraiber et al. 2012), and therefore we did not evaluate expression differences between individuals within crossing type in this study. Furthermore, random segregation induces variation in heterozygosity over the genome (even within an individual). However, this variation is also expected to be marginal in relation to the overall heterozygosity effect of the selfing treatment. In line with this reasoning, the significantly differentially expressed genes were distributed all over the genome (fig. 3).
In both populations, stress response-related genes were overexpressed in the selfed compared with the outcrossed progeny. That selfing causes a stress-related expression response and affects metabolic functions is in agreement with studies in D. melanogaster (Kristensen et al. 2005; Ayroles et al. 2009). It is likely that many of these genes are upregulated to alleviate the negative effects of exposure of other deleterious alleles, paralleling the role of heat shock proteins documented both in Drosophila (Kristensen et al. 2010) and Arabidopsis (Seki et al. 2002). The upregulation of stress genes is thus not necessarily a direct cause of inbreeding depression, but could rather represent a secondary response to the expression of the genetic load that helps restore protein function and stability. In Drosophila, there is considerable overlap in genes found to be upregulated on inbreeding versus genes upregulated by other “internal stresses,” such as oxidative stress and aging (Landis et al. 2004; Kristensen et al. 2010), and also by external factors, such as heat and chemicals (Sorensen et al. 2003). Also in Arabidopsis, there is overlap among stress-inducible genes responding to oxidative stress and different environmental factors such as drought, salinity, cold, and wounding (Seki et al. 2002; Swindell 2006). This indicates that a large part of the expression pattern induced by inbreeding represents a general stress response. This may be due to similar biological damage caused by different stresses, or a similar demand on metabolism and energetic resource allocation (Bubliy and Loeschcke 2005). In A. lyrata, there was a 50% growth reduction among inbred progeny still alive at the 12-week stage (Sletvold et al. 2013), indicating severe deleterious effects that would induce the expression of many metabolic pathways that control protein functionality.
The expression of resistance (R) genes can be associated with a cost (Tian et al. 2003), and the downregulation of some R genes in selfed progeny (fig. 5 and supplementary table S2, Supplementary Material online, protein ID 475633) may reflect their lower resource status. However, we also found some R genes to be upregulated among selfed progeny (e.g., cytochrome P450; supplementary table S2, Supplementary Material online), which is consistent with a study performed in ragged robin (Lychnis flos-cuculi) showing that inbreeding increases the activity of pathogenesis-related proteins such as kitinases and ß-1,3-glucanases (Leimu et al. 2012). Furthermore, there are parallels between pathogen recognition and self-incompatibility in the recognition and rejection of self and nonself. Both mechanisms largely rely on receptor-like kinases, like S-receptor kinases (SRKs) in the self-incompatible pathway, and pattern recognition receptors, which recognize pathogen-associated molecular patterns in innate immunity of plants (Sanabria et al. 2008). Previous studies (Pastuglia et al. 1997, 2002) found SRKs in Brassica and Arabidopsis to be upregulated upon response to biotic stress like wounding and pathogen infection, suggesting that they might play a role in processes other than self-incompatibility. In this study, such an SRK gene was upregulated in the selfed progeny of both populations. Because our data originated from vegetative tissue, it is possible that SRK has other functions in A. lyrata than its known role in the self-incompatibility system.
Photosynthesis was another major biological function affected by inbreeding in both populations, whereas effects on genes related to reproduction and pollination were observed only in the Norwegian population. The effect of inbreeding on the expression of photosynthesis-related genes differed between the two populations. Although selfed offspring had lower expression of photosynthesis-related genes in the Norwegian population, the opposite was true in the Swedish population. Effects of inbreeding on the expression of photosynthesis-related genes may reflect a reduced ability to match gene expression to prevailing environmental conditions, whereas the difference in the direction of the effect may be related to inherent differences in the seasonal pattern of growth between the two populations. The Norwegian population experiences a shorter growing season at its site of origin, mainly due to its higher altitude, and may at the 12-week stage slow down energy accumulation in preparation for winter. In addition, in the present experiment, a higher proportion of the 12-week plants may have been in transition to initiate the formation of reproductive structures in the Norwegian population compared with the Swedish population (8 weeks later, 26% of the Norwegian and 13% of the Swedish plants had produced flowers in the outcross treatment; Sletvold et al. 2013). This could potentially contribute to the difference observed in the expression of photosynthesis-related genes, but also explain why an effect of selfing on the expression of genes related to reproduction and pollination was observed only in the Norwegian population.
Conclusion
Whether deleterious recessive alleles can be purged from a population, thereby reducing inbreeding depression and facilitating mating-system transitions from outcrossing to selfing, is a central question in conservation and evolutionary biology. Both purging and transition to selfing are less likely if inbreeding depression is caused by many genes with weakly deleterious effects (Charlesworth et al. 1990, 1993; Ayroles et al. 2009). We found that many genes were significantly differentially expressed after selfing in A. lyrata (507 genes in Norway and 195 in Sweden), and that at least half of them interacted closely with other genes. Moreover, some of the inbreeding-sensitive genes were identified as transcription factors and proteases and are therefore likely to be located upstream in functional pathways and have regulatory functions. This indicates that the observed differences in gene expression may reflect the accumulation of a large number of interacting and functionally important recessive mutations in these populations, which would in turn render purging and transition to selfing unlikely. Nevertheless, transition from self-incompatibility to self-fertility has occurred in several Arabidopsis lineages, during the formation of A. thaliana as well as in some North American populations of A. lyrata (Clauss and Koch 2006; Mable and Adam 2007). This indicates that some of the genetic load has been purged in those lineages. Thus, it is possible that the wide inbreeding effects on gene expression that we have detected are at least partly driven by an extensive network of secondary responses, where pathways may be influenced by the expression of relatively few key genes with deleterious mutations. In Drosophila, it was found that at least 38% of the differentially expressed genes during inbreeding were affected by such indirect network effects (Ayroles et al. 2009). This highlights the difficulties in inferring the actual number of genes that are directly affected by inbreeding from transcriptome-wide gene expression analyses.
The similar magnitude of inbreeding depression (Sletvold et al. 2013) suggests that the opportunity for deleterious mutations to accumulate has been similar in the two self-incompatible study populations. In line with this, we found considerable population overlap in the expression of genes associated with stress response and general metabolic activities. The genes that were significantly affected by inbreeding in the same direction in both populations are particularly strong candidates for being inbreeding-sensitive genes in A. lyrata. We also found striking differences between the populations in the patterns of gene expression affected by selfing, where more genes were significantly affected by inbreeding in Norway, and photosynthesis-related genes were upregulated in Norway but downregulated in Sweden. Thus, this study indicates that the genetic basis of inbreeding depression partly differs between the two populations, despite very similar effects of inbreeding on fitness (Sletvold et al. 2013).
Materials and Methods
Study Species, Crossings, and RNA Extraction
Arabidopsis lyrata ssp. petraea has a disjunct, patchy distribution in Europe (Jalas and Suominen 1994), and occurs across a wide altitudinal range in habitats characterized by moderate soil disturbance. In 2007, seeds were collected from 50 plants in each of the two large Scandinavian A. lyrata ssp. petraea populations (>2,000 individuals): An alpine population in Norway (Spiterstulen: 1,100 m a.s.l., 61 °41′N, 8 °25′E) and a coastal population in Sweden (Stubbsand: <5 m a.s.l., 63 °13′N, 18 °58′E). The populations are differentiated at microsatellite loci (Gaudeul et al. 2007), as well as in phenotypic traits such as flowering phenology (Sandring et al. 2007), leaf trichome production (Løe et al. 2007), and tolerance to drought (Sletvold and Ågren 2012). Progeny raised from field-collected seeds were grown for one generation and randomly outcrossed in the greenhouse. Detailed descriptions of the laboratory conditions are given elsewhere (Sletvold et al. 2013) and will only be briefly mentioned here.
In 2008, seeds were produced through controlled self-pollination and cross-pollination, respectively. To circumvent the self-incompatibility system, flowers were self-pollinated at the bud stage (Cabin et al. 1996; Kärkkäinen et al. 1999; Kachroo et al. 2002). Each cross-pollinated flower received pollen from two random donors within the source population. In 2009, 9 replicates per cross type of 24 maternal parents per population were planted in 5.5 × 5.5 cm pots. In each pot, two seeds were planted. The pots were stratified for 4 days at 4 °C in a dark cold room and subsequently transferred to a growth room and kept at 20 °C 16 h day (150 µE/m2/s)/16 °C 8 h night. After 3 weeks, seedlings were thinned to one per pot. After ca. 12 weeks, one rosette leaf per experimental plant was collected in RNALater (Qiagen), and the sample was frozen. At this stage, all plants were still vegetative; outbred progeny were on average twice the size of inbred progeny in both populations (∂ = 0.50; effect of crosstype P < 0.0001 in two-way analysis of variance), but there was no significant difference between populations in plant size (effect of population P = 0.30, population × cross-type P = 0.95; Sletvold et al. 2013).
To verify the success of experimental selfing at the bud stage, maternal plants and their progeny were genotyped at seven microsatellite loci (Clauss et al. 2002; Woodhead et al. 2007; Sletvold et al. 2013). DNA was extracted and the microsatellites PCR amplified in two touchdown multiplexes using Qiagen Multiplex PCR Kit (PCR conditions given in Sletvold et al. 2013). The PCR products from the two multiplexes were pooled and then separated based on length and primer labeling using ABI Prism 3730 capillary Sequencer (Applied Biosystems). Genotypes were scored with GeneMapper 4.0 (Applied Biosystems). Parentage analyses were performed to verify the experimental selfing by matching the genotype at the seven loci of each progeny to the genotype of the mother. To verify selfing, we used a strict criterion, that is, no mismatches were allowed between the progeny and the mother at any loci. Among the outcrossed progeny not a single individual had a perfect match to their mothers, which indicates high power to detect contamination during the selfing experiment with this set of microsatellites. The overall proportion of contaminants in the whole greenhouse population (consisting of more than a thousand plants of which 848 were tested) was 0.15, and ranged from 0 to 0.78 within maternal lines.
RNA Extraction and Sequencing
In total, 24 individual seedlings (progeny) that were the products of outcrossing (12 individuals: 5 Norwegian and 7 Swedish) or of verified selfing (12 individuals: 5 Norwegian and 7 Swedish), respectively, were selected for RNA sequencing. Total RNA extraction, DNAse treatment, and rRNA reduction, as well as RNA quality control were performed by LGC Genomics (Berlin, Germany). LGC Genomics also conducted mRNA isolation and cDNA synthesis, as well as Illumina library preparation, indexing, and library quality control. LGC genomics then performed cluster generation and sequencing using Illumina HiSeq2000 (100-bp paired end).
Finding Differentially Expressed Genes
Raw reads of the transcriptome were assembled for each sample using TopHat version 2.04 (Trapnell et al. 2009), a spliced read mapper, by using default parameters and specifying the A. lyrata reference genome and the corresponding gene model annotation file (both obtained from Joint Genome Institute, JGI; Grigoriev et al. 2012). Later, read counts were extracted from the TopHat output using the python script HTSeq (http://www-huber.embl.de/users/anders/HTSeq/) and analysis of differential expression was performed using the Bioconductor package DESeq (Anders and Huber 2010) as outlined in the manual performing the following comparisons: Norway selfed versus Norway outcrossed and Sweden selfed versus Sweden outcrossed.
HTSeq counts the number of reads aligning to the mRNA of a gene by using the coordinates of the annotation file as guidance. In the approach used by DESeq to determine differential expression, the analysis is based on the assumption that the more reads are counted for one mRNA, the higher the corresponding gene is expressed. First, DESeq estimates the effective library size of each sample and divides the read counts for each gene of a sample by the value of this sample’s effective library size to obtain a common scale and enable comparison. In the next step, the dispersion for replicates of one condition is estimated, which is used as an estimate of the square of the coefficient for biological variation. The final step is the call for differential expression between two conditions (here: Selfed vs. outcrossed; Anders and Huber 2010), which is followed by filtering for significant genes (here a false discovery rate [FDR] of 0.05 was chosen as threshold for significance).
A detailed description of all analysis steps and computer code can be found in supplementary material S1A–C, Supplementary Material online.
Annotation and GO Term Enrichment
In our functional annotation analyses, we first considered genes that were significantly differentially expressed in both Norway and Sweden. Then, we examined the functional annotation of genes being differentially expressed in one population only.
The functional annotation analysis was performed using the standalone version of InterProScan version 5RC1 (Zdobnov and Apweiler 2001) as shown in supplementary material S1D, Supplementary Material online, and by extracting information from The Arabidopsis Information Resource (http://www.arabidopsis.org/, last accessed March 29, 2015). For GO annotation of the selected genes, BLAST2GO (Conesa et al. 2005) was used and GO term enrichment analysis was performed using the Cytoscape version 2.8.3 (Shannon et al. 2003) plugins BiNGO (Maere et al. 2005) and EnrichmentMap (Merico et al. 2010). The annotation file used for BiNGO was obtained from JGI (Araly1_goinfo_FilteredModels6.tab) (Grigoriev et al. 2012) and modified according to the plugin’s requirements. Hypergeometric test and FDR of 0.05 were chosen for enrichment analysis. An enrichment map of the GO terms was generated using the plugin EnrichmentMap with the following statistical parameters: P value cut-off of 0.001, Q value cut-off of 0.05, and Jaccard coefficient with an overlap coefficient cut-off of 0.25.
Pathway Visualization
For pathway visualization, we used MapMan version 3.5.1 (Thimm et al. 2004), a program that assigns genes to functional categories (biological functions), so called BINs, which can then be displayed in pathway images. Because a mapping file needed for the functional categorization does not exist for A. lyrata but for the close relative A. thaliana, A. lyrata genes were assigned to their A. thaliana orthologs. A fasta file containing focal genes’ protein sequences was uploaded to orthoMCL (web version; http://orthomcl.org/cgi-bin/OrthoMclWeb.cgi?rm=proteomeUploadForm, last accessed March 29, 2015), which uses a Markov clustering algorithm to find orthologs. Later, ortholog identifiers were translated into Arabidopsis Genome Initiative identifiers and together with their appertaining log2 fold changes for selfing orthologs loaded in MapMan for pathway mapping. Supplementary material S1E, Supplementary Material online, shows an example of the input mapping file.
Gene Interaction Networks
In order to identify genes that interact with the genes found to be differentially expressed in each of the populations, gene interaction networks were created. Differentially expressed A. lyrata genes were assigned to their orthologs in A. thaliana, because gene interactions are better documented for A. thaliana than for A. lyrata. The A. thaliana ortholog ID was used together with the expression fold changes (i.e., the expression ration between selfed and outcrossed progeny) from the present experiment as input for the PSICQUIC (Proteomics Standard Initiative Common Query InterfaCe, Aranda et al. 2011) client inbuilt in Cytoscape. PSICQUIC allows the search of multiple resources for molecular interactions. For our data, IntAct (Kerrien et al. 2012), The Bio-Analytic Resource for Plant Biology, BIND (Bader et al. 2003), MINT (Licata et al. 2012), and UniProt (UniProt Consortium 2014) were queried and from the database output a union network was constructed and interactions were restricted to first neighbor interactions. Nodes, representing the differentially expressed genes, were colored by fold change.
Distribution of Differentially Expressed Genes on the Arabidopsis lyrata Genome
In order to display the distribution of the differentially expressed genes of each population on the A. lyrata genome, their chromosomal locations were retrieved by BioMart (Smedley et al. 2009) and plotted in Circos version 0.67-5 (Krzywinski et al. 2009).
Statistical Analysis
If not stated otherwise, statistical analyses were performed in R (http://www.r-project.org/, last accessed March 29, 2015).
Supplementary Material
Supplementary figures S1–S6, tables S1–S8, and materials S1 and S2 are available at Molecular Biology and Evolution online (http://www.mbe.oxfordjournals.org/).
Acknowledgments
The authors acknowledge the Swedish Research council for funding to B.H. and J.Å.; LGC Berlin for extraction, library preparation, and sequencing; UPPMAX for computational support; and Björn Canbäck and Dag Ahrén for bioinformatics support.
References
- Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11:R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aranda B, Blankenburg H, Kerrien S, Brinkman FS, Ceol A, Chautard E, Dana JM, De Las Rivas J, Dumousseau M, Galeota E, et al. PSICQUIC and PSISCORE: accessing and scoring molecular interactions. Nat Methods. 2011;8:528–529. doi: 10.1038/nmeth.1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayroles JF, Hughes KA, Rowe KC, Reedy MM, Rodriguez-Zas SL, Drnevich JM, Caceres CE, Paige KN. A genomewide assessment of inbreeding depression: gene number, function, and mode of action. Conserv Biol. 2009;23:920–930. doi: 10.1111/j.1523-1739.2009.01186.x. [DOI] [PubMed] [Google Scholar]
- Bader GD, Betel D, Hogue CW. BIND: the Biomolecular Interaction Network Database. Nucleic Acids Res. 2003;31:248–250. doi: 10.1093/nar/gkg056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett SCH, Charlesworth D. Effects of a change in the level of inbreeding on the genetic load. Nature. 1991;325:522–524. doi: 10.1038/352522a0. [DOI] [PubMed] [Google Scholar]
- Bijlsma R, Bundgaard J, Boerema AC. Does inbreeding affect the extinction risk of small populations? Predictions from Drosophila. J Evol Biol. 2000;13:502–514. [Google Scholar]
- Bubliy OA, Loeschcke V. Correlated responses to selection for stress resistance and longevity in a laboratory population of Drosophila melanogaster. J Evol Biol. 2005;18:789–803. doi: 10.1111/j.1420-9101.2005.00928.x. [DOI] [PubMed] [Google Scholar]
- Cabin RJ, Evans AS, Jennings DL, Marshall DL, Mitchell RJ, Sher AA. Using bud pollinations to avoid self-incompatibility: implications from studies of three mustards. Can J Bot. 1996;74:285–289. [Google Scholar]
- Chakraborty R. The distribution of the number of heterozygous loci in an individual in natural populations. Genetics. 1981;98:461–466. doi: 10.1093/genetics/98.2.461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth D, Charlesworth B. Inbreeding depression and its evolutionary consequences. Annu Rev Ecol Syst. 1987;18:237–268. [Google Scholar]
- Charlesworth D, Morgan MT, Charlesworth B. Inbreeding depression, genetic load, and the evolution of outcrossing rates in a multilocus system with no linkage. Evolution. 1990;44:1469–1489. doi: 10.1111/j.1558-5646.1990.tb03839.x. [DOI] [PubMed] [Google Scholar]
- Charlesworth D, Morgan MT, Charlesworth B. Mutation accumulation in finite outbreeding and inbreeding populations. Genet Res. 1993;61:39–56. [Google Scholar]
- Charlesworth D, Willis JH. The genetics of inbreeding depression. Nat Rev Genet. 2009;10:783–796. doi: 10.1038/nrg2664. [DOI] [PubMed] [Google Scholar]
- Clauss MJ, Cobban H, Mitchell-Olds T. Cross-species microsatellite markers for elucidating population genetic structure in Arabidopsis and Arabis (Brassicaeae) Mol Ecol. 2002;11:591–601. doi: 10.1046/j.0962-1083.2002.01465.x. [DOI] [PubMed] [Google Scholar]
- Clauss MJ, Koch MA. Poorly known relatives of Arabidopsis thaliana. Trends Plant Sci. 2006;11:449–459. doi: 10.1016/j.tplants.2006.07.005. [DOI] [PubMed] [Google Scholar]
- Conesa A, Gotz S, Garcia-Gomez JM, Terol J, Talon M, Robles M. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21:3674–3676. doi: 10.1093/bioinformatics/bti610. [DOI] [PubMed] [Google Scholar]
- Darwin CR. The effects of cross and self fertilization in the vegetable kingdom. London: John Murray; 1876. [Google Scholar]
- Fisher RA. Average excess and average effect of a gene substitution. Ann Hum Genet. 1941;11:53–63. [Google Scholar]
- Gaudeul M, Stenøien HK, Ågren J. Landscape structure, clonal propagation, and genetic diversity in Scandinavian populations of Arabidopsis lyrata (Brassicaceae) Am J Bot. 2007;94:1146–1155. doi: 10.3732/ajb.94.7.1146. [DOI] [PubMed] [Google Scholar]
- Gething MJ, Sambrook J. Protein folding in the cell. Nature. 1992;355:33–45. doi: 10.1038/355033a0. [DOI] [PubMed] [Google Scholar]
- Grigoriev IV, Nordberg H, Shabalov I, Aerts A, Cantor M, Goodstein D, Kuo A, Minovitsky S, Nikitin R, Ohm RA, et al. The genome portal of the Department of Energy Joint Genome Institute. Nucleic Acids Res. 2012;40:D26–D32. doi: 10.1093/nar/gkr947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansson B, Naurin S, Hasselquist D. Does inbreeding affect gene expression in birds? Biol Lett. 2014 doi: 10.1098/rsbl.2014.0648. 10:20140648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansson B, Westerberg L. Heterozygosity-fitness correlations within inbreeding classes: local or genome-wide effects? Conserv Genet. 2008;9:73–83. [Google Scholar]
- Hill WG, Weir BS. Variation in actual relationship as a consequence of Mendelian sampling and linkage. Genetics Res. 2011;93:47–64. doi: 10.1017/S0016672310000480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Husband BC, Schemske DW. Evolution of the magnitude and timing of inbreeding depression in plants. Evolution. 1996;50:54–70. doi: 10.1111/j.1558-5646.1996.tb04472.x. [DOI] [PubMed] [Google Scholar]
- Jalas J, Suominen J. Atlas florae Europaeae: distribution of vascular plants in Europe. Vol. 1–10. Helsinki (Finland): The Committee for Mapping the Flora of Europe and Societas Biologica Fennica Vanamo; 1994. [Google Scholar]
- Jarne P, Charlesworth D. The evolution of the selfing rate in functionally hermaphrodite plants and animals. Annu Rev Ecol Syst. 1993;24:441–466. [Google Scholar]
- Kachroo A, Nasrallah ME, Nasrallah JB. Self-incompatibility in the Brassicaceae: receptor-ligand signaling and cell-to-cell communication. Plant Cell. 2002;14:S227–S238. doi: 10.1105/tpc.010440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kärkkäinen K, Kuittinen H, van Treuren R, Vogl C, Oikarinen S, Savolainen O. Genetic basis of inbreeding depression in Arabis petraea. Evolution. 1999;53:1354–1365. doi: 10.1111/j.1558-5646.1999.tb05400.x. [DOI] [PubMed] [Google Scholar]
- Kaul S, Koo HL, Jenkins J, Rizzo M, Rooney T, Tallon LJ, Feldblyum T, Nierman W, Benito MI, Lin XY, et al. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature. 2000;408:796–815. doi: 10.1038/35048692. [DOI] [PubMed] [Google Scholar]
- Keller LF, Waller DM. Inbreeding effects in wild populations. Trends Ecol Evol. 2002;17:230–241. [Google Scholar]
- Kerrien S, Aranda B, Breuza L, Bridge A, Broackes-Carter F, Chen C, Duesbury M, Dumousseau M, Feuermann M, Hinz U, et al. The IntAct molecular interaction database in 2012. Nucleic Acids Res. 2012;40:D841–D846. doi: 10.1093/nar/gkr1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristensen TN, Pedersen KS, Vermeulen CJ, Loeschcke V. Research on inbreeding in the ‘omic’ era. Trends Ecol Evol. 2010;25:44–52. doi: 10.1016/j.tree.2009.06.014. [DOI] [PubMed] [Google Scholar]
- Kristensen TN, Sorensen P, Kruhoffer M, Pedersen KS, Loeschcke V. Genome-wide analysis on inbreeding effects on gene expression in Drosophila melanogaster. Genetics. 2005;171:157–167. doi: 10.1534/genetics.104.039610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristensen TN, Sorensen P, Pedersen KS, Kruhoffer M, Loeschcke V. Inbreeding by environmental interactions affect gene expression in Drosophila melanogaster. Genetics. 2006;173:1329–1336. doi: 10.1534/genetics.105.054486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, Jones SJ, Marra MA. Circos: an information aesthetic for comparative genomics. Genome Res. 2009;19:1639–1645. doi: 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landis GN, Abdueva D, Skvortsov D, Yang J, Rabin BE, Carrick J, Tavare S, Tower J. Similar gene expression patterns characterize aging and oxidative stress in Drosophila melanogaster. Proc Natl Acad Sci U S A. 2004;101:7663–7668. doi: 10.1073/pnas.0307605101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leimu R, Kloss L, Fischer M. Inbreeding alters activities of the stress-related enzymes chitinases and beta-1,3-glucanases. PLoS One. 2012;7:e42326. doi: 10.1371/journal.pone.0042326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Licata L, Briganti L, Peluso D, Perfetto L, Iannuccelli M, Galeota E, Sacco F, Palma A, Nardozza AP, Santonico E, et al. MINT, the molecular interaction database: 2012 update. Nucleic Acids Res. 2012;40:D857–D861. doi: 10.1093/nar/gkr930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Løe G, Toräng P, Gaudeul M, Ågren J. Trichome production and spatiotemporal variation in herbivory in the perennial herb Arabidopsis lyrata. Oikos. 2007;116:134–142. [Google Scholar]
- Mable BK, Adam A. Patterns of genetic diversity in outcrossing and selfing populations of Arabidopsis lyrata. Mol Ecol. 2007;16:3565–3580. doi: 10.1111/j.1365-294X.2007.03416.x. [DOI] [PubMed] [Google Scholar]
- Maere S, Heymans K, Kuiper M. BiNGO: a Cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics. 2005;21:3448–3449. doi: 10.1093/bioinformatics/bti551. [DOI] [PubMed] [Google Scholar]
- Merico D, Isserlin R, Stueker O, Emili A, Bader GD. Enrichment map: a network-based method for gene-set enrichment visualization and interpretation. PLoS One. 2010;5:e13984. doi: 10.1371/journal.pone.0013984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastuglia M, Roby D, Dumas C, Cock JM. Rapid induction by wounding and bacterial infection of an S gene family receptor-like kinase gene in Brassica oleracea. Plant Cell. 1997;9:49–60. doi: 10.1105/tpc.9.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastuglia M, Swarup R, Rocher A, Saindrenan P, Roby D, Dumas C, Cock JM. Comparison of the expression patterns of two small gene families of S gene family receptor kinase genes during the defence response in Brassica oleracea and Arabidopsis thaliana. Gene. 2002;282:215–225. doi: 10.1016/s0378-1119(01)00821-6. [DOI] [PubMed] [Google Scholar]
- Sanabria N, Goring D, Nurnberger T, Dubery I. Self/nonself perception and recognition mechanisms in plants: a comparison of self-incompatibility and innate immunity. New Phytol. 2008;178:503–513. doi: 10.1111/j.1469-8137.2008.02403.x. [DOI] [PubMed] [Google Scholar]
- Sandring S, Riihimäki MA, Savolainen O, Ågren J. Selection on flowering time and floral display in an alpine and a lowland population of Arabidopsis lyrata. J Evol Biol. 2007;20:558–567. doi: 10.1111/j.1420-9101.2006.01260.x. [DOI] [PubMed] [Google Scholar]
- Schraiber JG, Shih S, Slatkin M. Genomic tests of variation in inbreeding among individuals and among chromosomes. Genetics. 2012;192:1477–1482. doi: 10.1534/genetics.112.145367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seki M, Narusaka M, Ishida J, Nanjo T, Fujita M, Oono Y, Kamiya A, Nakajima M, Enju A, Sakurai T, et al. Monitoring the expression profiles of 7000 Arabidopsis genes under drought, cold and high-salinity stresses using a full-length cDNA microarray. Plant J. 2002;31:279–292. doi: 10.1046/j.1365-313x.2002.01359.x. [DOI] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sletvold N, Ågren J. Variation in tolerance to drought among Scandinavian populations of Arabidopsis lyrata. Evol Ecol. 2012;26:559–577. [Google Scholar]
- Sletvold N, Mousset M, Hagenblad J, Hansson B, Ågren J. Strong inbreeding depression in two Scandinavian populations of the self-incompatible perennial herb Arabidopsis lyrata. Evolution. 2013;67:2876–2888. doi: 10.1111/evo.12174. [DOI] [PubMed] [Google Scholar]
- Smedley D, Haider S, Ballester B, Holland R, London D, Thorisson G, Kasprzyk A. BioMart—biological queries made easy. BMC Genomics. 2009;10:22. doi: 10.1186/1471-2164-10-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorensen JG, Kristensen TN, Loeschcke V. The evolutionary and ecological role of heat shock proteins. Ecol Lett. 2003;6:1025–1037. [Google Scholar]
- Swindell WR. The association among gene expression responses to nine abiotic stress treatments in Arabidopsis thaliana. Genetics. 2006;174:1811–1824. doi: 10.1534/genetics.106.061374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thimm O, Blasing O, Gibon Y, Nagel A, Meyer S, Kruger P, Selbig J, Muller LA, Rhee SY, Stitt M. MAPMAN: a user-driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. Plant J. 2004;37:914–939. doi: 10.1111/j.1365-313x.2004.02016.x. [DOI] [PubMed] [Google Scholar]
- Tian D, Traw MB, Chen JQ, Kreitman M, Bergelson J. Fitness costs of R-gene-mediated resistance in Arabidopsis thaliana. Nature. 2003;423:74–77. doi: 10.1038/nature01588. [DOI] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- UniProt Consortium. Activities at the Universal Protein Resource (UniProt) Nucleic Acids Res. 2014;42:D191–D198. doi: 10.1093/nar/gkt1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winn AA, Elle E, Kalisz S, Cheptou PO, Eckert CG, Goodwillie C, Johnston MO, Moeller DA, Ree RH, Sargent RD, et al. Analysis of inbreeding depression in mixed-mating plants provides evidence for selective interference and stable mixed mating. Evolution. 2011;65:3339–3359. doi: 10.1111/j.1558-5646.2011.01462.x. [DOI] [PubMed] [Google Scholar]
- Woodhead M, Russell J, Squirrell J, Hollingsworth PM, Cardle L, Gibby M, Powell W. Development of EST-derived microsatellite markers for Arabidopsis lyrata subspecies petraea (L.) Mol Ecol Notes. 2007;7:631–634. [Google Scholar]
- Zdobnov EM, Apweiler R. InterProScan—an integration platform for the signature-recognition methods in InterPro. Bioinformatics. 2001;17:847–848. doi: 10.1093/bioinformatics/17.9.847. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.