

Abstract
Aims
The aims of this study were to develop a population pharmacokinetic (PK) model to describe the PK of nalmefene in healthy subjects and to relate the exposure of nalmefene to the μ‐opioid receptor occupancy by simulations in the target population.
Methods
Data from nine phase I studies (243 subjects) with extensive blood sampling were pooled and used for the population PK model building. Data from four other phase I studies (85 subjects) were pooled and used as an external validation dataset. Eight subjects from an imaging study contributed occupancy data and the pharmacokinetic/pharmacodynamic (PK/PD) relationship was modelled. Combining the population PK model and the PK/PD relationship enabled simulations to predict μ‐opioid occupancy.
Results
A two compartment model with first order absorption best described the nalmefene PK data. The typical subject in the population was estimated to have a systemic clearance of 60.4 l h−1 and a central volume of distribution of 266 l. Absolute oral bioavailability was estimated to 41% without food intake and with food about 53%. Simulation of the μ‐opioid receptor occupancy shows that the 95% confidence bound is within or above 60–90% occupancy for up to 22–24 h after a single dose of 20 mg nalmefene.
Conclusions
A robust population PK model for nalmefene was developed. Based on the concentration–occupancy model the μ‐opioid receptor occupancy after a single 20 mg dose of nalmefene is predicted to be above the target therapeutic occupancy for about 24 h in about 95% of the target population.
Keywords: alcohol dependence, healthy subjects, nalmefene, opioid antagonist, pharmacodynamics, pharmacokinetics
What is Already Known About this Subject
Nalmefene is a new drug approved for reduction of alcohol consumption in adult patients with alcohol dependence.
Nalmefene is an opioid system modulator, with antagonist activity at the μ and δ receptors and partial agonist activity at the κ receptor.
What this Study Adds
A population pharmacokinetic model for nalmefene was developed and validated and related to occupancy of the μ‐opioid receptors.
By modelling/simulating the nalmefene pharmacokinetics/dynamics, it was shown that the occupancy of the μ‐opioid receptors by nalmefene will be within or above 60–90% for up to 22–24 h after a single dose of 20 mg nalmefene.
Introduction
Nalmefene is an opioid system modulator with antagonist activity at the μ and δ receptors and partial agonist activity at the κ receptor 1. In vitro studies have displayed that nalmefene has high and comparable affinity with the μ‐ and κ‐opioid receptors and a somewhat lower affinity to the δ‐opioid receptor. The opioid receptors are a subfamily of the family A G protein‐coupled opioid receptor superfamily and consist of μ (OPRM1), δ (OPRD1), and κ (OPRK1), all of which activate inhibitory G proteins 2. It is the first pharmacological treatment approved for the reduction of alcohol consumption in adult patients with alcohol dependence, who have a high drinking risk level (that is, an alcohol consumption >60 g day–1 for men and >40 g day–1 for women) and who do not require immediate detoxification 3. Nalmefene as‐needed has been shown to reduce the total amount of alcohol consumption and number of heavy drinking days (HDDs) and to improve liver function and clinical status in two published 6 month studies in patients with alcohol dependence and in one published 12 month study also in patients with alcohol dependence 4, 5, 6, 7. The proposed mechanism of action of nalmefene is to reduce the reinforcing effects of alcohol, helping the patient to reduce drinking.
The clinical PK of nalmefene have been reported in a couple of study specific papers, mainly using non‐compartmental analysis (NCA) 8, 9, 10, 11. However, no population PK analysis or meta‐analysis of nalmefene have been reported. Nalmefene is a high‐clearance drug with a relatively large volume of distribution.
In terms of pharmacodynamics (PD), the occupancy of the μ‐opioid receptor after administration of nalmefene has been investigated in a PET study using the radio ligand [11C]‐carfentanil 10, 12.
The primary objective of this paper is to describe the clinical PK of nalmefene in healthy subjects by means of a population PK analysis (non‐linear mixed effect modelling) and to assess the impact of subject specific covariates on the PK parameters. The secondary objective is to apply this model to relate the exposure of nalmefene (PK) to the μ‐opioid receptor occupancy (PD) by means of simulations.
Throughout this paper the doses of nalmefene are given as the salt nalmefene hydrochloride. Twenty mg nalmefene hydrochloride corresponds to 18.06 mg nalmefene.
Methods
Study designs and subject characteristics
Data from nine phase 1 studies in healthy subjects with extensive blood sampling were pooled (Table 1) and used for the population PK model building. These studies were conducted/reported from 1983 to 2010. Routes of administrations were intravenous (i.v), per oral solution and per oral tablets. Per oral administrations were performed both in connection with food intake and under fasting conditions. The dose ranges in the pooled dataset were 0.5–24 mg (i.v. single administration), 20–64 mg (oral single administration) and 20–80 mg (oral repeated administrations once daily for 7 days in the 20 mg group and 40 mg for 2 days followed by 80 mg for 5 days in the 80 mg group). The data set consisted of 243 healthy subjects, who contributed 4136 plasma concentrations. A summary of the characteristics for the subjects included in the pooled dataset is given in Table 2. The distribution of the formulations was i.v. (86 subjects), oral tablet (157 subjects) and oral solution (10 subjects). The distribution of food intake status was fasted (243 subjects) and fed (16 subjects).
Table 1.
Summary of included studies in the datasets for population pharmacokinetic analysis
| Study | Route of administration | Dose (mg) | n | Bioanalytical method |
|---|---|---|---|---|
| Analysis dataset studies | ||||
| A * | Oral solution + oral tablets | 50 as two SD | 6 | RIA |
| B | Oral tablets | 20 SD and 20 MD for 7 days | 12 | LCMS/MS |
| C | i.v. | 2, 6, 12, 24 as SD | 16 | RIA |
| D † | i.v. + oral solution | 2 i.v. 32 and 64 oral as 1 i.v SD. and 1 oral SD | 4 | RIA |
| E | i.v. | 0.5, 1, 2 as SD | 18 | RIA |
| F ‡ | Oral tablets | 20 as two SD | 16 | LCMS/MS |
| H | i.v. | 0.5, 1, 2 as SD | 36 | RIA |
| I | i.v. | 2 as SD | 12 | RIA |
| K | Oral tablets | Either 20 MD for 7 days or 40 MD for 2 days followed by 80 MD for 5 days | 123 | LCMS/MS |
| Validation dataset studies | ||||
| L | Oral solution | 20 SD | 6 | LCMS/MS |
| M | Oral tablets | 20 SD | 8 | LCMS/MS |
| N | Oral tablets | 20 or 40 MD for 5 days | 28 | LCMS/MS |
| O | Oral tablets | 20 SD | 43 | LCMS/MS |
Subjects were given nalmefene in fasting state except in study F.
Two single doses of nalmefene given in a crossover design of oral solution and oral tablets.
Two single doses of nalmefene given in a crossover design of i.v. administration and oral solution.
Two single doses of nalmefene given in a crossover design in fasted and fed condition.
i.v., intravenous; LCMS, liquid chromatography‐mass spectrometry; MD, multiple dose once daily; MS, mass spectrometry; RIA, radioimmunoassay; SD, single dose.
Table 2.
Summary of subject characteristics for the datasets for model building and external validation
| Mean | SD | Minimum | Median | Maximum | |
|---|---|---|---|---|---|
| Analysis dataset (n = 243) | |||||
| Age (years) | 35.2 | 16.9 | 18 | 28 | 80 |
| Weight (kg) | 72.29 | 11.44 | 51 | 71.4 | 109.6 |
| Height (cm) | 173.2 | 8.4 | 146 | 173 | 193 |
| BMI (kg m−2) | 24.01 | 2.69 | 19 | 23.9 | 34.8 |
| LBM (kg) | 55.65 | 8.46 | 37.3 | 56.3 | 74.2 |
| Men/women | 72%/28%: | ||||
| External validation dataset (n = 85) | |||||
| Age (years) | 38.74 | 9.6 | 20 | 39 | 69 |
| Weight (kg) | 72.37 | 10.96 | 48.9 | 71.2 | 106.6 |
| Height (cm) | 173.99 | 9.34 | 155 | 173 | 200 |
| BMI (kg m−2) | 23.65 | 2.52 | 18.7 | 23.9 | 29.4 |
| LBM (kg) | 54.93 | 9.04 | 38.5 | 54.69 | 80.9 |
| Men/women | 53%/47% | ||||
In addition to the pooled dataset used for model building, an external validation dataset was created. Data in this validation dataset were pooled from four different studies, three single dose and one repeated dose (5 days) study. These studies were conducted and reported in 2010 and 2011 and not included in the population PK model building. Route of administration was oral solution and oral tablets. The dose ranges in the external validation dataset were 20 mg (oral single administration) and 20–40 mg (oral repeated administrations). The external validation dataset consisted of 85 healthy subjects, who contributed 1522 plasma concentrations. A summary of the characteristics for the subjects included in the external validation dataset is given in Table 2. The distribution of the formulations was oral tablet (79 subjects) and oral solution (six subjects).
Ten of the 13 studies were performed according to the Helsinki Declaration. For the three oldest studies, no information is available. However, the studies were all approved by local Review Boards and included written consent to participate as specified in 21 CRF, Parts 50 and 56, respectively.
Drug analysis
Two different bioanalytical methods were used among the studies: radioimmunoassay (RIA) and liquid chromatography‐tandem mass spectrometry (LCMS/MS). During the early development of nalmefene, a specific radioimmunoassay (RIA) was developed to quantify nalmefene in human plasma 13. Assay specificity was achieved by extracting nalmefene from plasma at pH 9 into ether prior to RIA. The specificity of the RIA was established by demonstrating agreement with a less sensitive and more time‐consuming HPLC/ECD procedure which were used earlier. Three different RIA bioanalytical methods were used with a lover limit of quantification (LLOQ) in the range from 0.0625 to 2 ng ml−1 and a linear range from 0.0625 to 100 ng ml−1. In the most recent clinical studies, nalmefene and several of its metabolites have been simultaneously quantified in human plasma using liquid chromatography‐mass spectrometry (LC‐MS) techniques. All the bioanalytical methods using LC‐MS or LC‐MS/MS were validated in accordance with the United States Food and Drug Administration (FDA) guidance on bioanalytical method validation, and these methods all fulfilled the requirements for method accuracy and precision stated in the FDA guidance 14. Six different LC‐MS or LC‐MS/MS methods were used with a LLOQ in the range from 0.1 to 1 ng ml−1 and a linear range from 0.1 to 100 ng ml−1. 37.9% of the plasma concentrations were analyzed by RIA and 62.1% by LCMS/MS.
PK model development
Modelling/simulation strategy
A base structural population PK model was first developed after which the final model was established by adding the covariates to the base model one by one and tested by nonmem ® to determine if they were indeed statistically significant. The final model was evaluated and validated both by the model building data set and by an external data set. A PK/PD (occupancy) model was developed from one of the studies in the model building data set, and the established PK/PD model was used for simulation of concentration–time profiles of nalmefene and corresponding μ‐opioid receptor occupancies.
Structural and error models selection
Nalmefene concentration–time data were analyzed by non‐linear mixed effects modelling with the nonmem ® software to develop a base structural population PK model. The base model was identified by comparing different structural PK models. The initial structural model was a two compartment model consisting of central and peripheral compartments. Three compartment models were also tested. One compartment models were not examined, as plasma concentration–time curves showed an apparent multi‐exponential decline. A lag time in absorption after oral administration was not significant when tested. All processes were assumed to be first order. Inter‐individual variability (IIV) on the PK parameters was assumed a log‐normal distribution and described as
where Pj is the parameter P for the jth subject, θ is the estimate of the population mean, and nj is the deviation from the population mean for the jth subject under the assumption that η ~ N(0,ω2). Residual error was initially modelled as proportional. Since data after both i.v. and oral administration of nalmefene were included in the population PK analysis, the absorption was modelled as first order, whereas for the i.v. data, the population absorption rate constant (k a) and absolute bioavailability (F1) were fixed to 0.001 and 1, respectively.
Covariate analysis
A scatter plot correlation matrix was developed to examine the dependency among covariates. The distribution of covariates across subjects was examined. For covariates that were continuous in nature, scatter plots of PK parameter.estimates against covariates overlaid with a LOESS smoother were used to help identify functional relationships. For covariates that were categorical in nature, box and whisker plots of PK parameters for each of the groups were used to identify differences between groups.
Covariates that were continuous in nature were entered into the model in a median centered manner and tested in a linear manner or a power function normalized with the median value. The following covariates were tested in the population PK model building: age, weight, height, body mass index (BMI), lean body mass (LBM), gender, bioanalytical method and food intake. LBM were calculated according to:
An additional additive error model was evaluated for different bioanalytical methods. The additive and proportional error models were then compared with the proportional error model.
Covariates were added to the base model one by one and tested by nonmem ® to determine if they were indeed statistically significant. Covariates were incorporated into the base model or rejected based on the selection criteria previously described with a P value for statistical significance of 0.01. Covariates that demonstrated significant population PK model improvement were considered for the next iteration of covariate model development. The covariate model demonstrating the greatest improvement in the population PK model was incorporated into the base population PK model and remaining candidate covariates re‐evaluated incrementally. This process was repeated until none of the remaining candidate covariates provided significant improvement to the population PK model. To determine if all the covariates included in the fully parameterized population PK model continued to provide significant influence on the population model, the covariates included in the full model were sequentially removed and the resulting reduced model evaluated to determine if there was significant model degradation (stepwise elimination). The significance of the covariate was tested using the nested model criteria at a more stringent P value of 0.005 to avoid false positives.
Secondary parameters
The individual elimination half‐lives (t ½) of nalmefene for all subjects in the population PK dataset were estimated through empirical Bayesian estimates (EBE) of the final model parameters.
Model evaluation and validation
Diagnostic plots were used to assess the goodness‐of‐fit. In addition, the standard errors of the estimated parameter and the confidence intervals for the inter‐individual variabilities and the residual errors were checked.
The stability of the parameter estimates in the final model was compared with the stability of the parameter estimates in the base model using the condition number. The condition number is computed as the square root of the ratio of the largest eigenvalue to the smallest eigenvalue of the covariance matrix of the estimates. Condition numbers less than 1000 indicate acceptable stability of the parameter estimates 15. The standard error of the final model parameter values were estimated using the non‐parametric bootstrap approach. Two hundred bootstrap datasets were generated by repeated random sampling with replacement from the nonmem ® input data file, and the final nonmem ® model was fitted to the bootstrap datasets. The mean and 95% confidence intervals for the population PK parameters were derived and compared with the estimates for the original dataset. The percentile bootstrap 95% confidence interval was estimated by ranking the parameters from the bootstrap runs.
The shrinkage for the individually estimated concentrations (epsilon shrinkage) and parameters (eta shrinkage) was calculated 16. Although no formal criterion exists, a level of 0.2 was used to conclude ‘no shrinkage’.
External validation
The predictive performance of the final PK was also evaluated using the external validation dataset. Model‐ and simulation‐based diagnostics were performed by using normalized prediction distribution errors (NPDE) 17 and visual predictive checks (VPC). The NPDEs were produced by running the final PK model using the external validation dataset with all parameters, except the residual error, as fixed. The NPDEs are expected to follow a N (0, 1) distribution. The following plots were created: frequency histogram and qq‐plot of the NPDEs, NPDE vs. TIME and NPDE vs. population predicted concentration.
For the VPCs, 1000 simulations with 20 mg as a single dose and 1000 simulations with 20 mg as multiple dose for 5 days were performed based on the final PK model and mimicking the dosing length and PK sampling time points as for the study contributing with the external validation dataset. The median and 90% prediction intervals (5% and 95% percentiles) of simulated concentrations were plotted together with the observed concentrations in the external validation dataset and their median and 90% prediction intervals. The number of observed concentrations ‘outside’ the simulated 90% prediction intervals was counted. Different plots were created for day 1 and day 5 of dosing.
PK/PD (occupancy) modelling
Based on the last decade of research, the μ‐, δ‐ and κ‐antagonist [11C]‐diprenorphine, the μ‐agonist [11C]‐carfentanil and μ‐and κ‐antagonist [18F]‐fluoro‐cyclofoxy were the most widely applied tracers 18. Study B (Table 1) was an imaging study investigating central μ‐opioid receptor occupancy using positron emission tomography (PET) with the ligand [11C]‐carfentanil. As nalmefene in in vitro studies displayed the highest affinity to the μ opioid receptor and [11C]‐carfentanil has a pronounced selectivity for this receptor, [11C]‐carfentanil was chosen as the ligand. Eight healthy subjects were first administered a single dose of 20 mg nalmefene, followed by repeated dosing (20 mg orally, once daily over 7 days) after a drug‐free wash‐out period of at least 1 week and no more than 1 month. PET measurements and extensive blood sampling for PK analyses were performed after both dosings (i.e. single and repeated). The PK and occupancy results of the study have been published elsewhere 10, but without any PK/PD modelling. In order to simulate the μ‐opioid receptor occupancy in a large population, the relationship between the plasma concentrations of nalmefene and the occupancy for the eight subjects in study B was modelled using non‐linear mixed effect analysis using an Emax model on the form
where Occmax is the maximum μ‐opioid receptor occupancy, C PET the plasma concentration of nalmefene at the time of the PET measurement and EC 50 the plasma concentration of nalmefene necessary to give 50% occupancy. The impact of subject specific covariates was not tested due to the low number of subjects.
Model evaluation was based on goodness‐of‐fit plots together with assessment of standard errors of the estimated parameters. Random effects (IIV) on Occmax and EC 50 were assumed to follow a log‐normal distribution.
Simulations
Five thousand plasma concentration–time profiles of nalmefene and corresponding μ‐opioid receptor occupancies after a single dose of 20 mg nalmefene were simulated for the target population with a uniform age distribution (18–80 years) and a LBM distribution equal to the one found in the dataset used for population PK analysis (~N(56,72)). Half of the subjects were administered the tablet in connection with food and the other half in fasting conditions. Since no correlation between age and LBM could be found in the population PK analysis dataset (r = 0.13), no such correlation was included in the simulations.
Software
The population PK analysis was performed with the software nonmem ®, version VI, Level 1.0 (ICON Development Solutions, Ellicott City, Maryland, USA). Optimization was achieved using the FOCE method with interaction. The PK/PD analysis and simulations used version 7 of nonmem ®. Plots were produced with S‐PLUS®, version 6.2 (Insightful Corp., Seattle, Washington, USA).
Results
Population PK analysis
Visual inspection did not identify any outliers (Figure 1).
Figure 1.
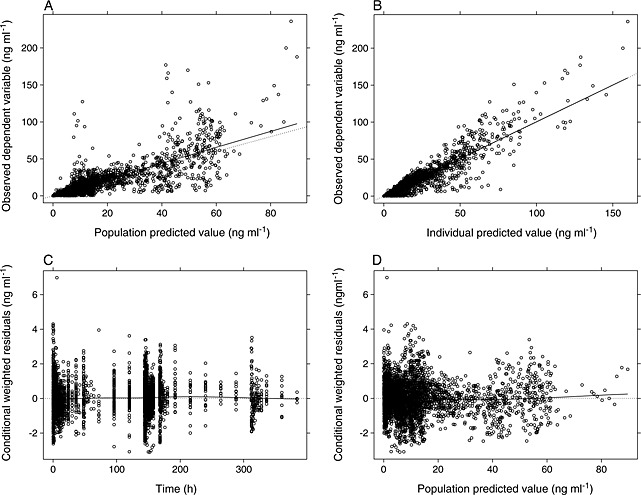
Goodness‐of‐fit plots for the final population pharmacokinetic model for nalmefene
A two compartment model best described the nalmefene PK data and was chosen as the structural model. The model was parameterized in terms of absorption rate constant (k a), clearance (CL), central volume of distribution (V2), inter‐compartmental clearance (Q), peripheral volume of distribution (V3) and bioavailability (F1). Inter‐individual variability was modelled as exponential terms on CL, V2, Q, V3, k a and F1. Following selection of the two compartment model as the structural model, food effect was tested on k a and F1 in the oral studies. Food effect on F1 produced a significant reduction (−67.689) in OFV compared with the reference model. An additional additive error model was also tested to describe residual variability for the different bioanalytical methods employed across all studies. An additional additive error model produced a significant reduction (−25.387) in OFV compared with the reference model. Therefore, food effect on F1 and an additional additive error model for the RIA method were included in the base model in order to obtain a better fit of the model to the data, and a significant reduction (−93.983) in OFV was observed compared with the reference. No significant correlation between CL and V2 was observed in the base model (Corr(V, CL) = 0.20).
The covariate modelling step revealed lean body mass (LBM) on CL (power model), formulation on k a and age on V2 (linear model) as significant covariate–parameter relationships. Therefore, the final model was a two compartment model with no lag time, inter‐individual variabilities expressed as exponential terms on CL, V2, Q, V3, k a and F1, food effect on F1, residual error modelled as proportional and an additional additive error model for the RIA bioanalytical method, with the following significant covariates: LBM (CL), formulation (k a) and age (V2).
Parameter values for the final model are given in Table 3 while goodness‐of‐fit plots are shown in Figure 1. The model for CL was as follows:
Table 3.
Parameter values for the final population pharmacokinetic model for nalmefene
| Model parameters | Parameter estimate |
|---|---|
| Clearance (CL) | |
| Estimate (l h−1) | 60.4 |
| RSE † (%) | 2.30 |
| IIV ‡ (%) | 18.7 |
| LBM on CL (power) | |
| Estimate | 0.626 |
| RSE † (%) | 19.3 |
| Volume of distribution, central compartment (V2) | |
| Estimate (l) | 266 |
| RSE † (%) | 13.9 |
| IIV ‡ (%) | 66.6 |
| Age on V2 (linear) | |
| Estimate (l) | −2.11 |
| RSE † (%) | 43.5 |
| Inter‐compartmental clearance (Q) | |
| Estimate (l h−1) | 109 |
| RSE † (%) | 5.38 |
| IIV ‡ (%) | 44.5 |
| Volume of distribution, peripheral compartment (V3) | |
| Estimate (l) | 537 |
| RSE † (%) | 5.05 |
| IIV ‡ (%) | 45.8 |
| Absorption rate constant (ka) for oral tablet | |
| Estimate (1 h−1) | 0.751 |
| RSE † (%) | 13.2 |
| IIV ‡ (%) | 69.9 |
| Absorption rate constant (ka) for oral solution | |
| Estimate (1 h−1) | 1.40 |
| RSE † (%) | 24.0 |
| Absolute bioavailability (F1) | |
| Estimate | 0.406 |
| RSE † (%) | 3.37 |
| IIV ‡ (%) | 20.1 |
| Food on F1 (fractional change) | |
| Estimate | 0.294 |
| RSE † (%) | 8.64 |
| Proportional residual error | |
| Estimate | 0.094 |
| CV (%) | 30.7 |
| RSE † (%) | 1.48 |
| Additive residual error for RIA | |
| Estimate | 0.00065 |
| SD | 0.0255 |
| RSE † (%) | 26.0 |
| Maximum effect (Emax) | |
| Estimate (%) | 99.4 |
| RSE † (%) | 2.4 |
| Concentration producing 50% of maximum effect (EC50) | |
| Estimate (ng ml−1) | 0.338 |
| RSE † (%) | 8.8 |
Relative standard error (RSE) was calculated as standard error/estimate* 100.
Inter‐individual variability.
The model for CL was as follows: CL = 60.4 * (LBM/56.28)0.626.
The model for V2 was as follows: V2 = 266–2.11 * (Age‐28).
The food effect on F1 was as follows: F1 = 0.406 * (1 + 0.294 *FED), where FED =0 under fasting condition, FED =1 under fed condition.
The error model for the bioanalytical method RIA was as follows: Y = IPRED + F * EPS(1) + EPS (2)
The error model for the bioanalytical method LC‐MS/MS was as follows: Y = IPRED + F * EPS(1).
The model for V2 was as follows:
The value for k a was 0.75 h−1 for the oral tablet and 1.40 h−1 for the oral solution and the value for V3 was 537 l. The typical subject in the population studied with a LBM of 56.28 kg and age of 28 years was estimated to have a central volume of distribution of 266 l and clearance of 60.4 l h−1.
The mean elimination half‐life (t ½) was 12.7 h with a standard deviation of 4.2 h. Absolute oral bioavailability was estimated as 41%. Food intake increased the oral bioavailability by approximately 30%, resulting in an oral bioavailability of 53%. Without food intake the oral values of CL/F, V2/F and V3/F are estimated to 149 l h−1, 655 l and 1323 l, respectively, whereas the values after food intake were estimated to 115 l h−1, 506 l and 1022 l, respectively.
Model performance and validation
The final model for nalmefene was considered stable with a condition number of 8.04. The base model had a condition number of 6.10, similar to the final model. In addition, results from the bootstrap analysis of 200 datasets (Table 4) indicated a stable final model with a difference of maximum ±3% between the parameter value from the final analysis and the bootstrap value. The eta shrinkages were 22% (CL), 19% (V2), 32% (Q), 22% (V3), 43% (k a) and 40% (F1), while epsilon shrinkage was 30%.
Table 4.
Bootstrap values and bootstrap confidence interval for the final pharmacokinetic model for nalmefene
| Model parameters | Bootstrap estimate |
|---|---|
| Clearance (CL) | |
| Estimate (l h−1) | 60.4 |
| RSE † (%) | 2.44 |
| 95% confidence interval | 57.4, 63.0 |
| LBM on CL (power) | |
| Estimate | 0.616 |
| RSE † (%) | 18.5 |
| 95% confidence interval | 0.375, 0.827 |
| Volume of distribution, central compartment (V2) | |
| Estimate (l) | 266 |
| RSE † (%) | 6.39 |
| 95% confidence interval | 228, 293 |
| Age on V2 (linear) | |
| Estimate (l) | −2.05 |
| RSE † (%) | 30.5 |
| 95% confidence interval | −3.23, −0.785 |
| Inter‐compartmental clearance (Q) | |
| Estimate (l h−1) | 108 |
| RSE † (%) | 6.55 |
| 95% confidence interval | 95.0, 124 |
| Volume of distribution, peripheral compartment (V3) | |
| Estimate (l) | 539 |
| RSE † (%) | 4.94 |
| 95% confidence interval | 490, 589 |
| Absorption rate constant (ka) for oral tablet | |
| Estimate (1 h−1) | 0.754 |
| RSE † (%) | 7.96 |
| 95% confidence interval | 0.638, 0.873 |
| Absorption rate constant (ka) for oral solution | |
| Estimate (1 h−1) | 1.36 |
| RSE † (%) | 22.3 |
| 95% confidence interval | 0.672, 1.92 |
| Absolute bioavailability (F1) | |
| Estimate | 0.406 |
| RSE † (%) | 3.94 |
| 95% confidence interval | 0.373, 0.438 |
| Food on F1 (fractional change) | |
| Estimate | 0.302 |
| RSE † (%) | 22. 4 |
| 95% confidence interval | 0.175, 0.424 |
Relative standard error (RSE) was calculated as standard error/estimate* 100.
The external validation dataset and simulations based on the final model were used to generate VPC plots (Figure 2). The number of observed concentrations outside the simulated 90% confidence bound was 7.8% for day 1 and 9.7% for day 5. The NPDEs were estimated and plotted (Figure 3) and followed very closely a N(0,1) distribution with no signs of dependency to time or estimated plasma concentrations.
Figure 2.
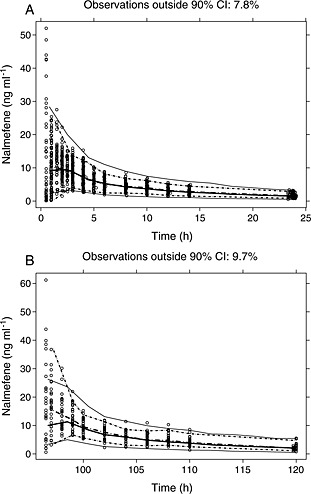
Visual predictive check (VPC) plots for the validation dataset for A) day 1 and B) day 5 of dosing. Full lines show median and percentiles (2.5% and 97.5%) of simulated data, while dotted lines show median and percentiles (2.5% and 97.5%) of observed data
Figure 3.
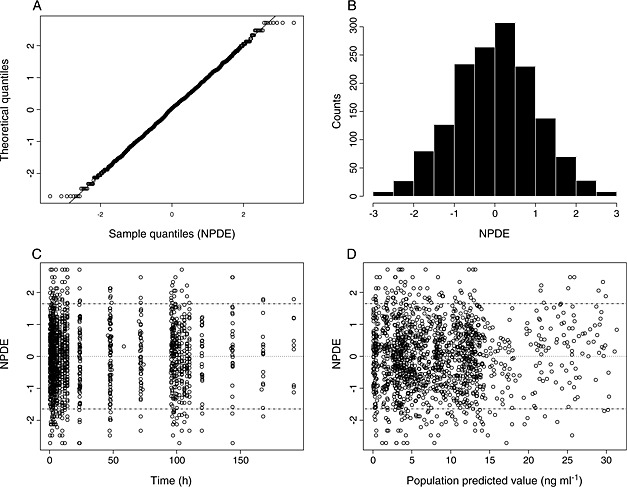
Normalized prediction distribution error (NPDE) plots for the validation dataset
PK/PD (occupancy) modelling
The relationship between the plasma concentration of nalmefene and the μ‐opioid receptor occupancy could be reliably described by an Emax model (see Figure 4). The parameter values for Emax and EC 50 were 99.4% and 0.338 ng ml−1, respectively, with relative standard errors of 2.4% and 8.8%, respectively.
Figure 4.
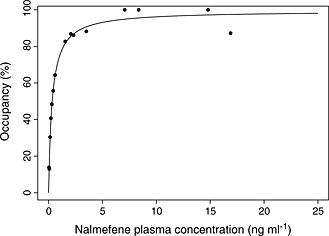
Plasma concentration of nalmefene vs. observed and fitted μ‐opioid receptor occupancy
Simulations
The developed PK/PD occupancy model was used to simulate plasma concentrations and corresponding μ‐opioid receptor occupancies during the first 24 h after a single dose of 20 mg nalmefene (Figure 5), both after fasting and fed conditions in connection with dosing. As can be seen in Figure 5, for up to 22–24 h after dosing the 95% confidence bound of simulated occupancies was within or above the 60–90% occupancy region.
Figure 5.
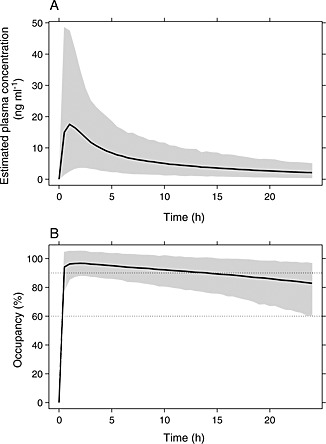
A) Simulated plasma concentration curves of nalmefene and B) simulated duration of μ‐opioid receptor occupancy after a single dose of 20 mg in fed state with 95% confidence bounds
Discussion
A reliable, robust and predictive population PK model for nalmefene has been developed. Non‐parametric bootstrap evaluation and external visual predictive check indicated that the final full model describes the data well and is predictive.
The PK of nalmefene were well described by a two compartment model without lag time and with first order absorption and elimination. Food effect and an additive error model for the RIA method were included in the base model in order to obtain a better fit of the model to the data.
Mean population clearance (CL) and volume of distribution (V) values in healthy subjects were in good agreement with those observed previously in healthy subjects. Mean population CL was 60.4 l h−1, similar to reported values for healthy subjects after i.v. administration of 2–24 mg nalmefene of 0.96 l h−1 kg−1 (67 l h−1 for a subject of 70 kg) 19 and 60–65 l h−1 8. Mean population volume of distribution, the sum of the central and peripheral compartment, was 803 l. Reported values for V ss after non‐compartmental analysis were 8.2 l kg−1 (574 l for a subject of 70 kg) 19 and 481–515 l 8. The inter‐individual variabilities were estimated to be 18.7% for CL and 66.6% for V2. Absolute oral bioavailability was estimated as 41% without food intake (with food about 53%). Earlier the absolute bioavailability has been reported to be in the range of 40–50% 8.
LBM and age were covariates found to statistically significantly affect CL and V2, respectively. Addition of LBM resulted in an overall decrease of 1.3% in inter‐individual variability of CL. Addition of age as a covariate helped reduce the inter‐individual variability in V2 by about 5.6%. A typical subject in the population studied with a LBM of 56.28 kg and age of 28 years old was estimated to have a V2 of 266 l and CL of 60.4 l h−1. As LBM increased from 37.3 to 74.2 kg in the population studied, the typical value of CL increased from 46.7 to 71.8 l h−1. V2 of nalmefene was found to decrease with age in a linear fashion. Over the age range of 18 to 80 years observed in the population studied, the typical value of V2 decreased from 287 to 156 l.
The mean elimination half‐life was estimated to 12.7 h, very similar to the 12.5 h given in the Summary of Product Characteristics for Selincro 18 mg film‐coated tablets 3. The elimination half‐life has previously been estimated to be around 10 h, both after i.v. and oral administration 8, 9, 11. Nalmefene is extensively metabolized by the liver and excreted predominantly in the urine (about 5% of an intravenous dose is excreted in the urine as intact nalmefene 8), but none of the metabolites are considered to be a major contributor to the pharmacological effect of nalmefene 3. Due to the relatively short half‐life, accumulation of nalmefene after multiple administrations once daily will be low (average accumulation index at steady‐state estimated to 1.37). After oral administration, nalmefene was rapidly absorbed with a mean peak concentration estimated around 1 to 1.5 h (see Figure 5). This is in accordance with previous reported values of peak concentrations occurring approximately 1 to 2.5 h after oral administration 9, 10.
The population PK model developed for nalmefene has used data from healthy subjects only since PK data from adults with alcohol dependence were not available. Neither are there any PK data of nalmefene in adults with alcohol dependence reported in the literature. One factor of importance for the PK that potentially could differ between the adult healthy population and adults with alcohol dependence is the liver function. The effect of liver disease on the disposition of nalmefene was studied in 12 patients with liver disease (four mild, five moderate and three severe) and 12 age‐, weight‐ and gender‐matched control subjects after a single i.v. administration of 2 mg 11. The patients with liver disease had on average 33% lower clearance, 32% longer half‐life and 39% higher AUC(0,∞) compared with the control subjects. There was also a significant relationship between disease severity (Pugh score) and clearance of nalmefene, with lower clearance for the more severe patients. Oral administration of a single dose of nalmefene 20 mg to patients with mild or moderate hepatic impairment increased exposure relative to that in healthy subjects 3 In patients with mild hepatic impairment, exposure increased 1.5 times and oral clearance decreased by approximately 35%. In patients with moderate hepatic impairment, exposure increased 2.9 times for AUC and 1.7 times for C max, while oral clearance decreased by approximately 60%. No clinically relevant changes were seen in t max or elimination half‐life for any of the groups. PK data after oral administration of nalmefene to patients with severe hepatic impairment are not available. No dose adjustment is recommended for patients with mild or moderate hepatic impairment, whereas nalmefene is contraindicated in patients with severe hepatic impairment.
Most of the patients targeted for nalmefene are expected to be without moderate to severe hepatic impairment. We therefore consider the healthy population as representative for the target population regarding the pharmacokinetics of nalmefene.
It has been concluded that ‘a persistent μ‐opioid receptor blockade can be induced by a single nalmefene tablet and supports the rational of administering nalmefene when needed before alcohol drinking’ 10. In the summary of product characteristics for nalmefene for the treatment of reduction of alcohol consumption it is stated that nalmefene ‘is to be taken as‐needed: on each day the patient perceives a risk of drinking alcohol, one tablet should be taken, preferably 1–2 h prior to the anticipated time of drinking’ 3. Antagonists for G‐protein coupled receptors (GPCR) require approximately 60–90% target occupancy to trigger therapeutic effects in patients 20. It has been shown that a single administration of 20 mg nalmefene resulted in μ‐opioid receptor occupancies of around 100% and 85% at 3 and 26 h after dosing, respectively 10. As can be seen in Figure 5, our simulations of the μ‐opioid receptor occupancy show that for 95% of the general population the occupancy remains within or above 60–90% for up to 22–24 h after a single administration of 20 mg nalmefene. That is, regardless if the tablet is taken at 10.00 h in the morning, at 16.00 h in the afternoon or at 22.00 h in the evening, the effect (μ‐opioid receptor occupancy) will still remain to at least the morning of the day after.
In summary a robust PK/PD model for nalmefene was developed. After a single 20 mg dose of nalmefene the μ‐opioid receptor occupancy was simulated to be within or above 60–90% for up to 22–24 h.
Competing Interests
All authors have completed the Unified Competing Interest form at http://www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare this analysis was funded by H. Lundbeck A/S. All authors are present or former employees of either H. Lundbeck A/S or of an organization contracted by H. Lundbeck A/S for this analysis and there are no other relationships or activities that could appear to have influenced the submitted work.
Supporting information
Table S1
Summary of forward step 1 of subject covariates
Table S2
Summary of forward step 2 of subject covariates
Table S3
Summary of forward step 3 of subject covariates
Table S4
Summary of forward section of subject covariates
Table S5
Summary of backward step of subject covariates
Supporting info item
Kyhl, L.‐E. B. , Li, S. , Faerch, K. U. , Soegaard, B. , Larsen, F. , and Areberg, J. (2016) Population pharmacokinetics of nalmefene in healthy subjects and its relation to μ‐opioid receptor occupancy. Br J Clin Pharmacol, 81: 290–300. doi: 10.1111/bcp.12805.
References
- 1. Bart G, Schluger JH, Borg L, Ho A, Bidlack JM, Kreek MJ. Nalmefene induced elevation in serum prolactin in normal human volunteers: partial kappa opioid agonist activity? Neuropsychopharmacology 2005; 30: 2254–62. [DOI] [PubMed] [Google Scholar]
- 2. Guerrero M, Urbano M, Brown SJ, Cayanan C, Ferguson J, Cameron M, Devi LA, Roberts E, Rosen H. Optimization and characterization of an opioid kappa receptor (OPRK1) antagonist. Probe Reports from the NIH Molecular Libraries Program [Internet]. Bethesda (MD): National Center for Biotechnology Information (US); 2010. 2013 Apr 15 [updated 2013 Nov 14]. [PubMed]
- 3. EMA . Selincro: EPAR prodution information. Annex I – summary of product characteristics. Available at www.ema.europa.eu [homepage on internet] (last accessed 2 October 2015).
- 4. Gual A, He Y, Torup L, van den Brick W, Mann K. A randomised, double‐blind, placebo‐controlled, efficacy study of nalmefene, as‐needed use, in patients with alcohol dependence. Eur Neuropsychopharmacol 2013; 23: 1432–42. [DOI] [PubMed] [Google Scholar]
- 5. Mann K, Bladström A, Torup L, Gual A, van den Brink W. Extending the treatment options in alcohol dependence: a randomized controlled study of as‐needed nalmefene. Biol Psychiatry 2013; 73: 706–13. [DOI] [PubMed] [Google Scholar]
- 6. van den Brink W, Aubin HJ, Bladström A, Torup L, Gual A, Mann K. Efficacy of as‐needed nalmefene in alcohol‐dependent patients with at least a high drinking risk level: results from a subgroup analysis of two randomized controlled 6‐month studies. Alcohol Alcohol 2013; 48: 570–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. van der Brink W, Sørensen P, Torup L, Mann K, Gual A. Long‐term efficacy, tolerability and safety of nalmefene as‐needed in patients with alcohol dependence: a 1‐year, randomised controlled study. J Psychopharmacol 2014; 28: 733–44. [DOI] [PubMed] [Google Scholar]
- 8. Dixon R, Howes J, Gentile J, Hsu HB, Hsiao J, Garg D, Weidler D, Meyer M, Tuttle R. Nalmefene: intravenous safety and kinetics of a new opioid antagonist. Clin Pharmacol Ther 1986; 39: 49–53. [DOI] [PubMed] [Google Scholar]
- 9. Dixon R, Gentile J, Hsu HB, Hsiao J, Howes J, Garg D, Weider D. Nalmefene: safety and kinetics after single and multiple oral doses of a new opiod antagonist. J Clin Pharmacol 1987; 27: 233–9. [DOI] [PubMed] [Google Scholar]
- 10. Ingman K, Hagelberg N, Aalto S, Någren K, Juhakoski A, Karhuvaara S, Kallio A, Oikonen V, Hietala J, Scheinin H. Prolonged central mu‐opioid receptor occupancy after single and repeated nalmefene dosing. Neuropsychopharmacology 2005; 30: 2245–53. [DOI] [PubMed] [Google Scholar]
- 11. Frye RF, Matzke GR, Schade R, Dixon R, Rabinovitz M. Effects of liver disease on the disposition of the opioid antagonist nalmefene. Clin Pharmacol Ther 1997; 61: 15–23. [DOI] [PubMed] [Google Scholar]
- 12. Kim S, Wagner HN Jr, Villemagne VL, Kao PF, Dannals RF, Ravert HT, Joh T, Dixon RB, Civelek AC. Longer occupancy of opioid receptors by nalmefene compared to naloxone as measured in vivo by a dual‐detector system. J Nucl Med 1997; 38: 1726–31. [PubMed] [Google Scholar]
- 13. Dixon R, Hsiao J, Taaffe W, Hahn E, Tuttle R. Nalmefene: radioimmunoassay for a new opioid antagonist. J Pharm Sci 1984; 73: 1645–6. [DOI] [PubMed] [Google Scholar]
- 14. United States Food and Drug Administration (FDA) . Guidance for Industry: Bioanalytical Method Validation. May 2001.
- 15. Bonate PL. Pharmacokinetic‐Pharmacodynamic Modeling and Simulation. USA: Springer, 2006. [Google Scholar]
- 16. Karlsson MO, Savic RM. Diagnosing model diagnostics. Clin Pharmacol Ther 2007; 82: 17–20. [DOI] [PubMed] [Google Scholar]
- 17. Mentré F, Escolano S. Prediction discrepancies for the evaluation of nonlinear mixed‐effects models. J Pharmacokinet Pharmacodyn 2006; 33: 345–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Henriksen G, Willoch F. Imaging of opioid receptors in the central nervous system. Brain 2008; 131: 1171–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Matzke GR, Frye RF, Alexander ACM, Reynolds R, Dixon R, Johnston J, Rault RM. The effect of renal insufficiency and hemodialysis on the pharmacokinetics of nalmefene. J Clin Pharmacol 1996; 36: 144–51. [DOI] [PubMed] [Google Scholar]
- 20. Grimwood S, Hartig PR. Target site occupancy: emerging generalizations from clinical and preclinical studies. Pharmacol Ther 2009; 122: 281–301. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1
Summary of forward step 1 of subject covariates
Table S2
Summary of forward step 2 of subject covariates
Table S3
Summary of forward step 3 of subject covariates
Table S4
Summary of forward section of subject covariates
Table S5
Summary of backward step of subject covariates
Supporting info item