

Abstract
Aim
Pridopidine, a new oral drug for treatment of patients with motor symptoms associated with Huntington's Disease (HD) is currently under development. In steady‐state conditions, pridopidine elimination is mediated primarily through renal excretion. This study evaluated single dose and steady‐state pharmacokinetics (PK) of a daily dose of pridopidine in subjects with mild and moderate renal impairment and matched healthy subjects.
Methods
Subjects with mild renal impairment (n = 12), moderate impairment (n = 12), or their matched healthy controls (n = 25) participated in this study. Subjects received a single dose of pridopidine (45 mg) on day 1 and a multiple dose cycle of 45 mg once daily on days 5–18. Blood and urine samples were collected on days 1 and 18 for PK analysis.
Results
Mild renal impairment did not affect the PK of pridopidine whilst an increase in exposure was seen in subjects with moderate renal impairment. Subjects with moderate impairment showed reduced plasma clearance (by 44%) and had 68% higher AUC (90% CI 1.22, 2.30) and 26% higher C max (90% CI 1.02, 1.56) values than those with normal renal function at steady‐state. Pridopidine was safe and well tolerated in healthy subjects and in subjects with mild and moderate renal impairment.
Conclusions
Mild renal impairment has no impact on exposure to pridopidine while moderately impaired renal function resulted in higher pridopidine concentrations.
Keywords: Huntington's disease, pharmacokinetics, pridopidine, renal impairment
What is already Known About this Subject
Pridopidine is a drug under clinical development for the treatment of motor symptoms in Huntington's disease.
Renal elimination is the most important elimination pathway for pridopidine at steady‐state. However the impact of renal impairment on its clinical pharmacokinetics have not been characterized.
This phase 1 study was performed to characterize the pharmacokinetics pridopidine in subjects with normal and mild or moderately impaired renal function, to enable appropriate dosing recommendations for these special patient populations.
What this Study Adds
Mild renal impairment does not affect pridopidine pharmacokinetics.
Moderate renal impairment resulted in lower total clearance and higher exposure to pridopidine.
Introduction
Huntington's disease (HD) is a rare neurodegenerative disorder of the central nervous system (CNS) with an autosomal dominant model of inheritance and a prevalence of 1/100 000 to 5/100 000 1, 2. HD is caused by mutant huntingtin gene (mHTT) protein aggregation which leads to progressive destruction of a wide variety of proteins, causing metabolic dysfunction and eventual cell death mainly in the striatum. The neurodegeneration in the striatal pathways leads to the characteristic symptoms such as involuntary choreatic movements and behavioural and psychiatric abnormalities, which aggravate with progression of the disease. Patients are on average between 30 and 50 years old at onset of the first symptoms 1, 2.
Pridopidine (4‐[3‐(methylsulfonyl)phenyl]‐1‐propylpidperidine, formerly known as ACR16), is a drug under clinical development for the treatment of motor symptoms in HD. It belongs to a new class of pharmaceutical agents, the dopidines which are considered to have dopamine stabilizing properties. Pridopidine presumably restores balance in the cortico‐striatal pathway that controls motor function disrupted in HD 3. In clinical trials conducted in HD patients, pridopidine at a dose of 45 mg twice daily non‐significantly improved the modified motor score (mMS) and significantly improved the total motor score (TMS) of the Unified Huntington's Disease Rating Scale (UHDRS). In both studies, pridopidine was considered safe and well tolerated with a benign adverse event (AE) profile 4, 5, 6.
Pridopidine is absorbed relatively rapidly after oral administration with a t max between 0.5 to 4 h 7, and a dose proportional increase in exposure for dose levels from 10 mg twice daily to 90 mg twice daily at steady‐state 8. Pridopidine's elimination half‐life after multiple doses is about 10–14 h 7 and it is approximately 30% bound to plasma proteins (data on file). Pridopidine is metabolized by the polymorphic enzyme CYP2D6 by N‐depropylation to TV‐45065 (previously named ACR30, a major metabolite assumed to be pharmacologically inactive based on in vitro and preclinical testing). The contribution of the CYP2D6 pathway to pridopidine's elimination after a single dose depends on the patient's 2D6 genotype (whether they are an extensive or poor CYP2D6 metabolizer) but over time pridopidine inhibits its own CYP2D6‐driven metabolism 9. Thus at steady‐state, for all patients and regardless of the CYP2D6 genotype, renal excretion becomes the most important route of elimination of pridopidine and TV‐45065 7.
This trial was conducted to assess the influence of renal function on the pharmacokinetics of pridopidine, by evaluating its pharmacokinetics in subjects with impaired vs. normal renal function, in order to provide appropriate dosing recommendation in the patient setting.
Methods
The study (EudraCT‐number 2009–013 158–34) was conducted at two sites in Germany in accordance with the Declaration of Helsinki 10 and the ethical principles of Good Clinical Practice 11. The competent authority, the leading ethics committee of the Medical Association of Baden‐Württemberg, Germany and the local ethics committee of the Medical Association of Schleswig‐Holstein, Germany approved the trial. All participants provided written informed consent before the start of the trial.
The study population consisted of male and female subjects, aged between 18 and 75 years, weighing at least 50 kg, and having a body mass index between 18 and 34 kg m–2. Renal function was assessed at screening based on creatinine clearance (CLcr) as an estimation of the glomerular filtration rate (eGFR), and calculated using the Modified Diet in Renal Disease (MDRD) equation 12. Renal function was classified as normal when CLcr was ≥90 ml min–1, as mildly impaired when CLcr was ≥60 to 89 ml min–1 and moderately impaired when CLcr ≥30 to 59 ml min–1. For each subject with renal impairment, a healthy subject of the same gender was included, matched by age (±5 years) and body weight (±10 kg). There were two groups of subjects with renal impairment (mild and moderately impaired) and two matching groups of healthy volunteers with normal renal function.
Study subjects were screened to exclude CYP2D6 poor metabolizers. The following alleles were analyzed: *2, *3, *4, *5, *6, *7, *8, *9, *10, *35, *41, *MxN and genotype was assigned according to four categories ((ultrarapid metabolizers (UM), extensive metabolizers (EM), intermediate metabolizer (IM) and poor metabolizers (PM)). EMs have at least one functional CYP2D6 allele, IMs have two CYP2D6 alleles with reduced function or one reduced and one non‐functional allele and PMs have two non‐functional alleles. Other key exclusion criteria included severe impairment of renal function or requiring dialysis, smoking more than five cigarettes day−1 or unable to abstain from smoking during confinement period, pregnant or nursing females, prolonged QTc interval at screening (defined as a QTc interval of >450 ms for males or >470 ms for females) or history of QTc prolongation or other ECG abnormalities including syncopes of unclear genesis in medical history, personal or familial history of seizures or significant cardiovascular events within 6 months. Subjects with renal impairment who required treatment for renal impairment or other chronic disease on a stable treatment plan for at least 2 months prior to dosing could be enrolled unless the treatment was excluded (inducers or inhibitors of CYP2D6, tricyclic antidepressants, or class I anti‐arrhythmic drugs).
Trial design
The trial design followed the recommendations of the FDA 13, 14 and EMA 15 guidances related to the investigation of pharmacokinetics in patients with impaired renal function. This study was open label and no randomization was required. As pridopidine exhibits non‐linear and time‐dependent pharmacokinetics, the study included assessments not only after single but also after multiple dose administration. Pridopidine exposure in subjects with mild renal impairment was evaluated before proceeding with the dosing of the moderate impairment group. No formal sample size estimation was performed. However a sample size of 12 subjects per group was considered to be adequate to meet the study goals and is commonly used in this kind of study.
Treatments were administered orally as 45 mg pridopidine capsules. The treatment period consisted of a single dose cycle (pridopidine administration on day 1) and a multiple dose cycle with 14 days of once daily administration (days 5 to 18). On days 1 and 18 (pharmacokinetic profile days), subjects were fasted for at least 10 h prior to dosing.
Blood and urine sampling
Blood samples for quantification of pridopidine and TV‐45065 were collected before and 0.5, 1, 1.5, 2, 2.5, 3, 4, 8, 12, 24, 32, 48 and 72 h after administration on day 1 and day 18. Blood samples for determination of trough concentrations were collected during the multiple dose period to assess steady‐state achievement. Urine samples for quantification of pridopidine and TV‐45065 concentrations were collected over the intervals 0–4, 4–8, 8–12, 12–24, 24–48, and 48–72 h after dosing on day 1 and day 18.
Concentrations of pridopidine and its metabolite, TV‐45065, were determined in potassium EDTA plasma and urine by validated methods using liquid chromatography and tandem mass spectrometric detection (LC‐MS/MS). For both plasma and urine, the samples were made basic by the addition of sodium hydroxide and the analytes were isolated using liquid–liquid extraction into diethyl ether. The analytes were resolved isocratically with a mobile phase composed of water : acetonitrile : methanol : concentrated ammonia, 1000 : 250 : 500 : 3, v/v with 0.5 g of ammonium acetate at a flow rate of 0.6 ml min on a Waters X‐Bridge Phenyl column (100 × 3 mm; 3.5 μm particle size). Quantification was performed using peak area ratios with respect to internal standards (d7‐pridopidine for the parent and a structural analogue, ACR481, for the metabolite). The analytes were monitored using electrospray ionization in the positive ion mode on a Sciex API 500. The mass transitions monitored were m/z 282.3 → 240.2 for pridopidine, m/z 240.3 → 161.0 for d7‐pridopidine, m/z 289.1 → 210.0 for TV‐45065, and m/z 258.3 → 179.3 for ACR481.
For plasma, weighted (1/concentration2) linear regression analysis was used to determine concentrations over a range of 1.849 to 397.194 ng ml–1, and 1.872 to 376.514 ng ml–1 for pridopidine and TV‐45065, respectively. During validation, the bias for pridopidine in plasma ranged from −3.30 to −0.39%, with a coefficient of variation (CV) of ≤6.65%. The bias of TV‐45065 ranged from −8.64 to 3.21%, with a CV of ≤13.61%. The urine method utilized weighted (1/concentration2) linear regression analysis to determine concentrations over a range of 0.062 to 25.465 μg ml–1 for pridopidine and 0.060 to 24.672 μg ml–1 for TV‐45065. During validation, the bias for pridopidine in urine ranged from 0.99 to 2.31%, with a CV of ≤3.51%. The bias of TV‐45065 ranged from 1.41 to 4.29%, with a CV of ≤6.32%. Methods were demonstrated to be selective for pridopidine and TV‐45065.
Calculation of pharmacokinetic parameters
Pharmacokinetic and statistical analyses were performed for pridopidine and TV‐45065 plasma and urine data from subjects with sufficient data for reliable analysis.
Pharmacokinetic parameters were calculated for both single and multiple doses from the individual concentrations by non‐compartmental procedures using SAS® version 9.2 (SAS Institute, Cry, NC, USA). Plasma concentrations below LLOQ observed between administration and first quantifiable concentration were set to zero for calculation of pharmacokinetic parameters. All other concentrations below LLOQ were excluded from calculations. Concentrations in urine below the LLOQ were set to zero for calculation of the amount excreted.
The plasma PK parameters calculated were the observed maximum plasma concentration (C max), the time from dosing to C max (t max), the area under the plasma concentration vs. time curve (AUC) extrapolated to infinity (for single dose, AUC(0,∞)) and over one dosing interval (AUC(0,τss) for multiple dose), which were calculated using the log‐linear trapezoidal formula. The apparent terminal half‐life (t 1/2) was calculated after single dose and at steady‐state from the slope of the terminal phase, apparent clearance (CL/F) was calculated as the quotient of AUC(0,∞) after single dose or AUC(0,τss) at steady‐state and dose and apparent volume of distribution (V z/F) was calculated as the quotient of CL/F and terminal slope. The accumulation index (Rac) was calculated as the quotient of AUC(0,τ) at steady‐state and AUC(0,24 h) after a single dose.
The calculated urine PK parameters included renal clearance (CLR) calculated as the ratio of the amount excreted over AUC (up to 72 h for single dose, and over a dosing interval for steady‐state assessments, respectively), and the fraction of dose excreted in urine (f e), calculated as amount excreted/dose in a similar way. Adjustment by the molecular weights (TV‐45065/pridopidine, i.e. 239.34/281.42) was applied when relevant.
Statistical analyses
An analysis of variance (anova) on trough plasma concentrations on days 15 to 18 was performed to confirm steady‐state. The effect of renal impairment on pridopidine pharmacokinetics was examined using anova of AUC(0,∞), C max (single dose) and AUC(0,τss), and C max,ss (steady‐state) (MIXED procedure, SAS) following logarithmic transformation. Each of the renally impaired groups was compared with the matching normal renal function group with a statistical significance level of alpha =0.05. Point estimates and corresponding 90% confidence interval (CI) for geometric means of ‘impaired/healthy’ ratios of pharmacokinetic parameters were separately calculated for comparison of each renal impairment group with the corresponding healthy matching subject group.
The relationships between CLcr and pharmacokinetic parameters C max and AUC were also investigated graphically by plotting the individual values of creatinine clearance against the log (ln) transformed pharmacokinetic variable.
Safety evaluation
Safety measurements of all subjects receiving at least one dose of study medication were based on incidence and severity of treatment‐emergent adverse events (TEAEs), vital signs, ECG, physical examination and safety laboratory tests at the follow‐up safety visit. Adverse events observed from first administration of pridopidine to the safety follow‐up visit (5 to 7 days after last administration) were considered for safety assessment. During outpatient periods, subjects were to report any adverse events to the investigator by phone.
Results
Forty‐nine subjects were enrolled and 48 completed the study and were part of the PK analysis (12 subjects on each group). One healthy subject matched to one mildly impaired subject was withdrawn due to an adverse event (renal colic due to urethral calculus) after the first dose of pridopidine and a replacement was recruited. All study subjects were extensive metabolizers except for four subjects who were intermediate metabolizers (one with normal renal function, two with mild renal impairment and one with moderate renal impairment). Demographic characteristics of the study population are presented in Table 1. On average, subjects with moderate renal impairment (and hence their matched controls) were older than those with mild renal impairment (and controls), with mean age of 61.5 and 58.2 years vs. 48.7 and 48.2 years.
Table 1.
Key demographic characteristics of study subjects included in the PK analysis
| Characteristic | Statistics | Healthy control of mild renal impairment | Mild renal impairment | Healthy control of moderate renal impairment | Moderate renal impairment |
|---|---|---|---|---|---|
| Number of subjects | n | 12 | 12 | 12 | 12 |
| Male/Female | n/n | 8/4 | 8/4 | 5/7 | 5/7 |
| Age (years) | Mean (range) | 48.2 (38–54) | 48.7 (41–53) | 58.2 (39–70) | 61.5 (36–74) |
| Median | 47 | 46.0 | 58.5 | 59.0 | |
| Body mass index (kg m–2) | Mean (range) | 25.1 (20.3–29.2) | 25.5 (20–31) | 26.9 (23.5–31.2) | 27.5 (24–32) |
| Median | 25.4 | 25.8 | 25.9 | 27.9 | |
| CLcr (MDRD equation) (ml min–11.73 m–2) | Mean (range) | 105.8 (94–122) | 77.7 (63–89) | 102.1 (90–119) | 46.3 (34–59) |
| Median | 108 | 76.5 | 97.5 | 48.0 |
CLcr: estimated glomerular filtration rate, calculated at screening using the modified diet in renal disease equation (175 x serum creatinine‐1.154 (mg dl–1) x age‐0.203 × 1.212 if patient is Black and × 0.742 if patient is female).
Pharmacokinetic results
After a single dose of 45 mg pridopidine, mean plasma concentration–time profiles of pridopidine and its metabolite TV‐45065 were comparable between subjects with mild renal impairment and their matched healthy subjects. In subjects with moderate renal impairment, the mean plasma concentration profiles of pridopidine were higher than in matched healthy subjects (Figure 1A). Steady‐state was confirmed after 14 days of dosing as point estimates of ratios day 15 : day 16, day 16 : day 17 and day 17 : day 18 were near 1.00 and all 90% confidence limits were within the equivalence range of 0.80–1.25. Plasma concentrations of pridopidine were higher at steady‐state compared with single dose in all groups, indicating accumulation (Figure 1B). The plasma concentration of pridopidine in subjects with moderate renal impairment was higher than in matched controls, the same trend as seen after a single dose. Plasma concentrations of TV‐45065 showed the same trend as the parent, and were slightly lower at steady‐state compared with single dose (Figure 2 panels A and B).
Figure 1.
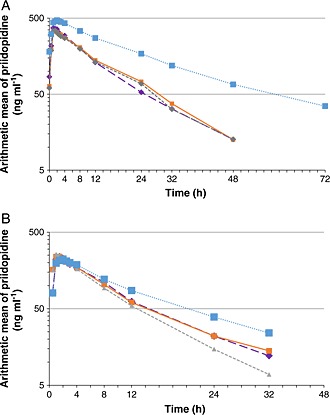
Semilogarithmic plots of the plasma concentrations of pridopidine after single and multiple dose administrations to healthy and renally‐impaired subjects.  normal matched to mild renal impairment,
normal matched to mild renal impairment,  normal matched to moderate renal impairment,
normal matched to moderate renal impairment,  mild renal impairment,
mild renal impairment,  moderate renal impairment
moderate renal impairment
Figure 2.
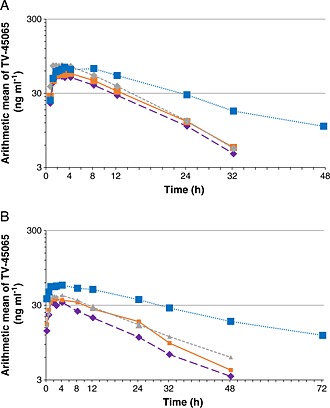
Semilogarithmic plots of the plasma concentrations of the metabolite TV45065 after single and multiple dose administrations of pridopidine to healthy and renally‐impaired subjects.  normal matched to mild renal impairment,
normal matched to mild renal impairment,  normal matched to moderate renal impairment,
normal matched to moderate renal impairment,  mild renal impairment,
mild renal impairment,  moderate renal impairment
moderate renal impairment
Single dose administration of pridopidine resulted in comparable PK parameters for the mild renally impaired subjects and their matched healthy controls (Table 2). In subjects with moderate renal impairment, exposure to pridopidine was higher than in the healthy control group, with about 50% higher AUC(0,∞) (3087 vs. 2059 ng ml–1 h) but similar C max (Table 3). Total CL/F of the subjects with moderate renal impairment was 25% lower than that observed in the control group (312 ml min–1 vs. 410 ml min–1), while CLR was almost the same (72.8 ml min–1 and 75 ml min–1, respectively). The lower CL/F resulted in a prolongation of the half‐life in subjects with moderate renal impairment compared with the healthy control group. The metabolite displayed similar findings to the parent compound.
Table 2.
Pharmacokinetic parameters of pridopidine and its metabolite TV‐45065 after single and multiple daily doses of 45 mg pridopidine to subjects with mild renal impairment and normal renal function
| Single dose | Steady‐state | ||||||
|---|---|---|---|---|---|---|---|
| Pridopidine | |||||||
| Parameter | Mild renal impairment (n = 12) | Healthy control of mild renal impairment (n = 12) | Mild renal impairment (n = 12) | Healthy control of mild renal impairment (n = 12) | |||
| AUC (ng ml–1 h) * | 2365 ± 1120 | 2365 ± 1264 | 3999 ± 1390 | 3851 ± 1557 | |||
| Cmax (ng ml–1) | 276 ± 66.6 | 263 ± 71.1 | 358 ± 87.3 | 426 ± 128 | |||
| tmax (h) † | 1.0 (0.5–3.0) | 1.2 (1.0–2.0) | 1.5 (0.5–3.0) | 1.3 (0.5–4.0) | |||
| t1/2 (h) | 6.9 ± 3.3 | 7.6 ± 3.6 | 9.1 ± 2.2 | 9.6 ± 3.0 | |||
| Vz/F (l) | 201 ± 42.3 | 219 ± 55.8 | 173 ± 45.55 | 156 ± 30.5 | |||
| CL/F (ml min–1) | 397 ± 185 | 388 ± 155 | 211 ± 75.6 | 232 ± 116 | |||
| CLR (ml min–1) | 101 ± 30.0 | 106 ± 32.1 | 99 ± 32.8 | 116 ± 31.1 | |||
| fe (%) | 29.8 ± 16.7 | 30.6 ± 14.7 | 49 ± 12.5 | 55 ± 16.3 | |||
| Rac | – | – | 2.04 ± 0.5 | 1.90 ± 0.3 | |||
| TV‐45065 | |||||||
| AUC (ng ml–1 h) * | 932 ± 305 | 804 ± 279 | 651 ± 255 | 514 ± 243 | |||
| Cmax (ng ml–1) | 59.4 ± 29.9 | 54.5 ± 25.9 | 38.6 ± 15.1 | 37.5 ± 25.2 | |||
| tmax (h) † | 2.5 (1.0–8.0) | 2.5 (1.5–8.0) | 2.5 (1.0–24.0) | 1.75 (1.0–8.0) | |||
| t1/2 (h) | 10.2 ± 4.7 | 8.5 ± 3.0 | 16.1 ± 7.0 | 13.6 ± 5.7 | |||
| CLR (ml min–1) | 455.5 ± 102.9 | 496.3 ± 112.4 | 444.8 ± 108.9 | 517.8 ± 130.8 | |||
| fe (%) | 42.7 ± 13.7 | 40.7 ± 10.9 | 30.7 ± 11.6 | 28.4 ± 11.6 | |||
AUC(0,∞) for single dose, AUC(0,τ) for multiple dose;
t max presented as median (range), all other parameters are presented as arithmetic mean ± SD.
Table 3.
Pharmacokinetic parameters of pridopidine and its metabolite TV‐45065 after single and multiple daily doses of 45 mg pridopidine to subjects with moderate renal impairment and normal renal function
| Single dose | Steady‐state | ||||||
|---|---|---|---|---|---|---|---|
| Pridopidine | |||||||
| Parameter | Moderate renal impairment (n = 12) | Healthy control of moderate renal impairment (n = 12) | Moderate renal impairment (n = 12) | Healthy control of moderate renal impairment (n = 12) | |||
| AUC (ng ml–1 h) * | 3087 ± 1667 | 2059 ± 711 | 7007 ± 4555 | 3853 ± 1459 | |||
| Cmax (ng ml–1) | 252 ± 53.8 | 278 ± 36.5 | 497 ± 215 | 375 ± 82.9 | |||
| tmax (h) † | 1.5 (0.5–4.0) | 1.0 (0.5–1.5) | 2.0 (0.5–2.5) | 1.5 (0.5–4.0) | |||
| t1/2 (h) | 9.6 ± 4.0 | 5.7 ± 1.3 | 13.6 ± 5.1 | 9.0 ± 2.4 | |||
| Vz/F (l) | 214 ± 48.47 | 192 ± 46.58 | 161 ± 39.0 | 161 ± 35.7 | |||
| CL/F (ml min–1) | 312 ± 148 | 410 ± 150 | 137 ± 56.5 | 218 ± 70.6 | |||
| CLR (ml min–1) | 72.8 ± 32.6 | 75.0 ± 21.3 | 67 ± 27.3 | 90 ± 32.4 | |||
| fe (%) | 24.6 ± 10.9 | 19.3 ± 6.4 | 50 ± 9.8 | 42 ± 11.9 | |||
| Rac | – | – | 2.8 ± 0.8 | 2.0 ± 0.3 | |||
| TV‐45065 | |||||||
| AUC (ng ml–1 h) * | 1728 ± 734 | 1066 ± 218 | 1116 ± 535 | 679 ± 259 | |||
| Cmax (ng ml–1) | 81.3 ± 45.0 | 79.8 ± 25.6 | 60.3 ± 23.8 | 43.0 ± 16.3 | |||
| tmax (h) † | 3.5 (1.5–12.0) | 2.25 (1.0–4.0) | 3.5 (1.0–12.0) | 3.25 (1.5–8.0) | |||
| t1/2 (h) | 12.5 ± 5.0 | 7.4 ± 1.8 | 21.8 ± 9.0 | 14.6 ± 4.9 | |||
| CLR (ml min–1) | 306.4 ± 160.7 | 391.2 ± 96.2 | 274.4 ± 127.1 | 415.8 ± 128.5 | |||
| fe (%) | 47.5 ± 10.0 | 43.9 ± 7.6 | 29.5 ± 7.8 | 28.9 ± 7.2 | |||
AUC(0,∞) for single dose, AUC(0,τ) for multiple dose;
t max presented as median (range), all other parameters are presented as arithmetic mean ± SD.
Multiple dose administration of pridopidine resulted in increased exposure to pridopidine in all study subjects compared with single dose due to drug accumulation (Tables 2 and 3). The extent of the changes seen was similar between the mild renal impairment group and its matched control group. Total clearance (CL/F ss) decreased by about 45% after multiple dosing compared with single dose) with a corresponding increase of 65% in AUC (from 2365 ng ml–1 h to about 3900 ng ml–1 h in both groups). C max increased with multiple dosing by about 30% in mild renal impairment and 60% in the matched control group. Renal clearance did not differ markedly between single and multiple dose administration.
The decrease in total clearance in subjects with moderate renal impairment with multiple dosing compared with single dose was only slightly higher than any other groups (−56%, from 312 to 137 ml min–1 Table 3). As a result of the change in clearance, and with drug accumulation over time, the difference in exposure between the moderate renally impaired group and its control was accentuated at steady‐state and AUC(0,τss) and C max,ss were approximately 80% and 30% higher in the impaired group as compared with the matched healthy group, respectively. One subject in the moderately impaired group had an AUC(0,τss) and C max,ss which were twice as high as the next highest subject. As the subject was the eldest amongst the moderate renally impaired subjects (subject was a 74‐year‐old female), this increased exposure could be also related to age (but not entirely).
Renal clearance after single and multiple dosing were approximately similar in subjects with moderate impairment and the matched control subjects. anova did not show significant differences between healthy subjects and subjects with mild renal impairment regarding pridopidine AUC and C max, with point estimates ranging from 0.86 to 1.06 at both dosing administrations (Table 4). In subjects with moderate renal impairment, both mean AUC and C max were higher compared with the means of the matched healthy group after multiple dosing, with point estimates of 1.26 and 1.68 for C max,ss and AUC(0,τss), respectively (statistically significant only for AUC(0,τss). Regression analysis of the ln‐transformed PK parameters also indicated a significant effect of CLcr on AUC(0,τss) (slope = −0.008, P < 0.001) but not on C max,ss (Figure 3).
Table 4.
Analysis of variance on the effect of renal impairment on pharmacokinetic parameters of pridopidine
| Point estimate | 90% confidence interval | P value | ||
|---|---|---|---|---|
| Mild/normal | C max after single dose | 1.06 | 0.85, 1.31 | 0.653 |
| AUC(0,∞) after single dose | 0.99 | 0.69, 1.43 | 0.977 | |
| C max,ss at steady‐state | 0.86 | 0.69, 1.06 | 0.222 | |
| AUC(0,τss) at steady‐state | 1.06 | 0.82, 1.37 | 0.671 | |
| Moderate/normal | C max after single dose | 0.90 | 0.80, 1.01 | 0.121 |
| AUC(0,∞) after single dose | 1.39 | 1.01, 1.92 | 0.092 | |
| C max,ss at steady‐state | 1.26 | 1.02, 1.56 | 0.071 | |
| AUC(0,τss) at steady‐state | 1.68 | 1.22, 2.30 | 0.014 | |
Figure 3.
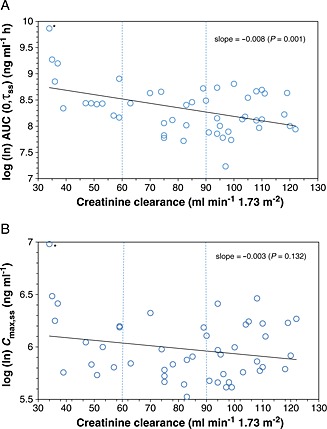
Log (ln)‐transformed regression analysis of AUC (A) and C max (B) at steady‐state. * This subject was the oldest subject in the trial (74‐year‐old female) and was only partially age‐matched to a 67‐year‐old subject
In all groups after multiple dosing, the fraction of the parent compound excreted in urine as unchanged pridopidine rose compared with a single dose, ranging from about 30% to 50% of the administered dose, and matched by a respective decrease in the fraction of excreted metabolite (Tables 2 and 3). The total fraction excreted (pridopidine and metabolite) remained relatively constant, between 70% and 80% of the administered dose.
Safety
During the study, 56 AEs were observed in 25 of the 49 subjects (51%). The most common adverse events in this study were headache (14 events in nine subjects), dizziness (six events in five subjects), nasopharyngitis (six events in six subjects) and fatigue (five events in five subjects). Events occurring more than once are presented in Table 5; sixty percent (60%), 67% and 17% of subjects with normal, mild, and moderately impaired renal function reported adverse events, respectively. All adverse events were resolved at the end of the study. No deaths and no serious adverse events were observed. One subject (normal renal function, male, 49 years) was withdrawn from the trial 2 days after the first dose due to an adverse event (renal colic due to urethral calculus) not related to the study medication.
Table 5.
Adverse events occurring more than once during the study
| Normal renal function (n = 25) | Mild renal impairment (n = 12) | Moderate renal impairment (n = 12) | Total (n = 49) | |
|---|---|---|---|---|
| Adverse events | F, n, % | F, n, % | F, n, % | F, n, % |
| Total | 29, 15, 60.0 | 25, 8, 66.7 | 2, 2, 16.7 | 56, 25, 51.0 |
| Headache | 8, 4, 16% | 6, 5, 42% | ‐ | 14, 9, 18% |
| Dizziness | ‐ | 5, 4, 33% | 1,1, 8% | 6, 5, 10% |
| Diarrhoea | 2, 2, 8% | ‐ | ‐ | 2, 2, 4% |
| Dry mouth | 2, 2, 8% | ‐ | ‐ | 2, 2, 4% |
| Fatigue | 4, 4, 16% | 1, 1, 8% | ‐ | 5, 5, 10% |
| Feeling hot | ‐ | 2, 2, 17% | ‐ | 2, 2, 4% |
| Nasopharyngitis | 5, 5, 20% | 1, 1, 8% | ‐ | 6, 6, 12% |
| Tinnitus | ‐ | 2, 1,8% | ‐ | 2,1, 2% |
| Vertigo | ‐ | 2, 2, 17% | ‐ | 2, 2, 4% |
| Sleep disorder | 1,1,4% | 1,1,8% | ‐ | 2,2,4% |
F, Number of events; n, number of subjects; %, the percent of treated subjects
Safety laboratory parameters, vital signs and ECG parameters showed no influence of pridopidine administration. None of the subjects with normal renal function showed clinically relevant changes in safety laboratory parameters. In the subjects with renal impairment, clinically relevant abnormal values in safety laboratory values were due to the underlying renal impairment. Overall, once daily treatment with 45 mg pridopidine over 14 days was safe and well tolerated in healthy subjects and in subjects with mild and moderate renal impairment.
Discussion
After oral absorption, pridopidine is eliminated partly by urinary excretion and partly by hepatic metabolism, primarily via the CYP2D6 pathway. Pridopidine is metabolized in extensive metabolizers by CYP2D6 to TV‐45065, while the contribution from other enzymatic pathways does not seem to be significant (data on file). Individuals who are poor CYP2D6 metabolizers depend on renal excretion as their main elimination pathway. It has been shown that during multiple dose administration, pridopidine can inhibit its own CYP2D6‐driven metabolism, meaning that upon repeated dosing, renal elimination becomes a more important elimination pathway than the CYP2D6‐driven metabolism 7. The present study assessed the influence of renal function on the pharmacokinetics of pridopidine and TV‐45065.
The pharmacokinetic parameters measured in the healthy controls in this group were consistent with those previously reported in extensive metabolizers, both after single and multiple dose 7, 16. Consistent with these studies, the apparent total clearance was about 50% lower at steady‐state than after single dose in all study subjects, while renal clearance was stable over time. In addition, exposure to the CYP2D6‐formed metabolite TV‐45065 was reduced in all groups following multiple doses. The decrease in total clearance can therefore be linked to the decrease in non‐renal clearance (i.e. inhibition rather than saturation of CYP2D6 metabolism) both in normal and renally impaired subjects. In agreement with this mechanism, the fraction of unchanged pridopidine excreted in urine increased over multiple dosing, together with a matching decrease of metabolite excretion, so that a total of about 70–80% of the dose was excreted in urine either as the parent or TV‐45065 after both single and multiple doses. Renal clearance of pridopidine for all subjects was close to the glomerular filtration rate (GFR), suggesting as previously reported that renal excretion of pridopidine occurs by passive filtration 7, 16.
In subjects with mild renal impairment (creatinine clearance ≥60 to 89 ml min–1), the pharmacokinetics of pridopidine was similar to that observed in the matched healthy subjects (creatinine clearance ≥90 ml min–1), indicating that mild renal impairment has no significant impact on pridopidine exposure, after either single or multiple dosing.
Subjects with moderate renal impairment (creatinine clearance ≥39 to 59 ml min–1) on the other hand exhibited lower total clearance than their matched healthy controls, which resulted in higher pridopidine exposure. A 74‐year‐old female subject with a CLcr of 34 ml min–1 exhibited a pridopidine AUC and C max that was almost double that of the next highest individual. However it is unknown whether her extreme exposure was due to the low CLcr or age. If the latter, then the true impact of moderate renal impairment could be lower than expected. Interestingly, the increase in plasma exposure was not fully reflected by differences in renal clearance in the respective groups (Table 3). A potential reason for this could be the difficulty related to the urine measurements as opposed to plasma (imprecision in measuring renal clearance, possible inhibition of other elimination paths by renal impairment‐related toxins, small number of subjects).
As expected, C max was less affected by renal impairment than AUC, as reflected both by the anova analysis and correlation analyses with eGFR. Moderate impairment can further be distinguished between Stage 3a (GFR 45–59 ml min–1) and Stage 3b (GFR 30–44 ml min–1). Although the impact of renal impairment was not analyzed for each moderate subgroup, Figure 3 shows that both subgroups were represented in the study (with five subjects falling into the 3a and seven subjects falling into the 3b category), and provides a relationship between the renal clearance rates and the C max and AUC parameters for the full evaluated GFR range.
Renal elimination of unchanged pridopidine is the main route of elimination in both CYP2D6 EMs and PMs at steady‐state. In PMs, renal clearance accounts for nearly the total clearance, while in EMs it accounts for about 70% of the total clearance, with the remaining CL/F mediated by the residual CYP2D6 activity not inhibited at steady‐state 7. This study excluded CYP2D6 PMs in order to reduce variability and eliminate confounding factors. Most subjects were EMs, and from review of the data the four enrolled IM subjects did not affect the conclusions. Also, the evaluation described in this work was conducted at 45 mg once daily, whilst clinical trials with pridopidine are being conducted with doses up to 112.5 mg twice daily 17. As the inhibition of CYP2D6 in EMs results in enzyme inactivation and a steady‐state pharmacokinetic profile comparable with those of the CYP2D6 PMs 7, the results of steady‐state pharmacokinetics obtained for CYP2D6 EMs in this study could also be cautiously extrapolated to the poor metabolizer population. The most conservative scenario for consideration when extrapolating these steady‐state results to higher daily doses of pridopidine and/or poor metabolizers would be a complete knock‐out of non‐CLR, whether due to non‐functional CYP2D6, greater auto‐inhibition at higher doses and/or accumulation at steady‐state. In such a case, CL/F could approximate CLR and dosing could be sought to be adjusted accordingly. Population pharmacokinetic analyses including data generated at higher doses and in individuals with different degrees of CYP2D6 activity will enable the prediction of exposure for specific populations. These analyses could also address the potential impact of age on pridopidine pharmacokinetics.
Finally, despite the increased exposure in subjects with moderately impaired renal function, no obvious relationship between renal function and number and nature of adverse events or other safety parameters was observed. In this study, pridopidine was well tolerated in healthy subjects as well as in those with renal impairment.
Competing Interests
All authors have completed the Unified Competing Interest form at www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare that Laura Rabinovich‐Guilatt (first/corresponding author) and Ofer Spiegelstein had support from Teva Pharmaceuticals for the submitted work and are employees of Teva Pharmaceuticals. Dr Karl Ernst Siegler, DrArmin Schultz and Dr Atef Halabi had support from CRS Clinical Research Services Mannheim GmbH. CRS Clinical Research was sponsored by Neurosearch to perform the study described in this paper. Asa Rembratt had support from Neurosearch for the submitted work. No other relationships or activities appear to have influenced the submitted work.
Contributors
Neurosearch A/S (author Asa Rembratt) sponsored the trial and participated in the interpretation of trial data. Trial conduct and data collection was performed by CRS Clinical Research Services Mannheim GmbH (authors Karl Ernst Siegler and Armin Schultz) and CRS Clinical Research Services Kiel GmbH (author Atef Halabi). Dr Armin Schultz and Dr Atef Halabi were clinical study investigators in the study. Sample analysis, data evaluation and manuscript writing were performed by CRS Clinical Research Services Mannheim GmbH. Teva Pharmaceuticals participated in data evaluation and manuscript writing (authors Laura Rabinovich‐Guilatt and Ofer Spiegelstein).
The authors thank Pippa Loupe PhD, of Teva Pharmaceuticals for assistance with manuscript development.
This trial was funded by Neurosearch A/S, Ballerup, Denmark. Teva Pharmaceuticals Industries acquired pridopidine in October 2012.
Rabinovich‐Guilatt, L. , Siegler, K. E. , Schultz, A. , Halabi, A. , Rembratt, A. , and Spiegelstein, O. (2016) The effect of mild and moderate renal impairment on the pharmacokinetics of pridopidine, a new drug for Huntington's disease. Br J Clin Pharmacol, 81: 246–255. doi: 10.1111/bcp.12792.
References
- 1. Mahant N, McCusker EA, Byth K, Graham S; Huntington Study Group . Huntington's disease: clinical correlates of disability and progression. Neurology 2003; 61: 1085–92. [DOI] [PubMed] [Google Scholar]
- 2. Roos RA. Huntington's disease: a clinical review. Orphanet J Rare Dis 2010; 5: 40. doi:10.1186/1750‐1172‐5‐40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Pettersson F, Sonesson C, Waters S, Waters N. In vivo pharmacology of the dopaminergic stabilizer pridopidine. Eur J Pharmacol 2010; 644: 88–95. [DOI] [PubMed] [Google Scholar]
- 4. Garcia de Yebenes J, Landwehrmeyer B, Squitieri F, Reilmann R, Rosser A, Barker R, Saft C, Magnet M, Sword A, Rembratt A, Tedroff J for the MermaiHD study investigators . Pridopidine for the treatment of motor function in patients with Huntington's disease (MermaiHD): a phase 3, randomised, double‐blind, placebo‐controlled trial. Lancet Neurol 2011; 10: 1049–57. [DOI] [PubMed] [Google Scholar]
- 5. Squitieri F, Landwehrmeyer B, Reilmann R, Rosser A, de Yebenes JG, Prang A, Ivkovic J, Bright J, Rembratt A. One‐year safety and tolerability profile of pridopidine in patients with Huntington disease. Neurology 2013; 80: 1086–94. doi:10.1212/WNL.0b013e3182886965. [DOI] [PubMed] [Google Scholar]
- 6. The Huntington Study Group HART Investigators . A randomized, double‐blind, placebo‐controlled trial of pridopidine in Huntington's disease. Mov Disord 2013. Sep; 28: 1407–15. [DOI] [PubMed] [Google Scholar]
- 7. Lindskov Krog P, Osterberg O, Gundorf Drewes P, Rembratt Å, Schultz A, Timmer W. Pharmacokinetic and tolerability profile of pridopidine in healthy‐volunteer poor and extensive CYP2D6 metabolizers, following single and multiple dosing. Eur J Drug Metab Pharmacokinet 2013; 38: 43–51. doi:10.1007/s13318-012-0100-2. [DOI] [PubMed] [Google Scholar]
- 8. Østerberg O, Sundgreen C, Ivkovic J, Muglia P, Prang A, Darpo B. A single center, randomized, placebo‐controlled, double‐blind study to evaluate the safety, tolerability, and pharmacokinetics of multiple‐ascending doses of pridopidine in healthy volunteers. Presented at The 7th European Huntington's Disease Network Plenary Meeting. Stockholm, Sweden 14–16 September, 2012.
- 9. Rabinovich‐Guilatt L, Spiegelstein O, Wickenberg A, Bassan M. A drug‐drug interaction study of pridopidine, a new drug for treatment of Huntington's disease, and metoprolol, a cytochrome P450 2D6 substrate. Presented at the World Congress on Huntington's Disease, Rio de Janeiro Brazil September 15–18 2013.
- 10. WORLD MEDICAL ASSOCIATION DECLARATION OF HELSINKI . Ethical Principles for Medical Research Involving Human Subjects. Available at http://www.wma.net/en/30publications/10policies/b3/17c.pdf (last accessed 27 October 2015).
- 11. Guidance for Industry E6 Good Clinical Practice: Consolidated Guidance . http://www.fda.gov/downloads/Drugs/Guidances/ucm073122.pdf (last accessed 27 October 2015).
- 12. Levey AS. Coresh J, Greene T, marsh J, Stevens LA, Kusek JW, Van lente F; chronic kidney disease epidemiology collaboration. Expressing the modification of diet in renal disease study equation for estimating glomerular filtration rate with standardized serum creatinine values. Clin Chem 2007; 53: 766–72. [DOI] [PubMed] [Google Scholar]
- 13. US Food and Drug Administration . Guidance for Industry: Pharmacokinetics in Patients with Impaired Renal Function ‐ Study Design, Data Analysis, and Impact on Dosing and Labeling. Rockville, MD. May 1998. [Google Scholar]
- 14. Guidance for Industry Pharmacokinetics in Patients with Impaired Renal Function Study Design, Data Analysis, and Impact on Dosing and Labeling (March 2010). Available at http://www.fda.gov/downloads/Drugs/Guidances/UCM204959.pdf (last accessed 27 October 2015).
- 15. European Medicines Agency . Note for Guidance on the Evaluation of the Pharmacokinetics of Medicinal Products in Patients with Impaired Renal Function (CHMP/EWP/225/02). London, UK 2004. Available at http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003123.pdf (last accessed 27 October 2015).
- 16. Helldén A, Panagiotidis G, Johansson P, Waters N, Waters S, Tedroff J, Bertilsson L. The dopaminergic stabilizer pridopidine is to a major extent N‐depropylated by CYP2D6 in humans. Eur J Clin Pharmacol 2012; 68: 1281–6. [DOI] [PubMed] [Google Scholar]
- 17. Landwehrmeyer GB, Reilmann R, Kieburtz K, Eyal E, Wickenberg A, Bassan M. Design of the dose‐range finding (DRF), randomized, double‐blind, placebo‐controlled study, evaluating the safety and efficacy of pridopidine for symptomatic treatment in patients with Huntington's disease [abstract]. Mov Disord 2014; 29: 571. [Google Scholar]