

Abstract
Although 60 years have passed since it became widely available on the therapeutic market, paracetamol dosage in patients with liver disease remains a controversial subject. Fulminant hepatic failure has been a well documented consequence of paracetamol overdose since its introduction, while short and long term use have both been associated with elevation of liver transaminases, a surrogate marker for acute liver injury. From these reports it has been assumed that paracetamol use should be restricted or the dosage reduced in patients with chronic liver disease. We review the factors that have been purported to increase risk of hepatocellular injury from paracetamol and the pharmacokinetic alterations in different pathologies of chronic liver disease which may affect this risk. We postulate that inadvertent under‐dosing may result in concentrations too low to enable efficacy. Specific research to improve the evidence base for prescribing paracetamol in patients with different aetiologies of chronic liver disease is needed.
Keywords: analgesia, cirrhosis, NAFLD, pharmacokinetics
Introduction
‘The oft‐cited warning that drugs known to produce hepatic injury should not be given to patients with liver disease has little foundation in fact’– Hy Zimmerman, 1999.
Paracetamol, (also known as acetaminophen, N‐acetyl‐p‐aminophenol or APAP) is a commonly used mild analgesic and antipyretic available in numerous prescription and over‐the‐counter formulations. It is considered a first line analgesic for many patients with chronic liver disease (CLD), primarily due to concern about the side effects of non‐steroidal anti‐inflammatory (NSAID) and opioid‐derived agents 2, 3. However its association with alanine transaminase (ALT) elevation, considered a surrogate marker for liver injury, has led to uncertainty about its safety margin in patients with pre‐existing liver disease 4.
While the use of liver function tests (LFTs) and other surrogate markers for clinical outcomes are often required, ALT in particular as a surrogate marker of liver injury is problematic as this enzyme fluctuates throughout the day in healthy individuals, in relation to exercise, concurrent intake of vitamins and some medications 5. It is also upregulated in other disease states such as diabetes and heart failure 5. Therefore minor transient elevations have not been strongly correlated with hepatotoxicity. This is in contrast to the very high transaminase concentrations seen in patients with drug induced liver injury (DILI). Case reports of DILI in people using ‘therapeutic’ doses of paracetamol are predominantly in patients who are heavy drinkers, fasting, malnourished, underweight or have concurrent febrile illness 6, 7, 8, 9, 10, 11, 12, 13, 14. We have not found any case reports of hepatotoxicity secondary to therapeutic doses of paracetamol in adults with pre‐existing CLD who did not have at least one of these risk factors. One large systematic review inclusive of over 40 000 patients found that the rate of increased serum transaminases and hepatotoxicity was higher in retrospective studies compared with prospective trials, suggesting that some retrospective cases may have been accidental overdoses 15.
While it is noteworthy that a double‐blind, two period crossover study of 20 patients with chronic stable liver disease (eight with cirrhosis) tolerated paracetamol at a dosage of 4 g day−1 for 13 days without adverse effects 16, there is a general lack of prospective long term studies examining its use in CLD. The limited evidence base in addition to the complex pharmacokinetics and pharmacodynamics within the heterogeneous CLD group has contributed to inconsistent paracetamol dosage recommendations of varying magnitude, frequency and daily maximum for acute and chronic pain. Reluctance to prescribe the standard dose of paracetamol may result in poorer efficacy and accelerate progression to alternative analgesics which have well‐documented side effects in CLD 2, 17.
For ethical reasons it is unlikely that any large randomized controlled trial (RCT) will be undertaken to understand paracetamol dose–response behaviour in liver disease, and therefore prescribing advice must be extrapolated from a combination of existing pharmacokinetic/pharmacodynamic data, simulated populations, surrogate and actual observed clinical outcomes. We therefore sought to review existing literature for evidence of pharmacokinetic alterations and paracetamol toxicity in different CLD aetiologies, to identify the limitations in the research and to make better‐informed recommendations on prescribing paracetamol in CLD.
The evolution of paracetamol in clinical care
Phenacetin and acetanilide, two aniline derivatives with analgesic and antipyretic properties, were first described by Harmon Morse in 1878 and Cahn & Hepp in 1886, respectively 18, 19. Both were much later found to be prodrugs for the active compound N‐acetyl‐p‐aminophenol 20, 21, 22, 23, 24. The drug was given the approved name ‘paracetamol’ in 1957 and has been included in the published British Pharmacopoeia since 1963 25.
In the early 1970s the US Food and Drug Administration (FDA) outlined a safe adult dosage of up to 1000 mg every 4−6 h, not to exceed 4000 mg day−1, for no longer than 10 days. The recommendation was based primarily on animal toxicology data, one bioavailability study (n = 15 healthy adult males) and four Industry sponsored placebo‐controlled post‐partum analgesia trials that investigated single dose efficacy over 4−6 h in 338 women (n = 112 paracetamol 1000 mg; n = 113 paracetamol 650 mg; n = 113 placebo) 26, 27, 28.
While most of the original data remains unpublished, the efficacy of this dosing regimen has subsequently been supported by clinical trials in a plethora of indications and extensive post‐marketing experience. Single dose trials and one large meta‐analysis inclusive of over 4000 patients have demonstrated that paracetamol dosed at 1000 mg provides superior analgesia compared with 650 mg 27, 29, 500 mg 30, 31, 32, 33 and placebo 27, 29, 34, but no less improvement than 2000 mg 35. Supra‐therapeutic doses of up to 8 g daily in healthy adults and 6 g daily in stroke patients have been shown to be relatively safe when used for up to 3 days 36, 37, although one case of chronic use at 6 g daily for 14 years has been associated with the later development of cirrhosis 38. Long term safety data indicated no adverse clinical or biochemical effects in 1039 individuals taking paracetamol 1950−4000 mg daily for up to 1 year 39 or 178 patients taking 2600 mg day−1 of paracetamol for 2 years in osteoarthritis trials 40.
Pharmacokinetics and metabolism
Paracetamol is almost completely metabolized by the liver. Pharmacology studies in healthy young men and women suggest that only a very small amount (2−9%) of paracetamol is excreted unchanged 36, 41. The remainder undergoes phase 1 sulphation predominately via SULT1A1 and SULT1A3/4 42, glucuronidation predominantly via UGT1A6 and UGT1A9 43, 44 and oxidation via the cytochrome P450 system (Figure 1). Glucuronide and sulphate conjugates are non‐toxic, inactive and readily excreted in the bile (minor) and urine (major) 42. Phase 1 oxidation via CYP2E1 and CYP2A6 results in the production of thiol and catechol metabolites, respectively 36. Isoenzymes CYP3A4 and CYP1A2 have been theorized to play a role in paracetamol metabolism based on data from in vitro studies. However a human model has found their involvement to be negligible compared with 2E1 45. The thiol produced via 2E1, N‐acetyl‐p‐benzoquinone imine (NAPQI), is a reactive oxidative intermediate that has been linked to hepatocellular injury due to its radical affinity and covalent binding to cysteine molecules on liver proteins 42. Under most circumstances a healthy liver is equipped to detoxify NAPQI by phase 2 conjugation with glutathione (GSH) spontaneously and possibly via glutathione‐s‐transferase pi 1 (GSTP1) to form inert mercapturate and cysteine metabolites 42. Alternatively NAPQI may be reduced back to paracetamol via NAD(P)H quinone oxidoreductase 1 (NQO1) 46, 47, 48.
Figure 1.
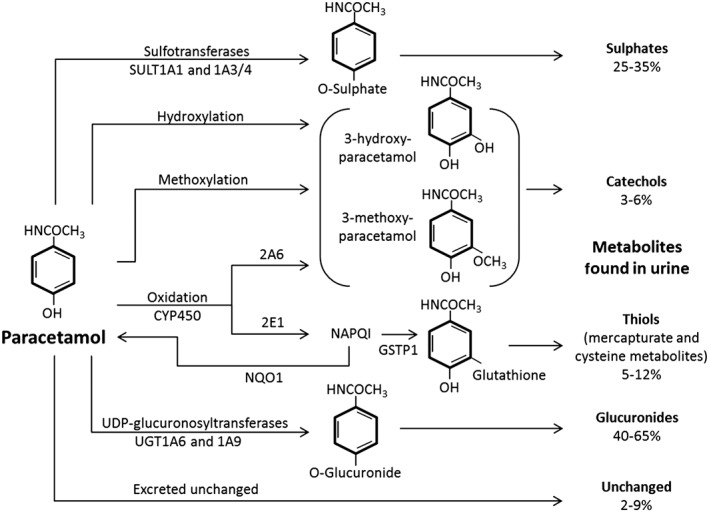
Pathways of paracetamol metabolism. GSTP1 glutathione‐s‐transferase pi 1; NQO1 NAD(P)H quinone oxidoreductase 1
Where creatinine clearance may be used to guide dose moderation in patients with renal insufficiency, the Child−Pugh and Model for End Stage Liver Disease (MELD) classification systems for liver disease severity cannot be used to predict changes in drug clearance. Furthermore, pharmacokinetic changes, whilst theoretically predictable, do not always translate into clinically significant toxicity. Several small studies have shown that the excretion of paracetamol in patients with CLD (mild, moderate and severe liver disease including hepatitis C, primary biliary cirrhosis and alcoholic cirrhosis) is slower than in healthy adults with a half‐life approximately 1–2 h longer 49, 50, 51, 52, 53. Existing biotransformation studies in this group are sparse (Table 1) although they indicate that paracetamol does not accumulate in CLD despite a higher plasma concentration 51. Theoretically this increased exposure may correspond with greater efficacy, but this has not been studied. The proportion of thiol metabolites also appears to remain unchanged with increasing severity of disease 51, 52, 54, 55, 56, suggesting that the presence of liver disease alone may not increase exposure to NAPQI and therefore does not increase risk of toxicity. It should be noted that these studies did not specifically address paracetamol metabolism in the setting of fatty liver disease or obesity, which are known to affect pharmacokinetics variably 57.
Table 1.
Half‐life (h ± SD) and 24 h metabolite recovery (as % of dose ± SD) following a single dose of paracetamol
| Study | Sample size | Paracetamol | Glucuronide | Sulphate | Thiol | Half‐life (t ½) |
|---|---|---|---|---|---|---|
| Forrest et al. 50 | Control n = 8 | 3.7 ± 0.2 | 54 ± 1.4 | 33 ± 1.2 | 8.6* | 2.4 ± 0.2 |
| Mild CLD n = 8 | 2.7 ± 0.3 | 59 ± 2.3 | 29 ± 1.9 | 8.7* | 2.2 ± 0.5 | |
| Severe CLD n = 7 | 4.6 ± 0.8 | 50 ± 3.7 | 35 ± 3.1 | 8.4* | 4.3 ± 1.2‡ | |
| Critchley et al. 54 | Control n = 10† | 3.6 ± 0.4 | 59.0 ± 1.0 | 28.8 ± 1.2 | 8.5* | 2.3 ± 0.1 |
| Ethanol n = 10† | 7.0 ± 2.1 | 58.1 ± 2.4 | 32.2 ± 1.3 | 2.8*§ | 2.6 ± 0.1 | |
| Leung et al. 55 | Control n = 26 | 4 ± 2 | 58 ± 10 | 31 ± 9 | 7* | – |
| Hepatitis B with cirrhosis n = 39 | 5 ± 2 | 58 ± 9 | 28 ± 7 | 10* | – | |
| HCC n = 19 | 4 ± 2 | 42±13¶ | 32 ± 11 | 22*** | – | |
| Zapater et al. 51 | Control n = 7 | 1.2 ± 0.6 | 59.9 ± 4.2 | 27.9 ± 4.4 | 9.7 ± 2.2 | 2.0 ± 0.4†† |
| Mild‐Moderate CLD n = 9 | 1.6 ± 1.2 | 57.1 ± 7.6 | 30.4 ± 7.1 | 9.0 ± 2.8 | 3.7 ± 1.3 | |
| Severe CLD n = 5 | 1.0 ± 0.5 | 53.5 ± 7.4 | 35.4 ± 7.9 | 9.2 ± 4.4 | 4.0 ± 0.6 | |
| Gelotte et al. 53 | Control n = 12 | 2.4 ± 0.8§§ | 49.0 ± 7.4§§ | 22.4 ± 7.0§§ | 5.5 ± 2.1§§ | 2.8 ± 1.4 |
| Mild‐Moderate CLD n = 12 | 2.9 ± 2.0 | 40.2 ± 13.3 | 27.4 ± 12.0 | 6.8 ± 2.5 | 3.4 ± 1.1 |
cysteine and mercapturate metabolites were reported separately, but are added here.
In a cohort study, participants (healthy adults n=5; heavy drinkers n=5) received paracetamol alone at baseline and then paracetamol with ethanol 1 week later. The authors reported similar metabolism between groups and therefore combined them.
half‐life in severe disease significantly greater than mild disease and control subjects (P < 0.001).
mercapturate and cysteine conjugates significantly reduced with co‐administration of ethanol (P < 0.001).
glucuronide conjugates significantly reduced compared with control and hepatitis B groups (P < 0.001).
significant increase in thiol metabolites compared with control and hepatitis B groups (P < 0.0001).
half‐life significantly less in control subjects compared with liver disease (combined mild‐moderate and severe) (P= 0.01).
Mean data from 11 participants due to sampling error.
Paracetamol toxicity
Side effects with paracetamol when it is used appropriately both short and long term in a healthy adult are very rare. A review of eight observational studies has suggested a dose–response increase in all‐cause mortality, cardiovascular, renal and gastrointestinal adverse events for people taking therapeutic doses of paracetamol, although half of the studies did not adjust for concomitant NSAID use 58. In excess, paracetamol is known to have a broad toxic spectrum ranging from nausea and anorexia to severe liver damage and death as the sulphation pathway becomes acutely saturated 59 and NAPQI is produced in excess. The sulphydryl donor N‐acetylcysteine (NAC), which serves as a precursor for glutathione and hence speeds the detoxification of NAPQI in this setting, is the only approved agent on the market for treatment of paracetamol overdose 42, 60, 61. Early diagnosis, modern medicine and use of NAC have considerably improved outcomes for patients who present early with toxicity 42, 60, 61.
While many of the first liver toxicity reports did not adequately report dose, involved large drug quantities or standard doses at what is now understood to be an inappropriate frequency, these cases nevertheless raised the concern that paracetamol should preferably be avoided or the dose reduced in patients with liver disease. This fear was compounded in 2006 when a RCT reported a statistically significant rise in ALT in healthy adult volunteers 62, the majority of whom were Hispanic, following 14 days of paracetamol administration (dosed at 1 g every 4−6 h) with or without co‐administration of an opioid (morphine, hydromorphone or oxycodone). ALT reached three times the upper limit of normal (ULN) in 41 of 106 paracetamol‐treated patients (39%) and none of 39 placebo‐treated subjects, irrespective of opioid. However, 8 years later the methodology and results of the study by Watkins et al. 62 have been challenged by an investigation 63 which demonstrated the time course of aminotransferase fluctuation in 205 healthy adult volunteers administered the same regimen of paracetamol over at least 16 days. In contrast to the earlier report, only three patients had an ALT greater than three times the ULN during the course of the study while 77% (n = 157) either had no ALT elevation or a minor elevation that resolved (ALT returned to <ULN) by day 16. The investigators followed 47 of the remaining 48 patients whose ALT persisted above 47 U l−1 at day 16. They continued to consume paracetamol until LFTs returned to normal. All but one patient (postulated to be due to systemic infection) had resolution by day 40. It has been suggested 63 that had the Watkins et al. 62 study protocol permitted data collection beyond 14 days and not discontinued treatment in patients with transaminases >three times the ULN, ALT may have returned to baseline. Indeed, given no patient exhibited clinical signs of toxicity in the Watkins et al. 62 paper, is possible that the ALT rise may have been a result of an interacting ethnic genetic variability in metabolism and was likely to be of little clinical significance 64, 65, 66, 67. Transient elevation of liver transaminases is not uncommon with numerous medications, including statins and some antibiotics, and marginal ALT elevations in patients who are institutionalized for studies have also been linked to institutionalization, not necessarily treatment intervention 68.
Nevertheless, the conservative recommendations that followed the publication of the Watkins et al. 62 paper made by leading bodies of ‘at risk’ populations persist. For example, the American Liver Foundation recommended patients not exceed 3 g of paracetamol daily and the American Geriatric Society suggested no more than 2−3 g daily in older patients with hepatic insufficiency or a history of alcohol abuse. McNeil Laboratories self‐initiated packaging changes to recommend a 3 g daily maximum on their Tylenol products, and the FDA's pursuit to discourage prescribers from recommending products that contain more than 325 mg paracetamol per dosage unit is ongoing. It must be noted that these generalized recommendations may not be applicable to all patients, and sub‐therapeutic dosing may accelerate progression to stronger analgesics.
Glucuronidation capacity
Most glucuronosyltransferases enzymes (UGTs) can metabolize paracetamol and unlike sulphotransferase enzymes, glucuronidation is a non‐saturable pathway 43, 69. A study of repeated paracetamol dosing at 4 g, 6 g and 8 g daily over 3 days in 24 healthy young men and women of normal weight has demonstrated a higher concentration of the glucuronide conjugate and a lower concentration of sulphate and thiol conjugates than was anticipated from single dose data, suggestive of time‐dependent changes in paracetamol metabolism 36, 70. As paracetamol may induce UGT1A6 36, chronic ingestion may reduce exposure to the reactive intermediate in healthy adults, as a greater proportion of the drug is metabolized via the non‐toxic phase 1 glucuronidation pathway. However, extrapolating these data into the CLD population is difficult as some metabolic pathways are variably up‐ or down‐regulated according to disease aetiology and severity, discussed in the Section entitled “Drug Interactions and Other Confounding Factors”.
Glutathione depletion
Detoxification of NAPQI by conjugation with hepatic glutathione may be impaired in the setting of malnutrition, recent fasting or advanced cirrhosis wherein the synthesis of glutathione may be impaired 71. Therefore these patients may theoretically be at higher risk of paracetamol toxicity. A number of small studies in healthy adults have indicated that nutritional deficiency decreases availability of glutathione precursors in muscle and plasma 72, reduces free glutathione but not total glutathione concentrations in the plasma 73 and decreases synthesis rate but not concentration of glutathione in red blood cells 74. A diet high in dairy products has been positively correlated with glutathione concentrations in the brain 75. Numerous pathways involved in the regulation of glutathione concentrations may be affected by the presence of liver disease, although the majority of studies have been performed in animal models 76.
Reduced glutathione concentrations and reduction in the GSH : GSSG ratio are suggestive of oxidative stress, reflecting reduced hepatic capacity to recover from insult 77. Measurement of human hepatic glutathione is achievable only through invasive tissue biopsy and thus is difficult to study on a large scale. Hepatic and peripheral plasma glutathione concentrations are correlated in rats 78, 79 and humans with various liver diseases 80, 81. Following paracetamol administration, a reduction in glutathione stores of at least 70% was required before liver damage became evident in mice 60, although no such study has been performed in humans. One study in alcoholic patients found that the median plasma glutathione concentration was not related to paracetamol intake, although a statistically significant inverse correlation was found between γ‐glutamyl transferase (GGT) and plasma glutathione 82.
Irrespective of causation, a plethora of studies have shown plasma and hepatic glutathione concentrations to be variably reduced or unchanged in patients with diverse liver diseases as outlined in Table 2 71, 83, 84, 85, 86, 87, 88, 89, 90, 91, 92, 93, 94. Although reduction of glutathione concentrations has been demonstrated in most liver disease aetiologies, there has been no change demonstrated in the ratio of thiol metabolites retrieved in urine, nor evidence of liver injury in these patients. Thus, in the absence of contributory factors, such as malnutrition, there is no strong evidence of impaired glutathione conjugation in CLD and therefore it is unlikely that glutathione store depletion alone increases risk of hepatotoxicity in the clinical setting. Indeed, case reports published on hepatotoxicity secondary to therapeutic doses of paracetamol 6, 11, 14 are often in severely underweight, malnourished or fasting patients suggesting that acute nutritional deficiency is likely a more important consideration than underlying liver disease when considering paracetamol for these patients.
Table 2.
Hepatic tissue and plasma glutathione concentrations in liver disease
| Study | Sample measured | Control | Liver disease | Data as mean ± 95% CI | P |
|---|---|---|---|---|---|
| Burgunder & Lauterburg 82 | Plasma GSH (mean ± SD μmol l−1) | 9.3 ± 2.4 (n = 8) | 3.6 ± 1.1 (alcoholic cirrhosis n = 8) | Control 9.3 ± 2.0 Alcoholic cirrhosis 3.6 ± 0.9 | P < 0.001 |
| Total GSH (mean ± SD μmol l−1) | 16.6 ± 6.2 (n = 8) | 7.1 ± 2.6 (alcoholic cirrhosis n = 8) | Control 16.6 ± 5.2 Alcoholic cirrhosis 7.1 ± 2.2 | P < 0.002 | |
| Altomare et al. 83 | Tissue GSH (mean ± SEM μmol g−1 liver) | 4.14 ± 0.1 (n = 15) | 2.77 ± 0.1 (NALD* n = 20) | Control 4.14 ± 0.2 NALD* 2.77 ± 0.2 | P < 0.001 |
| 2.55 ± 0.1 (ALD n = 35) | ALD 2.55 ± 0.2 | P < 0.001 | |||
| Bianchi et al. 70 | Plasma GSH (mean ± SD μmol l−1) | 5.91 ± 1.04 (n = 6) | 1.69 ± 1.06 (alcoholic and HCV cirrhosis n = 10) | Control 5.91 ± 1.09 Alcoholic and HCV cirrhosis 1.69 ± 0.76 | Not significant |
| Bernhard et al. 84 | Plasma GSH (mean ± 95%CI μmol l−1) | 14.1 ± 1.3 (n = 19) | 12.5 ± 1.6 (chronic hepatitis C n = 36) | Control 14.1 ± 1.3 Chronic hepatitis C 12.5 ± 1.6 | Not significant |
| Van de Casteele et al. 85 | Whole blood GSH (mean ± SEM μmol l−1) | 908 ± 43 (n = 21) | 952 ± 71 (alcoholism without cirrhosis n = 14) | Control 908 ± 90 Alcoholism without cirrhosis 952 ± 153 | Not significant |
| 823 ± 51 (alcoholic cirrhosis Child‐Pugh A n = 9) | Alcoholic cirrhosis Child‐Pugh A 823 ± 118 | Not significant | |||
| 909 ± 91 (alcoholic cirrhosis Child‐Pugh B n = 5) | Alcoholic cirrhosis Child‐Pugh B 909 ± 253 | Not significant | |||
| 500 ± 71 (alcoholic cirrhosis Child‐Pugh C n = 18) | Alcoholic cirrhosis Child‐Pugh C 500 ± 150 | P < 0.05 | |||
| 520 ± 90 (non‐alcoholic cirrhosis Child‐Pugh C n = 6) | Non‐alcoholic cirrhosis Child‐Pugh C 520 ± 231 | P < 0.05 | |||
| Saricam et al. 86 | Erythrocytic GSH (mean ± SD nmol g−1 Hb) | 56.90 ± 5.03 (n = 16) | 34.09 ± 2.19 (NAFLD n = 26) | Control 56.90 ± 2.68 NALFD 34.09 ± 0.88 | P < 0.001 |
| Cemek et al. 87 | Whole blood GSH (mean ± SD mg dl−1) | 34.38 ± 1.41 (n = 29) | 3.89 ± 1.59 (acute hepatitis A n = 19) | Control 34.38 ± 0.54 Hepatitis A 3.89 ± 0.77 | P < 0.001 |
| Czeczot et al. 88 | Tissue GSH (mean ± SD μmol mg−1 protein) | 5.52 ± 3.27 (n = 15) | 4.62 ± 2.94 (HCC n = 15) | Control 5.21 ± 1.81 HCC 4.62 ± 1.63 | P < 0.05 |
| 3.45 ± 2.11 (cirrhosis n = 15) | Cirrhosis 3.45 ± 1.17 | P < 0.05 | |||
| Kuffner et al. 89 | Plasma GSH (mean ± SD μmol l−1) | 2.17 ± 0.97 (n = 56) | 2.27 ± 0.85 (alcoholic patients, day 3 of detox while taking paracetamol n = 56) | Control 2.17 ± 0.26 Alcoholics taking paracetamol 2.27 ± 0.23 | Not significant |
| 1.90 ± 0.68 (n = 23) | 2.02 ± 0.74 (alcoholic patients, day 3 of detox while taking placebo n = 23) | Control 1.90 ± 0.29 Alcoholics taking placebo 2.02 ± 0.32 | Not significant | ||
| Lee et al. 90 | Blood GSH (mean ± SD μmol l−1) | 1294.3 ± 258.0 (n = 137) | 970.5 ± 321.7 (virus‐originated HCC n = 24) | Control 1294.3 ± 43.6 Virus‐originated HCC 970.5 ± 135.8 | P < 0.001 |
| Blood GSH : GSSG ratio (mean ± SD) | 20.3 ± 10.2 (n = 137) | 6.7 ± 4.6 (virus‐originated HCC n = 24) | Control 20.3 ± 1.7 Virus‐originated HCC 6.7 ± 1.9 | P < 0.001 | |
| Tissue GSH (mean ± SD μmol l−1g−1 protein) | 723.6 ± 215.0 (adjacent cancer‐free tissue n = 24) | 439.8 ± 198.4 (virus‐originated HCC n = 24) | Control 723.6 ± 90.8 Virus‐originated HCC 439.8 ± 83.8 | P < 0.001 | |
| Tissue GSH : GSSG ratio (mean ± SD) | 10.5 ± 3.7 (adjacent cancer‐free tissue n = 24) | 4.4 ± 1.9 (virus‐originated HCC n = 24) | Control 10.5 ± 1.6 Virus‐originated HCC 4.4 ± 0.8 | P < 0.001 | |
| Das et al. 91 | Tissue GSH (mean ± SD μg mg−1 protein) | 4.08 ± 0.59 (n = 38) | 3.64 ± 0.19 (NAFLD n = 35) | Control 4.08 ± 0.19 NAFLD 3.64 ± 0.07 | P < 0.01 |
| 3.19 ± 0.58 (ALD n = 38) | ALD 3.19 ± 0.19 | P < 0.001 | |||
| Narasimhan et al. 92 | Age adjusted whole blood GSH (mean ± SD μmol g−1 Hb) | 7.5 ± 1.4 (n = 50) | 6.2 ± 1.3 (NAFLD without T2DM n = 50) | Control 7.5 ± 0.40 NAFLD without T2DM 6.2 ± 0.37 | P < 0.05 |
| 4.0 ± 1.5 (NAFLD with T2DM n = 50) | NALFD with T2DM 4.0 ± 0.43 | P < 0.001 | |||
| 5.2 ± 1.5 (T2DM without NAFLD n = 50) | T2DM without NAFLD 5.2 ± 0.43 | P < 0.05 | |||
| Kaffe et al. 93 | Whole blood GSH (Median [range] μmol l−1) | 1101 [276‐5409] (n = 50) | 475 [4−2743] (AIC n = 49) | – | P ≤ 0.001 |
| 495 [4‐2743] (AIC without cirrhosis n = 43) | – | P ≤ 0.01 | |||
| 209 [89‐659] (AIC with cirrhosis n = 6) | – | P ≤ 0.001 | |||
| 1135 [293‐5409] (n = 41) | 512 [51‐5541] (AIH n = 36) | – | P ≤ 0.01 | ||
| 511 [88‐2977] (AIH without cirrhosis n = 26) | – | P ≤ 0.01 | |||
| 293 [51‐5541] (AIH with cirrhosis n = 10) | – | P ≤ 0.001 |
95% CI were calculated from original data in all studies except Bernhard et al., to facilitate comparison between studies.
NALD (non‐alcoholic liver disease) group included patients with chronic active hepatitis n = 7; chronic persisting hepatitis n = 3; steatosis n = 2; and cirrhosis n = 8.
AIC, autoimmune cholestatic liver disease;
AIH, autoimmune hepatitis;
ALD, alcoholic liver disease;
GSH, glutathione;
HCC, hepatocellular carcinoma;
NAFLD, non‐alcoholic fatty liver disease;
P = significance vs. control group;
T2DM, type 2 diabetes mellitus.
Ethanol consumption
There have been a handful of cases of hepatotoxicity following self‐reported therapeutic doses of paracetamol in heavy drinkers 10, 95. Irrespective of their body mass index, alcohol‐dependent subjects are frequently malnourished 96 affecting glutathione availability. Ethanol is also an inducer of CYP2E1, the same enzyme that produces the toxic metabolite NAPQI. Significant CYP2E1 induction measured using the chlorzoxazone test was demonstrated in healthy male volunteers within 1 week of consuming 40 g of ethanol per day and continued to increase for at least 4 weeks while ethanol consumption remained constant 97. However, as ethanol is a competitive substrate for the enzyme and also a stimulator of NQO1 47, 98, 99, 100, concurrent intake of ethanol and paracetamol has actually been shown to reduce paracetamol‐induced toxicity in mice 47, 101, 102 and rats 99, 103 and has been associated with a decrease in the elimination of thiol metabolites in humans 55, 100. Furthermore, concurrent acute alcohol ingestion with paracetamol overdose appears protective against hepatotoxicity irrespective of paracetamol dose 104.
However there is an increased risk of NAPQI formation during the acute alcohol withdrawal phase, as 2E1 activity can take between 3 to 8 days to return to normal 97, 105. Without a competitive substrate, production of thioether metabolites has been shown to transition from suppressed to over‐expressed within 8 h of ethanol cessation 106. While small, single dose studies have reported conflicting results of unchanged or increased production of thiol metabolites in heavy drinkers with or without cirrhosis who consumed paracetamol within 48 h of ethanol cessation 106, 107, stronger evidence from a number of RCTs has demonstrated that paracetamol ingestion during this ‘high risk’ withdrawal period does not predispose patients to liver injury 66, 90, 108, 109, 110, as summarized in Table 3. A meta‐analysis of published RCTs inclusive of 551 paracetamol‐treated heavy drinkers vs. 350 placebo‐treated heavy drinkers found no significant evidence of ALT elevation from baseline to day 4 111.
Table 3.
Controlled paracetamol consumption in chronic alcoholic patients
| Study | Ethanol intake | Paracetamol | Control | Duration | Result |
|---|---|---|---|---|---|
| Kuffner et al. 108 | Admission to alcohol detoxification facility, CAGE and MAST screen. Intake ceased during study. | 1 g four times daily (n = 102) | Placebo (n = 99) | 2 days (additional 2 days monitoring) | No significant difference in AST, ALT or INR |
| Kuffner et al. 89 | Positive alcohol on breathalyzer, CAGE and MAST screen. Intake ceased during study. | 1 g four times daily (n = 258) | Placebo (n = 114) | 3 days (additional 2 days monitoring) | No significant difference in AST, ALT, bilirubin or INR |
| Heard et al. 65 | Healthy volunteers who consume 1 to 3 alcoholic beverages daily. Intake continued throughout study. | 1 g four times daily (n = 100) | Placebo (n = 50) | 10 days (additional 1 day monitoring) | Mean ALT increased by 8.7 U l–1 in treatment group on day 11 (P = 0.04). No significant change in AST, INR or bilirubin |
| Bartels et al. 109 | >6 drinks daily for >6 weeks. Alcohol consumption ceased 12 to 72 h prior to enrolment. Intake ceased during study. | 1.3 g every 8 h (n = 18) | Placebo (n = 17) | 11 doses (5 days total monitoring) | Serum α‐GST significantly lower on day 2 (P = 0.017) and day 3 (P = 0.02) in treatment group. No difference in ALT, AST or INR |
| Dart et al. 107 | Current drinking episode of ≥7 days. Intake ceased during study. | 1 g four times daily (n = 74) | Placebo (n = 68) | 5 days (additional 2 days monitoring) | Mean ALT increased by 11.7 U l–1 in treatment group (P = 0.04). No significant change in AST, INR or bilirubin |
It would thus appear that paracetamol is relatively safe to use in the average heavy drinker (who is not malnourished and either has no cirrhosis or early compensated cirrhosis) at doses up to 4 g daily when clinically indicated for short periods of time, but the lack of data in decompensated alcoholic liver disease (ALD) makes dosing recommendations difficult in this group. While 3−4 g daily may be reasonable short term for acute pain, a maximum daily dose of 2−3 g in alcoholic cirrhosis may be a safer recommendation if analgesia is required for more than 14 days 112. Definitive evidence to guide recommendations here is lacking, and alternative analgesics also have relative contraindications.
Non‐alcoholic liver diseases
Paracetamol has been studied in small groups of adults with liver disease of various aetiologies at different stages of disease progression (Table 4). The drug has similar metabolism and elimination without clinical or biochemical signs of accumulation or toxicity following both single and repeated dosing for up to 2 weeks 16, 50, 51, 52, 53, 113, but there are scant quality prospective studies to support its use beyond this time frame. There appears to be no significant increase in excretion of thiol metabolites, indicating production of NAPQI is not amplified in mild, moderate or severe liver disease 51, 52, 54. Similarly, two studies in patients with compensated and decompensated cirrhosis (total n = 549, including alcohol‐related liver disease, chronic hepatitis C infection and cryptogenic cirrhosis) admitted to tertiary care facilities found that use of over‐the‐counter analgesia including paracetamol was not associated with hospitalization for liver‐related events, while frusemide, lactulose and alcohol consumption were positively related 114, 115. Of 45 patients treated for chronic active hepatitis, there were no differences in LFTs or disease manageability between 17 patients who were taking paracetamol prior to admission and those who were not 116. However, cumulative paracetamol intake ≥7.5 g in the days prior to admission has been found to decrease prothrombin index and factor V activity, and increase bilirubin in acute viral hepatitis, specifically hepatitis A 117.
Table 4.
Controlled paracetamol consumption in other chronic liver diseases
| Study | Liver disease | Paracetamol | Control | Duration | Result |
|---|---|---|---|---|---|
| Benson et al. 15 | ALD (n = 6), Laennec's cirrhosis (n = 4), post‐necrotic cirrhosis (n = 1), chronic active hepatitis (n = 6), chronic persistent hepatitis (n = 4), primary biliary cirrhosis (n = 2) and unspecified cirrhosis (n = 1) | 1 g four times daily (n = 20) | Placebo (n = 20) | 13 day crossover (13 days paracetamol, 13 days placebo) | No significant change in bilirubin, ALP, AST or ALT |
| Dargère et al. 112 | Chronic HCV | 1 g three times daily (n = 17) | Placebo (n = 17) | 7 days (additional 3 days monitoring) | No change in ALT or viral load |
| Dart et al. 107 | Alcoholics with HCV | 1 g four times daily (n = 24) | Placebo (n = 26) | 5 days (additional 2 days monitoring) | No significant change in ALT, AST, INR or bilirubin |
Recognition of non‐alcoholic fatty liver disease (NAFLD) and non‐alcoholic steatohepatitis (NASH) is relatively new. Hence there are a lack of patient data in this group. Despite evidence of increased NQO1 activity in NAFLD and NASH 118, a review of paracetamol‐induced liver injury in seven rodent models of obesity and NAFLD suggested a similar or increased risk of toxicity compared with controls 119. Patients with pre‐existing NAFLD without cirrhosis were reported to have a higher risk of acute liver injury (OR 7.5) following paracetamol overdose 120. Obese patients without fatty liver disease have similar or reduced risk of severe acute liver injury following paracetamol overdose compared with non‐obese patients, although obese patients who developed acute liver failure had poorer outcomes 121, 122. There are no clinical studies of pharmacokinetics in paracetamol dosing in adults with NAFLD or data on long term therapeutic use.
Drug interactions and other confounding factors
During the progressive stages of liver disease xenobiotic metabolism is altered with significant overall reduction of CYP450 enzymes 123. NQO1 protein expression and activity has been shown to increase in NASH and NAFLD, while activity of GST, UGT and SULT are variably dysregulated 118, 124. Glutathione peroxidase, reductase and transferase activity is regulated differently and inconsistently in NAFLD, ALD, cirrhosis and hepatocellular carcinoma 89, 92 and glutathione stores are also altered (Table 2). The clinical consequence of these alterations on paracetamol metabolism in patients with these liver diseases has not been studied.
Increased protein expression and activity of CYP2A6 has been reported in patients with progressive NAFLD, hepatitis, primary biliary cirrhosis and alcoholic cirrhosis 125, 126, 127, 128. However increase in catechol metabolite production has not been demonstrated, suggesting that paracetamol metabolism via this pathway is not necessarily affected in these disease states.
CYP2E1 function appears to remain preserved despite reduced protein expression in progressive NAFLD 128. Similarly, activity remains unchanged while disease remains mild or compensated. In contrast, activity decreases with worsening disease (Pugh score > 6, Child Class B or C) and in cholestatic cirrhosis 123, 129. While this may suggest that NAPQI formation will be reduced in decompensated cirrhosis or in people with cholestatic liver disease, this has not been studied.
It has been proposed that direct and indirect drug interactions may affect risk of paracetamol toxicity by moderation of the CYP450 isozyme system or changes to gastric emptying. Induction of 2E1 by isoniazid has been well‐documented, but studies in healthy volunteers have shown that isoniazid may inhibit NAPQI formation as a competitive substrate for the enzyme in much the same way as alcohol 130, 131. Other small studies on enzyme inducers such as phenobarbital, phenytoin, rifampicin, omeprazole and carbamazepine have suggested that their effect on 2E1 is not significant, and thus they are unlikely to be responsible for excessive NAPQI formation when taken concurrently with paracetamol 45, 132, 133.
Some guidelines recommend that adult patients of low body weight (<50 kg) receive a reduced maximum daily dose of paracetamol (15 mg kg−1 four times a day) 134. However weight as an independent risk factor for paracetamol toxicity, in the absence of chronic disease and malnutrition, has not been well studied.
Prescribing in other groups
The complex pharmacokinetic–pharmacodynamic relationship of drug–patient and drug–disease interactions causes debate around appropriate paracetamol prescribing in children, older persons and some other chronic disease states. Metabolism and clearance of paracetamol in the healthy elderly population has been found to be comparable with young adults, though persons over 80 years have a greater overall exposure following intravenous administration 135, 136, 137, 138. One study has demonstrated an increase in glucuronide conjugates in 12 male children with NAFLD, but no significant changes in terms of clearance, half‐life, AUC or C max 139. The nature of the pain being treated should also be taken into consideration regardless of population, as recent evidence suggests paracetamol used in patients (aged 45 ± 16 years) without CLD may be no better than placebo for acute low back pain 140.
Future research
A helpful future study would be a large, long term (> 1 month) prospective observational study of CLD patients with need for therapeutic analgesia, to provide correlation data between dose, concentration, efficacy and toxicity with paracetamol. This will improve dose guidance for individual patients. Furthermore, whilst glutathione content has been relatively well‐examined in numerous hepatic diseases, there remains scant knowledge of enzyme dysregulation (including CYP, UGT, SULT, NQO1) in most CLD aetiologies. Further research in the latter area will assist with development of recommendations regarding paracetamol dosing.
It is noted that one of the mechanisms of action of paracetamol is an inhibitory effect on COX enzymes, which may be linked to long term renal and cardiovascular outcomes. While this review has primarily focussed on the need for studies on hepatic toxicity with normal dosing in liver disease, studies on cardiovascular health with paracetamol are also needed.
Conclusion
Recent reviews have concluded that paracetamol is a safe and effective first line agent in almost all patients regardless of liver disease aetiology. Although the need for dose reduction in the healthy population seems largely unnecessary, it may be warranted in certain severe or decompensated hepatic disease states, particularly if patients are malnourished, are not eating or have a dry weight less than 50 kg. Whilst a cautious and conservative approach has previously been recommended for all CLD patients, prescribers should be encouraged to consider appropriate dosing for each individual patient, taking into account their underlying disease state and the pharmacological covariates.
As the prevalence of lifestyle related liver diseases such as ALD and NAFLD is likely to increase over the coming decades, it is important that clinicians are able to use existing analgesics safely and effectively. To that effect, studies aimed at improving our understanding of changes to paracetamol metabolism, efficacy and toxicity will be invaluable.
Competing Interests
All authors have completed the Unified Competing Interest form at ww.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare no support from any organization for the submitted work, no financial relationships with any organizations that might have an interest in the submitted work in the previous 3 years and no other relationships or activities that could appear to have influenced the submitted work.
Hayward, K. L. , Powell, E. E. , Irvine, K. M. , and Martin, J. H. (2016) Can paracetamol (acetaminophen) be administered to patients with liver impairment?. Br J Clin Pharmacol, 81: 210–222. doi: 10.1111/bcp.12802.
References
- 1. Zimmerman HJ. Hepatotoxicity: the adverse effects of drugs and other chemicals on the liver, Second Edition, (eds) John J. Seigafuse S. Teston D. Goldsbury CJ,Philadelphia: Lippincott Williams and Wilkins, 1999; 744. [Google Scholar]
- 2. Dwyer JP, Jayasekera C, Nicoll A. Analgesia for the cirrhotic patient: a literature review and recommendations. J Gastroenterol Hepatol 2014; 29: 1356–60. [DOI] [PubMed] [Google Scholar]
- 3. Imani F, Motavaf M, Safari S, Alavian SM. The therapeutic use of analgesics in patients with liver cirrhosis: a literature review and evidence‐based recommendations. Hepat Mon 2014; 14: e23539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Acetaminophen information: U.S . Food and Drug Administration; 2015. [Updated 08 May 2015; Cited 19 May 2015]. Available from: http://www.fda.gov/Drugs/DrugSafety/InformationbyDrugClass/ucm165107.htm.
- 5. Giboney PT. Mildly elevated liver transaminase levels in the asymptomatic patient. Am Fam Physician 2005; 71: 1105–10. [PubMed] [Google Scholar]
- 6. Lee PJ, Shen M, Wang S, Spiegler P, Caraccio T, DeMuro JP, Malone B. Possible hepatotoxicity associated with intravenous acetaminophen in a 36‐year‐old female patient. P T 2015; 40: 123–32. [PMC free article] [PubMed] [Google Scholar]
- 7. Gumbrevicius G, Sveikata A, Sveikatiene R, Stankevicius E. Paracetamol and simvastatin: a potential interaction resulting in hepatotoxicity. Medicina 2012; 48: 379–81. [PubMed] [Google Scholar]
- 8. Moling O, Cairon E, Rimenti G, Rizza F, Pristera R, Mian P. Severe hepatotoxicity after therapeutic doses of acetaminophen. Clin Ther 2006; 28: 755–60. [DOI] [PubMed] [Google Scholar]
- 9. Pearce B, Grant IS. Acute liver failure following therapeutic paracetamol administration in patients with muscular dystrophies. Anaesthesia 2008; 63: 89–91. [DOI] [PubMed] [Google Scholar]
- 10. Zimmerman HJ, Maddrey WC. Acetaminophen (paracetamol) hepatotoxicity with regular intake of alcohol: analysis of instances of therapeutic misadventure. Hepatology 1995; 22: 767–73. [PubMed] [Google Scholar]
- 11. Eriksson LS, Broome U, Kalin M, Lindholm M. Hepatotoxicity due to repeated intake of low doses of paracetamol. J Intern Med 1992; 231: 567–70. [DOI] [PubMed] [Google Scholar]
- 12. Horsmans Y, Sempoux C, Detry R, Geubel AP. Paracetamol‐induced liver toxicity after intravenous administration. Liver 1998; 18: 294–5. [DOI] [PubMed] [Google Scholar]
- 13. Mortensen ME, Cullen JL. Comment: hepatotoxicity associated with chronic acetaminophen administration in patients without risk factors. Ann Pharmacother 2002; 36: 1481–2. [DOI] [PubMed] [Google Scholar]
- 14. Claridge LC, Eksteen B, Smith A, Shah T, Holt AP. Acute liver failure after administration of paracetamol at the maximum recommended daily dose in adults. BMJ 2010; 341: 1269–70. [DOI] [PubMed] [Google Scholar]
- 15. Dart RC, Bailey E. Does therapeutic use of acetaminophen cause acute liver failure? Pharmacotherapy 2007; 27: 1219–30. [DOI] [PubMed] [Google Scholar]
- 16. Benson GD. Acetaminophen in chronic liver disease. Clin Pharmacol Ther 1983; 33: 95–101. [DOI] [PubMed] [Google Scholar]
- 17. Hirschfield GM, Kumagi T, Heathcote EJ. Preventative hepatology: minimising symptoms and optimising care. Liver Int 2008; 28: 922–34. [DOI] [PubMed] [Google Scholar]
- 18. Morse H. Ueber eine neue darstellungsmethode der acetylamidophenole [about a new method of synthesis of acetylamidophenole]. Ber Deutscher Chem Ges 1878; 11: 232–3. [Google Scholar]
- 19. Cahn A, Hepp P. Das antifebrin, ein neues fiebermittel [antifebrin, a new antipyretic]. Zentralbl Klin Med 1886; 7: 561–4. [Google Scholar]
- 20. Smith JN, Williams RT. Studies in detoxication: the metabolism of acetanilide in the rabbit. Biochem J 1948; 42: 538–44. [PubMed] [Google Scholar]
- 21. Smith JN, Williams RT. Studies in detoxication: the metabolism of phenacetin (p‐ethoxyacetanilide) in the rabbit and a further observation on acetanilide metabolism. Biochem J 1949; 44: 239–42. [PubMed] [Google Scholar]
- 22. Brodie BB, Axelrod J. The estimation of acetanilide and its metabolic products, aniline, N‐acetyl p‐aminophenol and p‐amino‐phenol (free and total conjugated) in biological fluids and tissues. J Pharmacol Exp Ther 1948; 94: 22–8. [PubMed] [Google Scholar]
- 23. Brodie BB, Axelrod J. The fate of acetanilide in man. J Pharmacol Exp Ther 1948; 94: 29–38. [PubMed] [Google Scholar]
- 24. Boreus LO, Sandberg F. A comparison of some pharmacological effects of acetophenetidin and N‐acetyl p‐aminophenol. Acta Physiol Scand 1953; 28: 261–5. [DOI] [PubMed] [Google Scholar]
- 25. Boyd EM, Bereczky GM. Liver necrosis from paracetamol. Br J Pharmacol Chemother 1966; 26: 606–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. U.S. Food and Drug Administration Nonprescription Drugs Advisory Committee . An archeological review of the regulatory history of over‐the‐counter (OTC) single ingredient acetaminophen: U.S. Food and Drug Administration; 2002. [Updated 19 Sept 2002; Cited 14 Sept 2015]. Available from: http://www.fda.gov/ohrms/dockets/ac/02/briefing/3882b1_02_A-History%20of%20OTC%20Single%20Ingredient.pdf1.
- 27. Hopkinson JH SM, Bare WW, Levin HM, Posatko RJ. Acetaminophen (500 mg) versus acetaminophen (325 mg) for the relief of pain in episiotomy patients. Curr Ther Res 1974; 16: 194–200. [Google Scholar]
- 28. U.S. Food and Drug Administration Division of Nonprescription Clinical Evaluation , Chang C. Single‐ingredient acetaminophen dose–response data in adults: U.S. Food and Drug Administration; 2009. [Updated 29 June 2009; Cited 14 Sept 2015]. Available from: http://www.fda.gov/downloads/AdvisoryCommittees/drugs/drugsafetyandriskmanagementadvisorycommittee/ucm172043.pdf.
- 29. McQuay HJ, Edwards JE, Moore RA. Evaluating analgesia: the challenges. Am J Ther 2002; 9: 179–87. [DOI] [PubMed] [Google Scholar]
- 30. McQuay HJ, Poppleton P, Carroll D, Summerfield RJ, Bullingham RE, Moore RA. Ketorolac and acetaminophen for orthopedic postoperative pain. Clin Pharmacol Ther 1986; 39: 89–93. [DOI] [PubMed] [Google Scholar]
- 31. Seymour RA, Kelly PJ, Hawkesford JE. The efficacy of ketoprofen and paracetamol (acetaminophen) in postoperative pain after third molar surgery. Br J Clin Pharmacol 1996; 41: 581–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nystrom E, Gustafsson I, Quiding H. The pain intensity at analgesic intake, and the efficacy of diflunisal in single doses and effervescent acetaminophen in single and repeated doses. Pharmacotherapy 1988; 8: 201–9. [DOI] [PubMed] [Google Scholar]
- 33. Quiding H, Oikarinen V, Sane J, Sjoblad AM. Analgesic efficacy after single and repeated doses of codeine and acetaminophen. J Clin Pharmacol 1984; 24: 27–34. [DOI] [PubMed] [Google Scholar]
- 34. Yuan CS, Karrison T, Wu JA, Lowell TK, Lynch JP, Foss JF. Dose‐related effects of oral acetaminophen on cold‐induced pain: a double‐blind, randomized, placebo‐controlled trial. Clin Pharmacol Ther 1998; 63: 379–83. [DOI] [PubMed] [Google Scholar]
- 35. Skoglund LA, Skjelbred P, Fyllingen G. Analgesic efficacy of acetaminophen 1000 mg, acetaminophen 2000 mg, and the combination of acetaminophen 1000 mg and codeine phosphate 60 mg versus placebo in acute postoperative pain. Pharmacotherapy 1991; 11: 364–9. [PubMed] [Google Scholar]
- 36. Gelotte CK, Auiler JF, Lynch JM, Temple AR, Slattery JT. Disposition of acetaminophen at 4, 6, and 8 g/day for 3 days in healthy young adults. Clin Pharmacol Ther 2007; 81: 840–8. [DOI] [PubMed] [Google Scholar]
- 37. den Hertog HM, van der Worp HB, van Gemert HM, Algra A, Kappelle LJ, van Gijn J, Koudstaal PJ, Dippel DW. The paracetamol (acetaminophen) in stroke (PAIS) trial: a multicentre, randomised, placebo‐controlled, phase III trial. Lancet Neurol 2009; 8: 434–40. [DOI] [PubMed] [Google Scholar]
- 38. Watelet J, Laurent V, Bressenot A, Bronowicki JP, Larrey D, Peyrin‐Biroulet L. Toxicity of chronic paracetamol ingestion. Aliment Pharmacol Ther 2007; 26: 1543–4. [DOI] [PubMed] [Google Scholar]
- 39. Kuffner EK, Temple AR, Cooper KM, Baggish JS, Parenti DL. Retrospective analysis of transient elevations in alanine aminotransferase during long‐term treatment with acetaminophen in osteoarthritis clinical trials. Curr Med Res Opin 2006; 22: 2137–48. [DOI] [PubMed] [Google Scholar]
- 40. Williams HJ, Ward JR, Egger MJ, Neuner R, Brooks RH, Clegg DO, Field EH, Skosey JL, Alarcon GS, Willkens RF, Paulus HE, Russell IJ, Sharp JT. Comparison of naproxen and acetaminophen in a two‐year study of treatment of osteoarthritis of the knee. Arthritis Rheum 1993; 36: 1196–206. [DOI] [PubMed] [Google Scholar]
- 41. Miners JO, Osborne NJ, Tonkin AL, Birkett DJ. Perturbation of paracetamol urinary metabolic ratios by urine flow rate. Br J Clin Pharmacol 1992; 34: 359–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. McGill MR, Jaeschke H. Metabolism and disposition of acetaminophen: recent advances in relation to hepatotoxicity and diagnosis. Pharm Res 2013; 30: 2174–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mutlib AE, Goosen TC, Bauman JN, Williams JA, Kulkarni S, Kostrubsky S. Kinetics of acetaminophen glucuronidation by UDP‐glucuronosyltransferases 1A1, 1A6, 1A9 and 2B15. Potential implications in acetaminophen‐induced hepatotoxicity. Chem Res Toxicol 2006; 19: 701–9. [DOI] [PubMed] [Google Scholar]
- 44. Court MH, Duan SX, von Moltke LL, Greenblatt DJ, Patten CJ, Miners JO, Mackenzie PI. Interindividual variability in acetaminophen glucuronidation by human liver microsomes: identification of relevant acetaminophen UDP‐glucuronosyltransferase isoforms. J Pharmacol Exp Ther 2001; 299: 998–1006. [PubMed] [Google Scholar]
- 45. Manyike PT, Kharasch ED, Kalhorn TF, Slattery JT. Contribution of CYP2E1 and CYP3A to acetaminophen reactive metabolite formation. Clin Pharmacol Ther 2000; 67: 275–82. [DOI] [PubMed] [Google Scholar]
- 46. Aleksunes LM, Goedken M, Manautou JE. Up‐regulation of NAD(P)H quinone oxidoreductase 1 during human liver injury. World J Gastroenterol 2006; 12: 1937–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lee SM, Cho TS, Kim DJ, Cha YN. Protective effect of ethanol against acetaminophen‐induced hepatotoxicity in mice: role of NADH:quinone reductase. Biochem Pharmacol 1999; 58: 1547–55. [DOI] [PubMed] [Google Scholar]
- 48. Moffit JS, Aleksunes LM, Kardas MJ, Slitt AL, Klaassen CD, Manautou JE. Role of NAD(P)H:quinone oxidoreductase 1 in clofibrate‐mediated hepatoprotection from acetaminophen. Toxicology 2007; 230: 197–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Arnman R, Olsson R. Elimination of paracetamol in chronic liver disease. Acta Hepatogastroenterol 1978; 25: 283–6. [PubMed] [Google Scholar]
- 50. Andreasen PB, Hutters L. Paracetamol (acetaminophen) clearance in patients with cirrhosis of the liver. Acta Med Scand Suppl 1979; 624: 99–105. [DOI] [PubMed] [Google Scholar]
- 51. Forrest JA, Adriaenssens P, Finlayson ND, Prescott LF. Paracetamol metabolism in chronic liver disease. Eur J Clin Pharmacol 1979; 15: 427–31. [DOI] [PubMed] [Google Scholar]
- 52. Zapater P, Lasso de la Vega MC, Horga JF, Such J, Frances R, Esteban A, Palazon JM, Carnicer F, Pascual S, Perez‐Mateo M. Pharmacokinetic variations of acetaminophen according to liver dysfunction and portal hypertension status. Aliment Pharmacol Ther 2004; 20: 29–36. [DOI] [PubMed] [Google Scholar]
- 53. Jorup‐Ronstrom C, Beermann B, Wahlin‐Boll E, Melander A, Britton S. Reduction of paracetamol and aspirin metabolism during viral hepatitis. Clin Pharmacokinet 1986; 11: 250–6. [DOI] [PubMed] [Google Scholar]
- 54.Tylenol(R) (acetaminophen) professional product information. McNeil Consumer Healthcare; 2010.
- 55. Critchley JA, Dyson EH, Scott AW, Jarvie DR, Prescott LF. Is there a place for cimetidine or ethanol in the treatment of paracetamol poisoning? Lancet 1983; 1: 1375–6. [DOI] [PubMed] [Google Scholar]
- 56. Leung NW, Critchley JA. Increased oxidative metabolism of paracetamol in patients with hepatocellular carcinoma. Cancer Lett 1991; 57: 45–8. [DOI] [PubMed] [Google Scholar]
- 57. Hanley MJ, Abernethy DR, Greenblatt DJ. Effect of obesity on the pharmacokinetics of drugs in humans. Clin Pharmacokinet 2010; 49: 71–87. [DOI] [PubMed] [Google Scholar]
- 58. Roberts E, Delgado Nunes V, Buckner S, Latchem S, Constanti M, Miller P, Doherty M, Zhang W, Birrell F, Porcheret M, Dziedzic K, Bernstein I, Wise E, Conaghan PG. Paracetamol: not as safe as we thought? A systematic literature review of observational studies. Ann Rheum Dis 2015. doi:10.1136/annrheumdis-2014-206914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Slattery JT, Levy G. Acetaminophen kinetics in acutely poisoned patients. Clin Pharmacol Ther 1979; 25: 184–95. [DOI] [PubMed] [Google Scholar]
- 60. Mitchell JR, Jollow DJ, Potter WZ, Gillette JR, Brodie BB. Acetaminophen‐induced hepatic necrosis. IV. Protective role of glutathione. J Pharmacol Exp Ther 1973; 187: 211–7. [PubMed] [Google Scholar]
- 61. Prescott LF, Park J, Ballantyne A, Adriaenssens P, Proudfoot AT. Treatment of paracetamol (acetaminophen) poisoning with N‐acetylcysteine. Lancet 1977; 2: 432–4. [DOI] [PubMed] [Google Scholar]
- 62. Watkins PB, Kaplowitz N, Slattery JT, Colonese CR, Colucci SV, Stewart PW, Harris SC. Aminotransferase elevations in healthy adults receiving 4 grams of acetaminophen daily: a randomized controlled trial. JAMA 2006; 296: 87–93. [DOI] [PubMed] [Google Scholar]
- 63. Heard K, Green JL, Anderson V, Bucher‐Bartelson B, Dart RC. A randomized, placebo‐controlled trial to determine the course of aminotransferase elevation during prolonged acetaminophen administration. BMC Pharmacol Toxicol 2014; 15: 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Yuan Y, Thabane M, Thabane L, Hunt RH. Acetaminophen and aminotransferase elevations. JAMA 2006; 296: 2798. [DOI] [PubMed] [Google Scholar]
- 65. Krenzelok EP, Royal MA. Confusion: acetaminophen dosing changes based on no evidence in adults. Drugs R&D 2012; 12: 45–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Heard K, Green JL, Bailey JE, Bogdan GM, Dart RC. A randomized trial to determine the change in alanine aminotransferase during 10 days of paracetamol (acetaminophen) administration in subjects who consume moderate amounts of alcohol. Aliment Pharmacol Ther 2007; 26: 283–90. [DOI] [PubMed] [Google Scholar]
- 67. Vuppalanchi R, Chalasani N. Therapeutic doses of acetaminophen frequently cause elevated aminotransferases in healthy volunteers: is it significant? Gastroenterology 2007; 132: 1624–6. [DOI] [PubMed] [Google Scholar]
- 68. Narjes H, Nehmiz G. Effect of hospitalisation on liver enzymes in healthy subjects. Eur J Clin Pharmacol 2000; 56: 329–33. [DOI] [PubMed] [Google Scholar]
- 69. Prescott LF. Drug conjugation in clinical toxicology. Biochem Soc Trans 1984; 12: 96–9. [DOI] [PubMed] [Google Scholar]
- 70. Hendrix‐Treacy S, Wallace SM, Hindmarsh KW, Wyant GM, Danilkewich A. The effect of acetaminophen administration on its disposition and body stores of sulphate. Eur J Clin Pharmacol 1986; 30: 273–8. [DOI] [PubMed] [Google Scholar]
- 71. Bianchi G, Bugianesi E, Ronchi M, Fabbri A, Zoli M, Marchesini G. Glutathione kinetics in normal man and in patients with liver cirrhosis. J Hepatol 1997; 26: 606–13. [DOI] [PubMed] [Google Scholar]
- 72. Hammarqvist F, Andersson K, Luo JL, Wernerman J. Free amino acid and glutathione concentrations in muscle during short‐term starvation and refeeding. Clin Nutr 2005; 24: 236–43. [DOI] [PubMed] [Google Scholar]
- 73. Martensson J. The effect of fasting on leukocyte and plasma glutathione and sulfur amino acid concentrations. Metabolism 1986; 35: 118–21. [DOI] [PubMed] [Google Scholar]
- 74. Lamers Y, O'Rourke B, Gilbert LR, Keeling C, Matthews DE, Stacpoole PW, Gregory JF. Vitamin B‐6 restriction tends to reduce the red blood cell glutathione synthesis rate without affecting red blood cell or plasma glutathione concentrations in healthy men and women. Am J Clin Nutr 2009; 90: 336–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Choi IY, Lee P, Denney DR, Spaeth K, Nast O, Ptomey L, Roth AK, Lierman JA, Sullivan DK. Dairy intake is associated with brain glutathione concentration in older adults. Am J Clin Nutr 2015; 101: 287–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Lu SC. Glutathione synthesis. Biochim Biophys Acta 1830; 2013: 3143–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Owen JB, Butterfield DA. Measurement of oxidized/reduced glutathione ratio. Methods Mol Biol 2010; 648: 269–77. [DOI] [PubMed] [Google Scholar]
- 78. Adams JD Jr, Lauterburg BH, Mitchell JR. Plasma glutathione and glutathione disulfide in the rat: regulation and response to oxidative stress. J Pharmacol Exp Ther 1983; 227: 749–54. [PubMed] [Google Scholar]
- 79. Koksal GM, Sayilgan C, Aydin S, Oz H, Uzun H. Correlation of plasma and tissue oxidative stresses in intra‐abdominal sepsis. J Surg Res 2004; 122: 180–3. [DOI] [PubMed] [Google Scholar]
- 80. Shigesawa T, Sato C, Marumo F. Significance of plasma glutathione determination in patients with alcoholic and non‐alcoholic liver disease. J Gastroenterol Hepatol 1992; 7: 7–11. [DOI] [PubMed] [Google Scholar]
- 81. Barbaro G, Di Lorenzo G, Ribersani M, Soldini M, Giancaspro G, Bellomo G, Belloni G, Grisorio B, Barbarini G. Serum ferritin and hepatic glutathione concentrations in chronic hepatitis C patients related to the hepatitis C virus genotype. J Hepatol 1999; 30: 774–82. [DOI] [PubMed] [Google Scholar]
- 82. Seifert CF, Anderson DC. Acetaminophen usage patterns and concentrations of glutathione and gamma‐glutamyl transferase in alcoholic subjects. Pharmacotherapy 2007; 27: 1473–82. [DOI] [PubMed] [Google Scholar]
- 83. Burgunder JM, Lauterburg BH. Decreased production of glutathione in patients with cirrhosis. Eur J Clin Invest 1987; 17: 408–14. [DOI] [PubMed] [Google Scholar]
- 84. Altomare E, Vendemiale G, Albano O. Hepatic glutathione content in patients with alcoholic and non alcoholic liver diseases. Life Sci 1988; 43: 991–8. [DOI] [PubMed] [Google Scholar]
- 85. Bernhard MC, Junker E, Hettinger A, Lauterburg BH. Time course of total cysteine, glutathione and homocysteine in plasma of patients with chronic hepatitis C treated with interferon‐alpha with and without supplementation with N‐acetylcysteine. J Hepatol 1998; 28: 751–5. [DOI] [PubMed] [Google Scholar]
- 86. Van de Casteele M, Zaman Z, Zeegers M, Servaes R, Fevery J, Nevens F. Blood antioxidant levels in patients with alcoholic liver disease correlate with the degree of liver impairment and are not specific to alcoholic liver injury itself. Aliment Pharmacol Ther 2002; 16: 985–92. [DOI] [PubMed] [Google Scholar]
- 87. Saricam T, Kircali B, Koken T. Assessment of lipid peroxidation and antioxidant capacity in non‐alcoholic fatty liver disease. Turk J Gastroenterol 2005; 16: 65–70. [PubMed] [Google Scholar]
- 88. Cemek M, Dede S, Bayiroglu F, Caksen H, Cemek F, Mert N. Relationship between antioxidant capacity and oxidative stress in children with acute hepatitis A. World J Gastroenterol 2006; 12: 6212–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Czeczot H, Scibior D, Skrzycki M, Podsiad M. Glutathione and GSH‐dependent enzymes in patients with liver cirrhosis and hepatocellular carcinoma. Acta Biochim Pol 2006; 53: 237–42. [PubMed] [Google Scholar]
- 90. Kuffner EK, Green JL, Bogdan GM, Knox PC, Palmer RB, Heard K, Slattery JT, Dart RC. The effect of acetaminophen (four grams a day for three consecutive days) on hepatic tests in alcoholic patients ‐ a multicenter randomized study. BMC Med 2007; 5: 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Lee KT, Tsai SM, Wang SN, Lin SK, Wu SH, Chuang SC, Wu SH, Ma H, Tsai LY. Glutathione status in the blood and tissues of patients with virus‐originated hepatocellular carcinoma. Clin Biochem 2007; 40: 1157–62. [DOI] [PubMed] [Google Scholar]
- 92. Das KS, Balakrishnan V, Mukherjee S, Vasudevan DM. Evaluation of blood oxidative stress‐related parameters in alcoholic liver disease and non‐alcoholic fatty liver disease. Scand J Clin Lab Invest 2008; 68: 323–34. [DOI] [PubMed] [Google Scholar]
- 93. Narasimhan S, Gokulakrishnan K, Sampathkumar R, Farooq S, Ravikumar R, Mohan V, Balasubramanyam M. Oxidative stress is independently associated with non‐alcoholic fatty liver disease (NAFLD) in subjects with and without type 2 diabetes. Clin Biochem 2010; 43: 815–21. [DOI] [PubMed] [Google Scholar]
- 94. Kaffe ET, Rigopoulou EI, Koukoulis GK, Dalekos GN, Moulas AN. Oxidative stress and antioxidant status in patients with autoimmune liver diseases. Redox Rep 2015; 20: 33–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Manchanda A, Cameron C, Robinson G. Beware of paracetamol use in alcohol abusers: a potential cause of acute liver injury. N Z Med J 2013; 126: 80–4. [PubMed] [Google Scholar]
- 96. Teixeira J, Mota T, Fernandes JC. Nutritional evaluation of alcoholic inpatients admitted for alcohol detoxification. Alcohol Alcohol 2011; 46: 558–60. [DOI] [PubMed] [Google Scholar]
- 97. Oneta CM, Lieber CS, Li J, Ruttimann S, Schmid B, Lattmann J, Rosman AS, Seitz HK. Dynamics of cytochrome P4502E1 activity in man: induction by ethanol and disappearance during withdrawal phase. J Hepatol 2002; 36: 47–52. [DOI] [PubMed] [Google Scholar]
- 98. Altomare E, Leo MA, Sato C, Vendemiale G, Lieber CS. Interaction of ethanol with acetaminophen metabolism in the baboon. Biochem Pharmacol 1984; 33: 2207–12. [DOI] [PubMed] [Google Scholar]
- 99. Sato C, Lieber CS. Mechanism of the preventive effect of ethanol on acetaminophen‐induced hepatoxicity. J Pharmacol Exp Ther 1981; 218: 811–5. [PubMed] [Google Scholar]
- 100. Banda PW, Quart BD. The effect of mild alcohol consumption on the metabolism of acetaminophen in man. Res Commun Chem Pathol Pharmacol 1982; 38: 57–70. [PubMed] [Google Scholar]
- 101. Wong LT, Whitehouse LW, Solomonraj G, Paul CJ. Effect of a concomitant single dose of ethanol on the hepatotoxicity and metabolism of acetaminophen in mice. Toxicology 1980; 17: 297–309. [DOI] [PubMed] [Google Scholar]
- 102. Tredger JM, Smith HM, Read RB, Portmann B, Williams R. Effects of ethanol ingestion on the hepatotoxicity and metabolism of paracetamol in mice. Toxicology 1985; 36: 341–52. [DOI] [PubMed] [Google Scholar]
- 103. Altomare E, Leo MA, Lieber CS. Interaction of acute ethanol administration with acetaminophen metabolism and toxicity in rats fed alcohol chronically. Alcohol Clin Exp Res 1984; 8: 405–8. [DOI] [PubMed] [Google Scholar]
- 104. Waring WS, Stephen AF, Malkowska AM, Robinson OD. Acute ethanol coingestion confers a lower risk of hepatotoxicity after deliberate acetaminophen overdose. Acad Emerg Med 2008; 15: 54–8. [DOI] [PubMed] [Google Scholar]
- 105. Dupont I, Berthou F, Bodenez P, Bardou L, Guirriec C, Stephan N, Dreano Y, Lucas D. Involvement of cytochromes P‐450 2E1 and 3A4 in the 5‐hydroxylation of salicylate in humans. Drug Metab Dispos 1999; 27: 322–6. [PubMed] [Google Scholar]
- 106. Thummel KE, Slattery JT, Ro H, Chien JY, Nelson SD, Lown KE, Watkins PB. Ethanol and production of the hepatotoxic metabolite of acetaminophen in healthy adults. Clin Pharmacol Ther 2000; 67: 591–9. [DOI] [PubMed] [Google Scholar]
- 107. Skinner MH, Matano R, Hazle W, Blaschke TF. Acetaminophen metabolism in recovering alcoholics. Methods Find Exp Clin Pharmacol 1990; 12: 513–5. [PubMed] [Google Scholar]
- 108. Dart RC, Green JL, Kuffner EK, Heard K, Sproule B, Brands B. The effects of paracetamol (acetaminophen) on hepatic tests in patients who chronically abuse alcohol ‐ a randomized study. Aliment Pharmacol Ther 2010; 32: 478–86. [DOI] [PubMed] [Google Scholar]
- 109. Kuffner EK, Dart RC, Bogdan GM, Hill RE, Casper E, Darton L. Effect of maximal daily doses of acetaminophen on the liver of alcoholic patients: a randomized, double‐blind, placebo‐controlled trial. Arch Intern Med 2001; 161: 2247–52. [DOI] [PubMed] [Google Scholar]
- 110. Bartels S, Sivilotti M, Crosby D, Richard J. Are recommended doses of acetaminophen hepatotoxic for recently abstinent alcoholics? A randomized trial. Clin Toxicol (Phila) 2008; 46: 243–9. [DOI] [PubMed] [Google Scholar]
- 111. Rumack B, Heard K, Green J, Albert D, Bucher‐Bartelson B, Bodmer M, Sivilotti ML, Dart RC. Effect of therapeutic doses of acetaminophen (up to 4 g/day) on serum alanine aminotransferase levels in subjects consuming ethanol: systematic review and meta‐analysis of randomized controlled trials. Pharmacotherapy 2012; 32: 784–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Chandok N, Watt KD. Pain management in the cirrhotic patient: the clinical challenge. Mayo Clin Proc 2010; 85: 451–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Dargère S, Collet T, Crampon D, Galais MP, Cohen D, Ollivier I, Fatome A, Lefilliatre P. Lack of toxicity of acetaminophen in patients with chronic hepatitis C: a randomized controlled trial. Gastroenterology 2000; 118: A947. [Google Scholar]
- 114. Khalid SK, Lane J, Navarro V, Garcia‐Tsao G. Use of over‐the‐counter analgesics is not associated with acute decompensation in patients with cirrhosis. Clin Gastroenterol Hepatol 2009; 7: 994–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Fenkel JM, Coron RN, Daskalakis C, Vega M, Rossi S, Herrine SK, Navarro VJ. Over‐the‐counter analgesics in cirrhotic patients: a case–control study examining the risk of hospitalization for liver‐associated events. Scand J Gastroenterol 2010; 45: 1101–9. [DOI] [PubMed] [Google Scholar]
- 116. Neuberger J, Davis M, Williams R. Long‐term ingestion of paracetamol and liver disease. J R Soc Med 1980; 73: 701–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Yaghi C, Honein K, Boujaoude J, Slim R, Moucari R, Sayegh R. Influence of acetaminophen at therapeutic doses on surrogate markers of severity of acute viral hepatitis. Gastroenterol Clin Biol 2006; 30: 763–8. [DOI] [PubMed] [Google Scholar]
- 118. Hardwick RN, Fisher CD, Canet MJ, Lake AD, Cherrington NJ. Diversity in antioxidant response enzymes in progressive stages of human nonalcoholic fatty liver disease. Drug Metab Dispos 2010; 38: 2293–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Michaut A, Moreau C, Robin MA, Fromenty B. Acetaminophen‐induced liver injury in obesity and nonalcoholic fatty liver disease. Liver Int 2014; 34: 171–9. [DOI] [PubMed] [Google Scholar]
- 120. Nguyen GC, Sam J, Thuluvath PJ. Hepatitis C is a predictor of acute liver injury among hospitalizations for acetaminophen overdose in the United States: a nationwide analysis. Hepatology 2008; 48: 1336–41. [DOI] [PubMed] [Google Scholar]
- 121. Rutherford A, Davern T, Hay JE, Murray NG, Hassanein T, Lee WM, Chung RT, Acute Liver Failure Study Group . Influence of high body mass index on outcome in acute liver failure. Clin Gastroenterol Hepatol 2006; 4: 1544–9. [DOI] [PubMed] [Google Scholar]
- 122. Radosevich JJ, Patanwala AE, Erstad BL. Hepatotoxicity in obese versus nonobese patients with acetaminophen poisoning who are treated with intravenous n‐acetylcysteine. Am J Ther 2013. doi:10.1097.mjt.0000434043.62372.00. [DOI] [PubMed] [Google Scholar]
- 123. George J, Murray M, Byth K, Farrell GC. Differential alterations of cytochrome P450 proteins in livers from patients with severe chronic liver disease. Hepatology 1995; 21: 120–8. [PubMed] [Google Scholar]
- 124. Hardwick RN, Ferreira DW, More VR, Lake AD, Lu Z, Manautou JE, Slitt AL, Cherrington NJ. Altered UDP‐glucuronosyltransferase and sulfotransferase expression and function during progressive stages of human nonalcoholic fatty liver disease. Drug Metab Dispos 2013; 41: 554–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Kirby GM, Batist G, Alpert L, Lamoureux E, Cameron RG, Alaoui‐Jamali MA. Overexpression of cytochrome P‐450 isoforms involved in aflatoxin B1 bioactivation in human liver with cirrhosis and hepatitis. Toxicol Pathol 1996; 24: 458–67. [DOI] [PubMed] [Google Scholar]
- 126. Bechtel YC, Haffen E, Lelouet H, Brientini MP, Paintaud G, Miguet JP, Bechtel PR. Relationship between the severity of alcoholic liver cirrhosis and the metabolism of caffeine in 226 patients. Int J Clin Pharmacol Ther 2000; 38: 467–75. [DOI] [PubMed] [Google Scholar]
- 127. Lelouet H, Bechtel YC, Paintaud G, Brientini MP, Miguet JP, Bechtel PR. Caffeine metabolism in a group of 67 patients with primary biliary cirrhosis. Int J Clin Pharmacol Ther 2001; 39: 25–32. [DOI] [PubMed] [Google Scholar]
- 128. Fisher CD, Lickteig AJ, Augustine LM, Ranger‐Moore J, Jackson JP, Ferguson SS, Cherrington NJ. Hepatic cytochrome P450 enzyme alterations in humans with progressive stages of nonalcoholic fatty liver disease. Drug Metab Dispos 2009; 37: 2087–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Frye RF, Zgheib NK, Matzke GR, Chaves‐Gnecco D, Rabinovitz M, Shaikh OS, Branch RA. Liver disease selectively modulates cytochrome P450‐mediated metabolism. Clin Pharmacol Ther 2006; 80: 235–45. [DOI] [PubMed] [Google Scholar]
- 130. Zand R, Nelson SD, Slattery JT, Thummel KE, Kalhorn TF, Adams SP, Wright JM. Inhibition and induction of cytochrome P4502E1‐catalyzed oxidation by isoniazid in humans. Clin Pharmacol Ther 1993; 54: 142–9. [DOI] [PubMed] [Google Scholar]
- 131. Epstein MM, Nelson SD, Slattery JT, Kalhorn TF, Wall RA, Wright JM. Inhibition of the metabolism of paracetamol by isoniazid. Br J Clin Pharmacol 1991; 31: 139–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Toes MJ, Jones AL, Prescott L. Drug interactions with paracetamol. Am J Ther 2005; 12: 56–66. [DOI] [PubMed] [Google Scholar]
- 133. Anderson GD. A mechanistic approach to antiepileptic drug interactions. Ann Pharmacother 1998; 32: 554–63. [DOI] [PubMed] [Google Scholar]
- 134.Ofirmev(R) (acetaminophen) Injection product information. Mallinckrodt Pharmaceuticals; 2014.
- 135. Miners JO, Penhall R, Robson RA, Birkett DJ. Comparison of paracetamol metabolism in young adult and elderly males. Eur J Clin Pharmacol 1988; 35: 157–60. [DOI] [PubMed] [Google Scholar]
- 136. Wynne HA, Cope LH, Herd B, Rawlins MD, James OF, Woodhouse KW. The association of age and frailty with paracetamol conjugation in man. Age Ageing 1990; 19: 419–24. [DOI] [PubMed] [Google Scholar]
- 137. Bannwarth B, Pehourcq F, Lagrange F, Matoga M, Maury S, Palisson M, Le Bars M. Single and multiple dose pharmacokinetics of acetaminophen (paracetamol) in polymedicated very old patients with rheumatic pain. J Rheumatol 2001; 28: 182–4. [PubMed] [Google Scholar]
- 138. Liukas A, Kuusniemi K, Aantaa R, Virolainen P, Niemi M, Neuvonen PJ, Olkkola KT. Pharmacokinetics of intravenous paracetamol in elderly patients. Clin Pharmacokinet 2011; 50: 121–9. [DOI] [PubMed] [Google Scholar]
- 139. Barshop NJ, Capparelli EV, Sirlin CB, Schwimmer JB, Lavine JE. Acetaminophen pharmacokinetics in children with nonalcoholic fatty liver disease. J Pediatr Gastroenterol Nutr 2011; 52: 198–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Williams CM, Maher CG, Latimer J, McLachlan AJ, Hancock MJ, Day RO, Lin CW. Efficacy of paracetamol for acute low‐back pain: a double‐blind, randomised controlled trial. Lancet 2014; 384: 1586–96. [DOI] [PubMed] [Google Scholar]