

Abstract
Aims
Ibrutinib, an inhibitor of Bruton's tyrosine kinase, is used in the treatment of mantle cell lymphoma or chronic lymphocytic leukaemia. Ibrutinib undergoes extensive rapid oxidative metabolism mediated by cytochrome P450 3A both at the level of first pass and clearance, which might result in low oral bioavailability. The present study was designed to investigate the absolute bioavailability (F) of ibrutinib in the fasting and fed state and assess the effect of grapefruit juice (GFJ) on the systemic exposure of ibrutinib in order to determine the fraction escaping the gut (Fg) and the fraction escaping hepatic extraction (Fh) in the fed state.
Methods
All participants received treatment A [560 mg oral ibrutinib, under fasting conditions], B (560 mg PO ibrutinib, fed, administered after drinking glucose drink) and C (140 mg oral ibrutinib, fed, with intake of GFJ before dosing). A single intravenous (i.v.) dose of 100 μg 13C6‐ibrutinib was administered 2 h after each oral dose.
Results
The estimated ‘F’ for treatments A, B and C was 3.9%, 8.4% and 15.9%, respectively. Fg and Fh in the fed state were 47.0% and 15.9%, respectively. Adverse events were mild to moderate in severity (Grade 1–2) and resolved without sequelae by the end of the study.
Conclusion
The absolute oral bioavailability of ibrutinib was low, ranging from 3.9% in the fasting state to 8.4% when administered 30 min before a standard breakfast without GFJ and 15.9% with GFJ. Ibrutinib was well tolerated following a single oral and i.v. dose, under both fasted and fed conditions and regardless of GFJ intake status.
Keywords: absolute bioavailability, Bruton's tyrosine kinase, CYP3A, grape fruit juice, ibrutinib, stable isotope‐labelled microdosing
What is Already Known About This Subject
The microdose approach for absolute bioavailability is already known, but usually carried out using a 14C microdose. Stable isotope‐labelled microdoses have been applied before but reports on their use have been published by only one company.
What This Study Adds
Determination of the bioavailability (F), gut bioavailability (Fg) and hepatic bioavailability (Fh) of ibrutinib
Use of an innovative study design by simultaneous administration of an i.v. stable isotope‐labelled microdose and an oral unlabelled dose
Use of an alternative approach for the calculation of Fh, by using GFJ to block gut metabolism as the hepatic clearance of ibrutinib is similar to the rate of blood flow through the liver.
Introduction
Chronic lymphocytic leukaemia (CLL) is the most common leukaemia in adults in Western countries. In participants with relapsed or refractory disease or in the elderly, because of the advanced age at onset, there is a need for tolerable effective treatment options for CLL 1, 2. Dysregulated Bruton's tyrosine kinase (BTK) leads to the maintenance and expansion of B‐cell malignancies, including CLL 3, 4, 5. Ibrutinib is a covalently binding inhibitor of BTK 6. It has been approved for treating patients with mantle cell lymphoma (MCL) and for treating patients with CLL 7, 8, 9, 10.
In vitro studies have shown that ibrutinib is metabolized primarily by the cytochrome P450 (CYP) family of enzymes, specifically CYP3A enzymes 6. Results from an earlier study in healthy participants demonstrated a ≥ 24‐fold mean increase in ibrutinib exposure following coadministration with the CYP3A inhibitor ketoconazole 11. Furthermore, as ibrutinib is not a permeability glycoprotein (P‐gp) substrate, it is confirmed that the increase in ibrutinib exposure observed when coadministered with ketoconazole was based on CYP3A4‐dependent interaction. This underscores that ibrutinib is a high‐extraction drug with blood flow‐limited clearance (CL) and a high first‐pass extraction 6. In a mass‐balance study, complete oral absorption of ibrutinib under fasting conditions was confirmed 12. In previous studies, it was observed that the administration of ibrutinib under fasting conditions resulted in approximately 60% of exposure [area under the plasma concentration–time curve (AUC) from time 0 to the time of the last quantifiable concentrations (AUClast)] compared with its administration either 30 min before or 2 h after a meal 13. Similar observations were made for propranolol, metoprolol, diprafenone and budesonide, the latter drug being exclusively cleared through CYP3A‐mediated metabolism 14, 15.
Several studies have examined the effect of grapefruit juice (GFJ) and its constituents on CYP3A‐mediated drug metabolism. It has been shown that the elevation in drug exposure that occurs with GFJ ingestion is the result of an irreversible inhibition of CYP3A activity by furanocoumarins present in the juice 16, 17, 18, 19, 20, 21, 22. The GFJ furanocoumarins inhibit CYP3A in the enterocyte cells lining the small intestine but do not significantly inhibit hepatic CYP3A enzyme activity (single‐strength GFJ). Therefore, to differentiate between gut and hepatic first‐pass metabolism of ibrutinib, single‐strength GFJ was used in the present study.
The traditional approach for the determination of the absolute bioavailability (F) of an orally dosed compound uses a crossover study design for oral and intravenous (i.v.) administration. This approach requires the development of an i.v. formulation, and thus for i.v. animal toxicology studies to be carried out to assess the risk associated with exposing healthy participants to a new drug product via a route not previously studied 23. An alternative approach to determining F that does not require additional i.v. animal toxicology studies is the use of a radio isotope‐labelled or stable isotope‐labelled (SIL) microdose (≤100 μg) for the i.v. component of the study 24. In this approach, the linearity of pharmacokinetics (PK) following a microdose relative to an oral therapeutic dose is ascertained by administering the i.v. microdose at the time taken (tmax) to reach peak level (Cmax) of the (unlabelled) oral dose 24. This approach is feasible as the body cannot distinguish between ibrutinib molecules originating from the i.v. microdose and the oral therapeutic dose when dosed at the same time, and therefore ‘sees’ the i.v. microdose as a therapeutic dose with identical PK characteristics as the oral therapeutic dose. The microdose approach requires an analytical method with a high sensitivity. 14C radioisotopes can be measured with high sensitivity using accelerated mass spectrometry (AMS) 24. Owing to significant improvements that have been made in the sensitivity of triple quadrupole mass spectrometry instrumentation over the past decade, it is now also possible to analyse nonradioactive drugs with high sensitivity, down to the low or sub‐pg ml–1 concentration range 25, 26. This makes it possible to apply the microdosing approach by administering a SIL drug rather than a 14C‐labelled drug and to use triple quadrupole mass spectrometry for the quantification of both the SIL‐labelled and unlabelled drug. Therefore, use of a SIL as the microdose was selected and was applied in the current study.
Methods
Participants
Healthy men and women (18–55 years of age, body mass index 18–30 kg m–2, body weight ≥ 50 kg) were included in the study. Women included in the study were postmenopausal or surgically sterile. Individuals were excluded from participating if they had a clinically relevant history of (or current) medical illness or other clinically relevant clinical laboratory, physical examination, vital sign or electrocardiogram (ECG) abnormalities, as determined by the investigator. Additionally, participants who had received an experimental drug or used an experimental medical device within 1 month or a period of less than ten times the drug's half‐life, whichever was longer, before the first dose of the study drug were excluded.
The study protocol and amendment(s) (EudraCT number: 2013–000 963–96) were reviewed and approved by the Ethics Committee of University Hospital, Antwerp (approved on 13 May 2013). The present study was conducted in accordance with the ethical principles originating in the Declaration of Helsinki and that are consistent with good clinical practices and applicable regulatory requirements. All participants provided their written consent to participate in the study.
Study design
This was an open‐label, single‐centre, sequential and two‐way crossover PK study, designed to assess the value of F for oral ibrutinib and the effect of GFJ on the PK of ibrutinib in healthy participants. The participants were screened within 21 days (day −21 to −2) prior to drug administration. Eligible participants were admitted to the study centre on Day −1. The treatments were as follows: treatment A: ibrutinib: 560 mg administered as four capsules (140 mg each) (fasting); treatment B: ibrutinib, 30 min after drinking 240 ml of a simple sugar drink (10% w/v glucose in water) to match the caloric intake in the arm with GFJ intake and followed by a standard breakfast. In treatments B and C, considered as the fed condition, breakfast was provided 30 min after dosing and had to be consumed within 20 min. In treatment C, participants drank 240 ml of GFJ on the evening before, and again 30 min before dosing. The intake of one glass of single‐strength GFJ on the evening before drug intake and on the day of dosing is known to block intestinal CYP3A metabolism completely, although this GFJ dose is sufficiently low to avoid any effect on hepatic CYP3A activity 31, 32. Hence, single‐strength GFJ was added to treatment C to differentiate the first‐pass effect caused by the liver from that taking place in the intestinal wall. Ibrutinib 140 mg (one capsule) was administered and followed by a standard breakfast 30 min after dosing. All participants received treatment A in period 1, and were randomized to treatments B and C in crossover fashion in periods 2 and 3. Thus, participants received treatments in either sequence A‐B‐C or sequence A‐C‐B.
The participants fasted overnight for at least 10 h prior to oral dosing in each period. Water was permitted until 1 h before oral dosing and again 2 h after oral dosing. Lunch and subsequent standard meals were to be provided, beginning 4 h after the oral dose. All capsules were administered with 240 ml of noncarbonated water.
A single i.v. dose of 100 μg 13C6‐ibrutinib was administered (bolus) 2 h after each oral dose (following the 2‐h PK sample). To the extent possible, participants were to remain seated throughout the morning (i.e. from 30 min before dosing until after lunch). Blood samples for the analysis of ibrutinib and its dihydrodiol metabolite PCI‐45227 and for the analysis of 13C6‐ibrutinib concentrations in the plasma were collected before dosing and over 72 h after dosing. Participants were released from the study centre after the 72‐h PK sample had been taken on Day 4 and safety procedures had been observed, and returned 3 days later.
Safety assessments were carried out from the time of consent until the end of the study, and included physical examination, ECGs, the reporting of adverse events (AEs), vital signs and clinical laboratory results. A follow‐up visit approximately 10 (±2) days after the last dose was performed to measure the white blood cell (WBC) count with differential, and to capture any additional AEs and concomitant medications used by the participants.
Prohibited and concomitant therapy
Throughout the study, prescription or nonprescription medication (including vitamins and herbal supplements) other than the study drug were prohibited, except for paracetamol (allowed until 3 days before the first study drug administration). A maximum of three doses per day of 500 mg, and no more than 3 g per week, paracetamol were allowed for the treatment of headache or other pain conditions.
Participants were not permitted to consume food or beverages containing alcohol, GFJ, Seville oranges or quinine (e.g. tonic water) from 24 h (72 h in the case of GFJ and Seville oranges) before each PK sample collection day, until after the last PK sample was collected in each period. GFJ was permitted as planned in the study, in treatment C only.
Treatment
The test drug ibrutinib was supplied as size ‘0’ hard gelatin capsules containing 140 mg of ibrutinib. All formulation excipients were compendial (commonly used in oral formulations). The reference drug 13C6‐ibrutinib was supplied as 0.1 mg ml–1 in water for injection.
Blood sampling
Blood samples for the determination of ibrutinib and PCI‐45227 concentrations in the plasma were collected at predose, and 30 min, 1 h and 1.5 h after the oral dose, and for the determination of ibrutinib, PCI‐45227 and 13C6‐ibrutinib they were collected at 2 h (i.e. just prior to the i.v. dose), 2 h and 2 min, 5 min, 10 min, 15 min and 30 min; then at 3 h, 3.5 h, 4 h, 5 h, 6 h, 8 h, 12 h, 16 h, 24 h, 36 h, 48 h and 72 h after the oral dose.
Pharmacokinetic parameters
All PK parameters were calculated using validated Phoenix WinNonlin® software versions 5.2 and 6.3 (Pharsight Corp, Certara, L.P., St Louis, MO, USA). The following plasma PK parameters were estimated for each participant using the actual times of blood sampling: Cmax, the plasma concentration following i.v. administration, extrapolated back to time zero (C0), tmax, AUC from time 0 to 22 h (AUC22), AUC from time 0 to 24 h (AUC24), AUClast, AUC from time 0 to infinite time (AUC∞), percentage of AUC∞ obtained by extrapolation (%AUC∞,ex), elimination half‐life associated with the terminal slope (λz) of the semi‐logarithmic drug concentration–time curve (t½,term), F, the fraction escaping the gut (Fg) and the fraction escaping hepatic first‐pass extraction (Fh), total CL and volume of distribution based on the terminal phase (Vdz) after i.v. administration. Fg was calculated as [AUCoral∞, (Treatment B)/D (Treatment B)]/[AUCoral∞ (Treatment C)/D (Treatment C)]×100. Fh was calculated as F in treatment C, as further detailed in the results section.
Synthesis and release of 13C6‐ibrutinib
SIL‐13C6‐ibrutinib was obtained through a small‐scale (2 g), multistep synthesis. The drug substance was released under Good Manufacturing Practice (GMP) procedures according to the guidance of exploratory clinical trials 33 with a focus on clinical aspects, whereas the synthesis was not done under GMP. The commercially available stable labelled 13C6 phenol was used as critical starting material to incorporate the desired isotopes.
Bioanalytical procedures
Unlabelled ibrutinib and PCI‐45227
Plasma concentrations of unlabelled ibrutinib and metabolite PCI‐45227 were determined using a validated liquid chromatography–tandem mass spectrometry (LC‐MS/MS) assay with a quantification range of 0.5–100 ng ml–1 for both ibrutinib and PCI‐45227 (Supporting Information).
13C6‐ibrutinib
Plasma concentrations of 13C6‐ibrutinib were determined using an ultrasensitive validated LC‐MS/MS method with a quantification range of 2–1000 pg ml–1, using 13C6‐ibrutinib as an internal standard (Supporting Information).
Naringenin, bergamottin, dihydroxybergamottin, GF‐I‐1 and GF‐I‐4
Naringenin, GF‐I‐1, bergamottin (GF‐I‐2), 6,7‐dihydroxybergamottin and GF‐I‐4 in GFJ are known or presumed to inhibit CYP3A 27. An analytical Liquid Chromatography with Ultraviolet detection and tandem Mass Spectrometric detection (LC‐UV‐MS/MS) method was set up to quantify these five analytes in GFJ. Naringenin was quantified using its LC‐MS/MS response and the other analytes were quantified using their response on Liquid Chromatography with Ultraviolet detection (LC‐UV). As no reference standards were available for GF‐I‐1 and GF‐I‐4, these compounds were quantified using an LC‐UV calibration curve for GF‐I‐2, assuming a similar UV response factor for GF‐I‐1, GF‐I‐4 and GF‐I‐2. GFJ was purchased from 20 different suppliers at local supermarkets and analysed for all five constituents. The GFJ brand with the highest concentration of the analysed compounds (Albert Heijn pink GFJ) was selected and used in the study. Concentrations of naringenin, GF‐I‐2, 6,7‐dihydroxybergamottin, GF‐I‐1 and GF‐I‐4 were found to be 66 ng ml–1, 20 500 ng ml–1, 20 600 ng ml–1, 2230 ng ml–1 and <100 ng ml–1, respectively. In comparison, published concentrations of GFJ constituents as determined in 28–58 different GFJs were1600–7300 ng ml–1 and 200–7700 ng ml–1 for bergamottin and 6,7‐dihydroxy bergamottin, respectively 28, and 321 ± 95 ng ml–1 and 296 ± 85 ng ml–1 for GF‐I‐1 and GF‐I‐4, respectively 29. The stability of the analysed compounds in GFJ for 16 days in the refrigerator was demonstrated by analysis of the used GFJ sample before and after the dosing period.
Safety evaluations
Safety assessments consisted of monitoring and recording AEs and serious AEs; vital signs; ECG; haematology; clinical chemistry; urinalysis; and other protocol‐specified tests that were deemed critical to the safety evaluation of the study drug. The severity of AEs/serious AEs was graded according to the National Cancer Institute – Common Terminology Criteria for Adverse Events (NCI‐CTCAE) grading system version 4.03. Clinical laboratory toxicity grading was based on NCI‐CTCAE, version 4.03 criteria.
The population evaluated for safety included all participants who received at least one dose of study drug and provided postbaseline safety data.
Statistical analysis
Sample size
Assuming an estimated intraparticipant coefficient of variation (CV) of 41% for ibrutinib AUC, a sample size of eight participants was considered sufficient for the ratio of dose‐normalized mean AUCs of ibrutinib for the oral formulation relative to the i.v. formulation to fall within 68% and 148% of the true value with 90% confidence.
Pharmacokinetic analyses
The graphic method and descriptive statistics method were used to summarize plasma ibrutinib, PCI‐45227 and 13C6‐ibrutinib concentrations at each sampling time point and for PK parameters.
The inferential statistical method was applied to construct the 90% confidence interval (CI) for geometric mean ratios (GMRs) relevant to F and the effect of GFJ on the systemic exposure of ibrutinib. Prior to analysis, all AUCs were log‐transformed, and the AUCs of the i.v. formulation were dose‐normalized to 560 mg. For each log‐transformed dose‐normalized PK parameter, a mixed‐effects model, with treatment as a fixed effect and participant as a random effect, was used to estimate the least‐squares means (LSMs) and intraparticipant variance. Using these estimated LSM and intraparticipant variance values, the point estimate and 90% CIs for the difference in means on a log scale were constructed. By using antilogarithm transformation, GMRs and the associated 90% CIs of the AUCs were constructed separately between following treatments per treatment period: ibrutinib oral capsule (treatment A) vs. the i.v. formulation (reference); ibrutinib oral capsule without GFJ (treatment B) vs. the i.v. formulation; ibrutinib oral capsule with GFJ (treatment C) vs. the i.v. formulation.
To evaluate the effect of the CYP3A inhibitor GFJ on ibrutinib absorption in the presence of food, exploratory statistical analysis was performed on the PK parameters Cmax, AUC24, AUClast and AUC∞ from treatment groups B and C for the ibrutinib oral capsule and i.v. formulations. Only the data from participants who completed both treatments was included in the statistical analysis. If one of the PK parameters of interest was not estimable for a given participant for one of the formulations, the participant's data was not included in the statistical analysis of that particular PK parameter. By using antilogarithmic transformation, the GMRs and the 90% CIs of PK parameters were constructed for the following treatments: ibrutinib oral capsule with GFJ (treatment C) vs. oral capsule without GFJ (treatment B); the i.v. formulation with GFJ (treatment C) vs. the i.v. formulation without GFJ (treatment B).
For each AE, the percentages of participants who experienced at least one occurrence of the given event were summarized by treatment group.
Results
The study was conducted between 5 July 2013 and 19 August 2013. All of the eight enrolled participants completed the study and were white [3 (37.5%) men] with median (range) age 49 (34–55) years, weight 69.7 (range 50.7–90.6) kg, height 170 (range 160–189) cm and body mass index 23.0 (range 19.6–27.4) kg m–2. No participant reported prior medication use. Two participants received concomitant therapy (diclofenac for the treatment of epicondylitis lateralis, and paracetamol for back injury).
PK
All eight enrolled participants were included in the PK analysis. Following oral administration of 560 mg ibrutinib and i.v. administration of 100 μg 13C6‐ibrutinib under fasting conditions, the mean plasma concentration–time profiles were much more variable following oral administration compared with i.v. administration (Figure 1). A secondary peak was observed approximately 4 h following oral administration under fasting conditions (treatment A). The secondary peak seen with treatment A was not observed in either of the ‘fed’ treatments (B and C) (Figures 2A and 2B). The mean concentrations for treatment A were lower than those for the other treatments (Table 1). The dose‐normalized Cmax and AUC∞ for ibrutinib were approximately 3.5‐fold and twofold higher, respectively, when administered with GFJ than when administered without GFJ (treatments C and B; Table 2). The apparent CL (total CL/F) and half‐life both decreased by half for the oral route when participants were pretreated with GFJ as compared with controls (treatment B). The intravenous CL, however, was unchanged by GFJ. Under fed conditions, the estimated CL was higher than under fasting conditions (Table 1).
Figure 1.
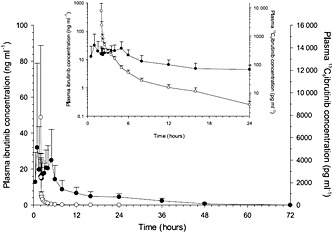
Mean (standard deviation) (n = 8) linear‐linear and logarithmic‐linear (insert) plasma concentration–time profile of ibrutinib following oral administration of 560 mg ibrutinib and intravenous (i.v.) administration of 100 μg 13C6‐ibrutinib.  Treatment A ibrutinib 560 mg (n = 8, oral);
Treatment A ibrutinib 560 mg (n = 8, oral);  Treatment A 13C6 ibrutinib 100 μg (n = 8, i.v.)
Treatment A 13C6 ibrutinib 100 μg (n = 8, i.v.)
Figure 2.
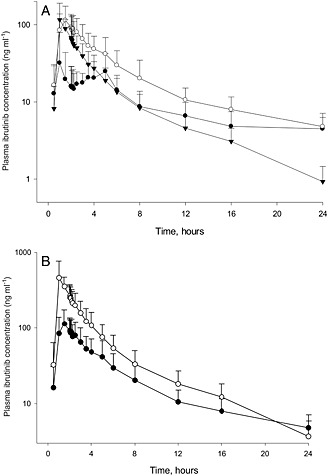
The mean plasma concentration‐time profiles of ibrutinib following (n=8) A. Oral administration of 560 mg ibrutinib under fasted (  Treatment A) and fed (
Treatment A) and fed (  Treatment B) conditions, and 140 mg ibrutinib under fed condition with grapefruit juice (
Treatment B) conditions, and 140 mg ibrutinib under fed condition with grapefruit juice (  Treatment C). B. Ibrutinib following oral administration of 560 mg ibrutinib without grapefruit juice (
Treatment C). B. Ibrutinib following oral administration of 560 mg ibrutinib without grapefruit juice (  Treatment B) and dose‐normalized ibrutinib with grapefruit juice (
Treatment B) and dose‐normalized ibrutinib with grapefruit juice (  Treatment C).
Treatment C).
Table 1.
Summary of ibrutinib pharmacokinetic parameters after three different treatments using a combination of a single oral and intravenous (i.v.) dose in eight healthy participants
| Mean (SD) | ||||||
|---|---|---|---|---|---|---|
| Oral | i.v. | |||||
| Treatment A | Treatment B | Treatment C | Treatment A | Treatment B | Treatment C | |
| Ibrutinib 560 mg (N = 8) | Ibrutinib 560 mg without GFJ (N = 8) | Ibrutinib 140 mg with GFJ (N = 8) | 13C6PCI‐32 765 100 μg (N = 8) | 13C6PCI‐32 765 100 μg (N = 8) | 13C6PCI‐32 765 100 μg (N = 8) | |
| tmax, h * | 3.75 (0.5–5.02) | 1.77 (1.5–5.0) | 1.5 (1.0–1.5) | 0.03 (0.03–0.08) | 0.03 (0.03–0.1) | 0.03 (0.03–0.08) |
| Cmax, ng ml–1 † | 45.2 (43.3) | 128 (45.6) | 125 (67.5) | 8.54 (5.39) | 8.11 (5.78) | 7.69 (6.29) |
| C0, ng ml–1 † | NA | NA | NA | 40.9 (33.8)† | 25.0 (18.7)† | 26.0 (22.4)§ |
| AUC24, ng h ml–1 § | 229 (107) | 544 (161) | 339 (96.6)† | 1.64 (0.18)‡ | 1.41 (0.35)†, ‡ | 1.44 (0.37)‡ |
| AUClast, ng h ml–1 § | 289 (117) | 606 (160) | 325 (103) | 1.64 (0.18) | 1.41 (0.33) | 1.44 (0.37) |
| AUC∞, ng h ml–1 § | 368 (80.1)§ | 659 (132)† | 334 (105) | 1.64 (0.17)† | 1.36 (0.25)† | 1.47 (0.37) |
| t1/2,term, h | 12.8 (4.90)§ | 9.51 (4.06)† | 5.83 (2.20) | 5.91 (1.38)† | 6.01 (2.21)† | 5.31 (0.80) |
| CL/F, l h–1 | 1572 (318)§ | 875 (144)† | 466 (176) | NA | NA | NA |
| CL, l h–1 | NA | NA | NA | 61.5 (7.00)† | 76.1 (15.6)† | 71.4 (14.2) |
| Vd/F, l | 30 464 (17 148)§ | 12 026 (5783)† | 3757 (1511) | NA | NA | NA |
| Vd, l | NA | NA | NA | 523 (121)† | 683 (375)† | 551 (148) |
AUC24, area under the plasma concentration–time curve from time zero to 24 h; AUC∞, area under the plasma concentration–time curve from time zero to infinite time, calculated as the sum of AUClast and Clast‐‐>/λz; AUClast, area under the plasma concentration–time from time zero to the time of the last quantifiable concentration Clast, the last observed plasma concentration; C0, plasma concentration following i.v. administration, extrapolated back to time zero; CL, total clearance of drug after i.v. administration; CL/F, total clearance of drug after extravascular administration, corrected for F; Cmax, maximum plasma concentration; F, absolute bioavailability; Fg, the fraction escaping the gut; Fh, the fraction escaping first‐pass metabolism in the liver; GFJ, grapefruit juice; NA, not applicable; SD, standard deviation; t1/2, term, terminal half‐life; tmax, time to reach the maximum plasma concentration; Vd, apparent volume of distribution; Vd/F, apparent volume of distribution after extravascular administration, corrected for F.
Median (range);
N = 7;
AUC22;
N = 4.
Table 2.
Geometric mean ratios and 90% confidence intervals (CIs) for comparison of oral capsule and intravenous (i.v.) formulation of ibrutinib, with and without food (treatment B vs. treatment A), and with and without grapefruit juice (treatment C vs. treatment B)
| Parameter* | Test treatment/reference treatment | N | Geometric mean | Ratio: test/reference (%) | 90% CI (%) | Intraparticipant CV (%) |
|---|---|---|---|---|---|---|
| Oral (treatment B vs. treatment A: with vs. without food) | ||||||
| Cmax (ng ml–1) | B | 8 | 121 | 352 | (213, 582) | 62 |
| A | 8 | 34 | ||||
| AUClast (ng h ml–1) | B | 8 | 588 | 223 | (167, 297) | 31 |
| A | 8 | 264 | ||||
| AUC∞ (ng h ml–1) | B | 4 | 677 | 187 | (137, 256) | 19 |
| A | 4 | 362 | ||||
| i.v. (treatment B vs. treatment A: with vs. without food | ||||||
| B | 8 | 5032 | 81 | (37, 176) | 99 | |
| Cmax (pg ml–1) | A | 8 | 6255 | |||
| B | 8 | 1380 | 85 | (72, 100) | 18 | |
| AUClast (pg h ml–1) | A | 8 | 1631 | |||
| B | 6 | 1329 | 82 | (68, 100) | 17 | |
| AUC∞ (pg h ml–1) | A | 6 | 1612 | |||
| Oral (treatment C vs. treatment B: with vs. without GFJ) | ||||||
| Cmax (ng ml–1) | C | 8 | 437 | 360 | (269, 483) | 31 |
| B | 8 | 121 | ||||
| AUClast (ng h ml–1) | C | 8 | 1236 | 210 | (182, 243) | 15 |
| B | 8 | 588 | ||||
| AUC∞ (ng h ml–1) | C | 7 | 1378 | 215 | (184, 250) | 14 |
| B | 7 | 643 | ||||
| i.v. (treatment C vs. treatment B: with vs. without GFJ) | ||||||
| Cmax (pg ml–1) | C | 8 | 5903 | 117 | (58, 238) | 84 |
| B | 8 | 5032 | ||||
| AUClast (pg h ml–1) | C | 8 | 1401 | 102 | (89, 116) | 14 |
| B | 8 | 1380 | ||||
| AUC∞ (pg h ml–1) | C | 7 | 1416 | 104 | (90, 120) | 13 |
| B | 7 | 1363 | ||||
AUC∞, area under the plasma concentration–time curve from time zero to infinite time, calculated as the sum of AUClast and Clast/λz; AUClast, area under the plasma concentration–time curve from time zero to the time of the last quantifiable concentration; Cmax, maximum plasma concentration; CV, coefficient of variation.
A mixed‐effects model with treatment as a fixed effect and subject as a random effect was used for analysis on a log scale and the results were presented at original scale after antilogarithmic transformation.
The traditional way to calculate Fh by CL theory is from the ratio of i.v. CL or hepatic clearance (CLh) to hepatic blood flow 30. However, when CLh is similar to the hepatic blood flow, the calculation could result in a large error for Fh. Therefore, in the present study, an alternative approach for the calculation of Fh was used, using the data from treatment C. For treatment C, Fg was considered as 100%. The fraction absorbed (Fa) was determined to be 100% in the human mass balance study under fasting conditions. Therefore, for treatment C, F = Fa × Fg × Fh reduces to F = Fh, so Fh equals F in treatment C. Using this approach, the fraction escaping first‐pass metabolism in the liver (Fh) under nonfasting conditions was calculated to be 15.9%. Fg was calculated to be 47.0% from the dose‐normalized AUC∞ GMR of treatments B and C.
For all estimations of F, the AUCs were dose normalized to 560 mg. Dose normalization was applied as ibrutinib PK has been shown to be dose independent between doses of 40 mg and 1400 mg. The F for oral dosing, based on the GMR for AUC∞, was 3.9%, 8.4% and 15.9% for treatments A, B and C, respectively (Table 3). The Cmax increased 3.5‐fold and the AUC∞ 2.2‐fold when ibrutinib was ingested with food as compared with fasting conditions (Table 2). The GMRs for Cmax and AUC were slightly but not significantly lower following i.v. administration 1.5 h after a meal compared with fasting conditions. The AUC∞ and Cmax following i.v. administration under fed conditions in the presence or absence of GFJ were not different but the 90% CI around the treatment ratios was very wide.
Table 3.
Geometric mean ratio and 90% CI for comparison of oral capsule vs. i.v. formulation of ibrutinib (treatment A ‐fasted, treatment B‐ fed without grapefruit juice, and treatment C ‐ fed with grapefruit juice) (pharmacokinetics data analysis set, dose‐normalized to 560 mg)
| Parameter* | Ibrutinib test treatment/reference treatment | N | Geometric mean | Ratio: test/reference (%) | 90% confidence interval (%) | Intra‐participant CV (%) |
|---|---|---|---|---|---|---|
| Treatment A | ||||||
| AUClast (ng h ml–1) | Oral | 8 | 264 | 2.9 | (2.12, 3.94) | 36.5 |
| i.v. | 8 | 9134 | ||||
| AUC∞ (ng h ml–1) | Oral | 3 | 330 | 3.9 | (3.06, 5.02) | 10.4 |
| i.v. | 3 | 8421 | ||||
| Treatment B | ||||||
| AUClast (ng h ml–1) | Oral | 8 | 588 | 7.6 | (6.41, 9.03) | 18.2 |
| i.v. | 8 | 7726 | ||||
| AUC∞ (ng h ml–1) | Oral | 6 | 666 | 8.4 | (7.32, 9.68) | 12.1 |
| i.v. | 6 | 7918 | ||||
| Treatment C | ||||||
| AUClast (ng h ml–1) | Oral | 8 | 1236 | 15.8 | (11.93, 20.79) | 29.9 |
| i.v. | 8 | 7847 | ||||
| AUC∞ (ng h ml–1) | Oral | 8 | 1270 | 15.9 | (12.07, 20.89) | 29.6 |
| i.v. | 8 | 8001 |
AUC∞ = area under the plasma concentration‐time curve from time zero to infinite time, calculated as the sum of AUClast and Clast/λz; AUClast = area under the plasma concentration‐time curve from time zero to the time of the last quantifiable concentration;
AUC22
Formation of the metabolite PCI‐45227 was delayed when ibrutinib was administered under fasting conditions. Thereafter, the distribution and elimination phases were similar to those under fed conditions (Figure 3A). The dose‐normalized PCI‐45227 concentrations following GFJ (treatment C) were comparable with concentrations without GFJ (treatment B) (Figure 3B). PCI‐45227 exposure (Cmax and AUC) increased approximately 1.6‐fold when ibrutinib was administered with food (treatment B) compared with fasting conditions (treatment A) and tmax occurred sooner with food (Table 4). With the addition of GFJ, the Cmax metabolite‐to‐parent ratio decreased from 1.03 to 0.32. The dose‐normalized AUC was slightly (~10%) lower in the presence of GFJ. The tmax and half‐life appeared to be slightly lower in the presence of GFJ compared with food alone. Metabolite‐to‐parent ratios for Cmax and AUC showed a decreasing trend when ibrutinib was administered in the fasting vs. fed vs. fed with GFJ conditions.
Figure 3.
Mean (SD) (n=8) plasma concentration‐time profile of PCI‐45227 following oral administration of A) 560 mg ibrutinib without grapefruit juice and 140 mg ibrutinib with grapefruit juice (  Treatment A: Ibrutinib 560 mg, fasted (n = 8);
Treatment A: Ibrutinib 560 mg, fasted (n = 8);  Treatment B: Ibrutinib 560 mg, fed without grapefruit juice (n = 8);
Treatment B: Ibrutinib 560 mg, fed without grapefruit juice (n = 8);  Treatment C: Ibrutinib 140 mg, fed with grapefruit juice (n = 8). B) 560 mg ibrutinib without grapefruit juice and dose‐normalized ibrutinib with grapefruit juice (
Treatment C: Ibrutinib 140 mg, fed with grapefruit juice (n = 8). B) 560 mg ibrutinib without grapefruit juice and dose‐normalized ibrutinib with grapefruit juice ( Treatment A: Ibrutinib 560 mg, fasted (n = 8);
Treatment A: Ibrutinib 560 mg, fasted (n = 8);  Treatment B: Ibrutinib 560 mg, fed without grapefruit juice (n = 8);
Treatment B: Ibrutinib 560 mg, fed without grapefruit juice (n = 8); 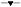 Treatment C: Ibrutinib dose‐normalized to 560 mg, fed with grapefruit juice (n = 8)
Treatment C: Ibrutinib dose‐normalized to 560 mg, fed with grapefruit juice (n = 8)
Table 4.
Summary of PCI‐45227 pharmacokinetic parameters after three different treatments using a single oral dose in eight healthy participants
| Mean (SD) | |||
|---|---|---|---|
| Treatment A Oral ibrutinib 560 mg, fasted (N = 8) | Treatment B Oral ibrutinib 560 mg without GFJ (N = 8) | Treatment C Oral ibrutinib 140 mg with GFJ (N = 8) | |
| Cmax, ng ml–1 | 84.3 (28.3) | 135 (42.0) | 33.9 (6.41) |
| Cmax ratio * | 2.41 (1.12) | 1.03 (0.31) | 0.32 (0.15) |
| tmax, h † | 4.5 (1.0–5.0) | 2.1 (1.5–8.0) | 1.5 (1.0–5.0) |
| AUC24, ng h ml–1 | 677 (152) | 1058 (386) | 252 (90.7) |
| AUC24 ratio * | 3.22 (1.35) | 1.87 (0.61) | 0.74 (0.31)‡ |
| AUClast, ng h ml–1 | 833 (192) | 1236 (414) | 274 (107) |
| AUClast ratio * | 3.09 (1.32) | 1.95 (0.67) | 0.84 (0.35) |
| AUC∞, ng h ml–1 | 854 (198) | 1257 (419) | 283 (108) |
| AUC∞ ratio * | 2.66 (0.83)‡ | 1.92 (0.72)§ | 0.84 (0.34) |
| t½, term, h | 11.4 (3.01) | 10.4 (2.32) | 7.45 (1.53) |
λz, terminal slope of the semi‐logarithmic drug; AUC24, area under the plasma concentration–time curve from time zero to 24 h; AUC∞, area under the plasma concentration–time curve from time zero to infinite time, calculated as the sum of AUClast and Clast/λz; AUClast, area under the plasma concentration–time curve from time zero to the time of the last quantifiable concentration; Cmax, maximum plasma concentration; GFJ, grapefruit juice; SD, standard deviation; t1/2, term, terminal half‐life; tmax, time to reach the maximum plasma concentration.
Ratio of metabolite/parent;
median (range);
N = 4;
N = 7.
Safety
Of the eight participants, three experienced ≥1 AE which all resolved without sequelae by the end of the study. None of the participants reported an AE of Grade ≥3. Two participants experienced three AEs of Grade 2 severity (one participant a experienced worsening of pre‐existing epicondylitis lateralis, and was treated with topical application of diclofenac gel, while the other had abdominal cramps (which the investigator considered to be probably related to the study drug) and worsening of pre‐existing back pain, and was treated with paracetamol; the investigator considered neither the epicondylitis lateralis nor the back pain to be related to the study drug) and all other AEs were of Grade 1 severity. The most common AEs (≥3 participants) reported were Grade 1 abdominal cramps and Grade 1 diarrhoea, each reported by three participants. Two participants had AEs considered to be at least possibly related to ibrutinib, including lightheadedness (n = 2), abdominal cramps (n = 2), diarrhoea (n = 1), nausea (n = 1) and hyperventilation (n = 1). There were no changes in laboratory parameters for haematology, chemistry or coagulation, or in the platelet function assay, that were deemed by the investigator to be clinically significant in relation to the administration of ibrutinib.
Discussion and conclusions
The current study used oral dosing with simultaneous microdosing of a stable isotope (13C6) of ibrutinib to determine F for ibrutinib. Under fasting conditions, F was 3.9%, using the GMR of the estimated AUC∞. A combination of food and GFJ pretreatment increased F to 15.9%. On average, 47.0% of orally ingested ibrutinib escaped first‐pass metabolism at the level of the small intestine (Fg) when the drug was taken with food. F under fed conditions increased by twofold to 8.4%. The fraction escaping liver CL on first pass (Fh) was estimated to be 15.9%.
AUC∞ is estimated using AUClast and extrapolation to infinity by using the estimated t½. Under fasting conditions, there is some uncertainty about the determination of AUC∞ because the observed secondary rise in concentration at around 4 h after drug intake hampered accurate t½ estimation. This secondary rise was not observed following an i.v. administration, or when ibrutinib was given 30 min prior to a standard breakfast. Therefore, F was also calculated using AUClast to evaluate if the estimate had sufficient certainty on the basis of the data from four participants. On the basis of AUClast, an alternative F was estimated at 2.9%, which is in good agreement with the AUC∞‐based F of 3.9%. For comparison, the AUClast‐ and AUC∞‐based F values in the nonfasting condition were 7.6% and 8.4%, respectively (Table 3). In addition, as ibrutinib is a high CL drug and has a high volume of distribution, it was difficult to estimate a C0 on the basis of i.v. concentration–time curves. Such estimation might also introduce some uncertainty around the accuracy of AUC estimations and thus might affect the determination of F. Even though there may remain some uncertainty around the F estimation, the low F value found is in good agreement with the observed significant effect of ketoconazole coadministration increasing ibrutinib Cmax and AUC by 29‐fold and 24‐fold, respectively 11. Finally, in spite of high interparticipant variability, the observed mean AUC in treatment A (fasting condition) was consistent with the AUCs observed in other clinical pharmacology studies with larger sample sizes, thus supporting the validity of the estimation of the value of F following oral dosing in the present study 11, 13.
The secondary rise in concentration observed under fasting conditions and coinciding with lunchtime (food intake) was also observed in a formal food effect study on ibrutinib 13. Bile secretion from the gallbladder triggered by the lunchtime meal, resulting in acceleration of the solubility of the drug already in the gastrointestinal lumen, is suggested as a possible explanation for this second peak 13. Enterohepatic recycling could be an alternative explanation but is unlikely as ibrutinib is primarily cleared via hepatic metabolism and excreted in the bile as metabolites, and no unchanged drug is found in the bile or faeces 12. The estimated apparent terminal half‐life after oral intake was much longer (~12 h) than after i.v. administration (~6 h), suggesting that the resultant absorption rate was slower than the elimination rate (flip‐flop kinetics) 13.
In the present study, food (i.e. a standard non‐high‐fat breakfast) was added to treatments B and C, at 30 min after administering oral ibrutinib, to mimic more closely the dosing conditions in clinical studies with ibrutinib. F was 8.4% when ibrutinib was administered 30 min before a meal (treatment B). The ~2‐fold increase in F compared with fasting conditions (treatment A) is consistent with another food effect study 13.
Food intake also had an effect on the i.v. concentration–time profile of ibrutinib. The mean CL was approximately 30% higher following i.v. administration 1.5 h after a meal compared with fasting conditions. Interestingly, such an increase in CL is somewhat lower but still comparable with the expected increase in average blood flow upon consumption of a meal, and such a higher blood flow would indeed predict a higher liver CL in the case of a drug with perfusion‐limited CL 14.
A less than fourfold maximum increase in exposure (both Cmax and AUC) by GFJ was predicted by simulations using SimCyp modelling and simulation software (v12). Therefore, a dose of 140 mg ibrutinib was used in treatment C to compensate for the potential impact of CYP3A inhibition, caused by GFJ, on Cmax and AUC. As ibrutinib PK has been shown to be dose independent between doses of 40 mg and 1400 mg, a comparison between treatments B and C was carried out on the basis of dose‐normalized parameters (to 560 mg) 34.
When ibrutinib was administered with GFJ, under fed conditions the dose‐normalized mean Cmax was 3.5‐fold higher and the mean AUC∞ twofold higher as compared with administration under fed conditions without GFJ. In the presence of food with GFJ, the estimated F was 15.9%. Apparent CL (total CL/F) and terminal elimination half‐life both decreased by half when GFJ was administered, indicating a twofold change in F or CL. However, the estimated intravenous CL was similar in the two treatments; therefore, GFJ does not appear to affect overall CL. By comparing the AUC∞ of ibrutinib with and without GFJ, it was estimated that 47.0% of ibrutinib is available after metabolism in the gut (Fg) when taken in combination with food.
When ibrutinib was administered under fasting conditions, formation of the metabolite PCI‐45227 was delayed compared with fed conditions. However, the distribution and elimination phases were similar to those under fed conditions. Dose‐normalized PCI‐45227 concentrations following oral administration of 140 mg ibrutinib with food and GFJ were comparable with concentrations following oral administration of 560 mg ibrutinib with food but without GFJ. The dose‐normalized AUC was similar or slightly (~10%) lower. The half‐life of PCI‐45227 appeared to be slightly lower in the presence of GFJ compared with food alone. However, this might have been an artefact of lower plasma concentrations associated with the lower ibrutinib dose, and hence more samples returning concentrations below the limit of quantitation at later time points. Visual inspection of the PK profiles did not suggest a difference. Metabolite‐to‐parent ratios for Cmax and AUC showed a decreasing trend with treatment A vs. B vs. C, which was in line with the decreased first‐pass metabolism.
In conclusion, F was 3.9% under fasting conditions, 8.4% under fed conditions and 15.9% under fed conditions with GFJ. In the presence of food, GFJ intake increased Cmax by 3.5‐fold and AUC by twofold. Fg and Fh in the fed state were 47.0% and 15.9%, respectively.
Competing Interests
All authors have completed the Unified Competing Interest form at http://www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare: RdV, JS, PH, JJ, JM, DS, JSn, MV, LdZ, EM and JdJ are employees of Janssen Research & Development LLC and hold company stocks. JSu is an employee of Pharmacyclics Inc. There are no financial relationships with any organizations that might have an interest in the submitted work in the previous 3 years and no other relationships or activities that could appear to have influenced the submitted work.
The authors thank the study participants, without whom this study would never have been accomplished. Jelke Backeljau, Luc Diels and Marc Verhemeldonck conducted the bioanalysis of the plasma samples and the grapefruit juice analysis. Dr Sangita P Patil (SIRO Clinpharm Pvt. Ltd.) provided writing assistance for this manuscript and Dr Namit Ghildyal (Janssen Research & Development, LLC) provided editorial support for the development of this manuscript.
Contributors
RdV contributed to bioanalytical method development and validation, and bioanalysis of plasma and grapefruit juice samples. JSm, JJ, JM, DS, JSn, JSu, EM, PH, LdZ and JdJ contributed to study design (protocol), review of analyses and data interpretation. MV contributed to the synthesis and release analysis strategy and execution of SIL drugs. Sofie Deleu MD (Clinical Pharmacology Unit, AZ Jan Palfijn, Merksem, Belgium) was the principal investigator, and was involved in the conduct of the study in the clinical pharmacology unit but was not involved in the clinical pharmacokinetics and bioanalysis of the study, and therefore was not included as an author.
Supporting information
Determination of plasma concentrations of 13C6‐ibrutinib, Ibrutinib and PCI‐45227
Supporting info item
de Vries, R. , Smit, J. W. , Hellemans, P. , Jiao, J. , Murphy, J. , Skee, D. , Snoeys, J. , Sukbuntherng, J. , Vliegen, M. , de Zwart, L. , Mannaert, E. , and de Jong, J. (2016) Stable isotope‐labelled intravenous microdose for absolute bioavailability and effect of grapefruit juice on ibrutinib in healthy adults. Br J Clin Pharmacol, 81: 235–245. doi: 10.1111/bcp.12787.
References
- 1. Brown JR. The treatment of relapsed refractory chronic lymphocytic leukemia. Hematology Am Soc Hematol Educ Program 2011; 2011: 110–8. [DOI] [PubMed] [Google Scholar]
- 2. Shanafelt T. Treatment of older patients with chronic lymphocytic leukemia: key questions and current answers. Hematology Am Soc Hematol Educ Program 2013; 2013: 158–67. [DOI] [PubMed] [Google Scholar]
- 3. Burger JA. Bruton's tyrosine kinase (BTK) inhibitors in clinical trials. Curr Hematol Malig Rep 2014; 9: 44–9. [DOI] [PubMed] [Google Scholar]
- 4. Wiestner A. Emerging role of kinase‐targeted strategies in chronic lymphocytic leukemia. Hematology Am Soc Hematol Educ Program 2012; 2012: 88–96. [DOI] [PubMed] [Google Scholar]
- 5. Woyach JA, Bojnik E, Ruppert AS, Stefanovski MR, Goettl VM, Smucker KA, Smith LL, Dubovsky JA, Towns WH, MacMurray J, Harrington BK, Davis ME, Gobessi S, Laurenti L, Chang BY, Buggy JJ, Efremov DG, Byrd JC, Johnson AJ. Bruton's tyrosine kinase (BTK) function is important to the development and expansion of chronic lymphocytic leukemia (CLL). Blood 2014; 123: 1207–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. IMBRUVICA® prescribing information. Pharmacyclics, Inc. Revised July 2014.
- 7. Advani RH, Buggy JJ, Sharman JP, Smith SM, Boyd TE, Grant B, Kolibaba KS, Furman RR, Rodriguez S, Chang BY, Sukbuntherng J, Izumi R, Hamdy A, Hedrick E, Fowler NH. Bruton tyrosine kinase inhibitor ibrutinib (PCI‐32765) has significant activity in patients with relapsed/refractory B‐cell malignancies. J Clin Oncol 2013; 31: 88–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Burger JA, Keating MJ, Wierda WG, Hartmann E, Hoellenriegel J, Rosin NY, de Weerdt I, Jeyakumar G, Ferrajoli A, Cardenas‐Turanzas M, Lerner S, Jorgensen JL, Nogueras‐Gonzalez GM, Zacharian G, Huang X, Kantarjian H, Garg N, Rosenwald A, O'Brien S. Safety and activity of ibrutinib plus rituximab for patients with high‐risk chronic lymphocytic leukaemia: a single‐arm, phase 2 study. Lancet Oncol 2014; 15: 1090–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Byrd JC, Furman RR, Coutre SE, Flinn IW, Burger JA, Blum KA, Grant B, Sharman JP, Coleman M, Wierda WG, Jones JA, Zhao W, Heerema NA, Johnson AJ, Sukbuntherng J, Chang BY, Clow F, Hedrick E, Buggy JJ, James DF, O'Brien S. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N Engl J Med 2013; 369: 32–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wang ML, Rule S, Martin P, Goy A, Auer R, Kahl BS, Jurczak W, Advani RH, Romaguera JE, Williams ME, Barrientos JC, Chmielowska E, Radford J, Stilgenbauer S, Dreyling M, Jedrzejczak WW, Johnson P, Spurgeon SE, Li L, Zhang L, Newberry K, Ou Z, Cheng N, Fang B, McGreivy J, Clow F, Buggy JJ, Chang BY, Beaupre DM, Kunkel LA, Blum KA. Targeting BTK with ibrutinib in relapsed or refractory mantle‐cell lymphoma. N Engl J Med 2013; 369: 507–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. de Jong J, Skee D, Murphy J, Sukbuntherng J, Hellemans P, Smit J, de Vries R, Jiao J. Effect of CYP3A perpetrators on ibrutinib exposure in normal healthy subjects. Pharma Res Per 2015; 3: e00156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Scheers E, Leclercq L, de Jong J, Bode N, Bockx M, Laenen A. Absorption, metabolism and excretion of oral 14C radiolabeled ibrutinib: an open‐label, phase I, single‐dose study in healthy men submitted to drug metabolism and disposition. Drug Metab Dispos 2015; 43: 289–97. [DOI] [PubMed] [Google Scholar]
- 13. de Jong J, Sukbuntherng J, Skee D, Murphy J, O'Brien S, Byrd JC, James D, Hellemans P, Loury DJ, Jiao J, Chauhan V, Mannaert E. The effect of food on the pharmacokinetics of oral ibrutinib in healthy participants and patients with chronic lymphocytic leukemia. Cancer Chemother Pharmacol 2015; 75: 907–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Marasanapalle VP, Boinpally RR, Zhu H, Grill A, Tang F. Correlation between the systemic clearance of drugs and their food effects in humans. Drug Dev Ind Pharm 2011; 37: 1311–7. [DOI] [PubMed] [Google Scholar]
- 15. Seidegard J, Nyberg L, Borga O. Differentiating mucosal and hepatic metabolism of budesonide by local pretreatment with increasing doses of ketoconazole in the proximal jejunum. Eur J Pharm Sci 2012; 46: 530–6. [DOI] [PubMed] [Google Scholar]
- 16. Greenblatt DJ, von Moltke LL, Harmatz JS, Chen G, Weemhoff JL, Jen C, Kelley CJ, LeDuc BW, Zinny MA. Time course of recovery of cytochrome p450 3A function after single doses of grapefruit juice. Clin Pharmacol Ther 2003; 74: 121–9. [DOI] [PubMed] [Google Scholar]
- 17. Guo LQ, Fukuda K, Ohta T, Yamazoe Y. Role of furanocoumarin derivatives on grapefruit juice‐mediated inhibition of human CYP3A activity. Drug Metab Dispos 2000; 28: 766–71. [PubMed] [Google Scholar]
- 18. Lundahl J, Regardh CG, Edgar B, Johnsson G. Relationship between time of intake of grapefruit juice and its effect on pharmacokinetics and pharmacodynamics of felodipine in healthy subjects. Eur J Clin Pharmacol 1995; 49: 61–7. [DOI] [PubMed] [Google Scholar]
- 19. Paine MF, Widmer WW, Hart HL, Pusek SN, Beavers KL, Criss AB, Brown SS, Thomas BF, Watkins PB. A furanocoumarin‐free grapefruit juice establishes furanocoumarins as the mediators of the grapefruit juice‐felodipine interaction. Am J Clin Nutr 2006; 83: 1097–105. [DOI] [PubMed] [Google Scholar]
- 20. Schmiedlin‐Ren P, Edwards DJ, Fitzsimmons ME, He K, Lown KS, Woster PM, Rahman A, Thummel KE, Fisher JM, Hollenberg PF, Watkins PB. Mechanisms of enhanced oral availability of CYP3A4 substrates by grapefruit constituents. Decreased enterocyte CYP3A4 concentration and mechanism‐based inactivation by furanocoumarins. Drug Metab Dispos 1997; 25: 1228–33. [PubMed] [Google Scholar]
- 21. Seidegard J, Randvall G, Nyberg L, Borga O. Grapefruit juice interaction with oral budesonide: equal effect on immediate‐release and delayed‐release formulations. Pharmazie 2009; 64: 461–5. [PubMed] [Google Scholar]
- 22. Takanaga H, Ohnishi A, Murakami H, Matsuo H, Higuchi S, Urae A, Irie S, Furuie H, Matsukuma K, Kimura M, Kawano K, Orii Y, Tanaka T, Sawada Y. Relationship between time after intake of grapefruit juice and the effect on pharmacokinetics and pharmacodynamics of nisoldipine in healthy subjects. Clin Pharmacol Ther 2000; 67: 201–14. [DOI] [PubMed] [Google Scholar]
- 23. ICH‐M3 . ICH Topic M3 (R2). Non‐clinical safety studies for the conduct of human clinical trials and marketing authorization for pharmaceuticals. International Conference on Harmonization of technical Requirements for Registration of Pharmacueticals for Human Use 2009.
- 24. Boulton DW, Kasichayanula S, Keung CF, Arnold ME, Christopher LJ, Xu XS, Lacreta F. Simultaneous oral therapeutic and intravenous (1)(4)C‐microdoses to determine the absolute oral bioavailability of saxagliptin and dapagliflozin. Br J Clin Pharmacol 2013; 75: 763–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gu H, Wang J, Aubry AF, Jiang H, Zeng J, Easter J, Wang JS, Dockens R, Bifano M, Burrell R, Arnold ME. Calculation and mitigation of isotopic interferences in liquid chromatography‐mass spectrometry/mass spectrometry assays and its application in supporting microdose absolute bioavailability studies. Anal Chem 2012; 84: 4844–50. [DOI] [PubMed] [Google Scholar]
- 26. Jiang H, Zeng J, Li W, Bifano M, Gu H, Titsch C, Easter J, Burrell R, Kandoussi H, Aubry AF, Arnold ME. Practical and efficient strategy for evaluating oral absolute bioavailability with an intravenous microdose of a stable isotopically‐labeled drug using a selected reaction monitoring mass spectrometry assay. Anal Chem 2012; 84: 10031–7. [DOI] [PubMed] [Google Scholar]
- 27. Hanley MJ, Cancalon P, Widmer WW, Greenblatt DJ. The effect of grapefruit juice on drug disposition. Expert Opin Drug Metab Toxicol 2011; 7: 267–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Widmer W, Haun C. Variation in furanocoumarin content and new furanocoumarin dimers in commercial grapefruit (Citrus paradisi Macf.) juices. J Food Sci 2005; 70: C307–12. [Google Scholar]
- 29. Fukuda K, Guo L, Ohashi N, Yoshikawa M, Yamazoe Y. Amounts and variation in grapefruit juice of the main components causing grapefruit–drug interaction. J Chromatogr B Biomed Sci Appl 2000; 741: 195–203. [DOI] [PubMed] [Google Scholar]
- 30. Kato M, Chiba K, Hisaka A, Ishigami M, Kayama M, Mizuno N, Nagata Y, Takakuwa S, Tsukamoto Y, Ueda K, Kusuhara H, Ito K, Sugiyama Y. The intestinal first‐pass metabolism of substrates of CYP3A4 and P‐glycoprotein‐quantitative analysis based on information from the literature. Drug Metab Pharmacokinet 2003; 18: 365–72. [DOI] [PubMed] [Google Scholar]
- 31. Rogers JD, Zhao J, Liu L, Amin RD, Gagliano KD, Porras AG, Blum RA, Wilson MF, Stepanavage M, Vega JM. Grapefruit juice has minimal effects on plasma concentrations of lovastatin‐derived 3‐hydroxy‐3‐methylglutaryl coenzyme A reductase inhibitors. Clin Pharmacol Ther 1999; 66: 358–66. [DOI] [PubMed] [Google Scholar]
- 32. Veronese ML, Gillen LP, Burke JP, Dorval EP, Hauck WW, Pequignot E, Waldman SA, Greenberg HE. Exposure‐dependent inhibition of intestinal and hepatic CYP3A4 in vivo by grapefruit juice. J Clin Pharmacol 2003; 43: 831–9. [DOI] [PubMed] [Google Scholar]
- 33.Guidance to the conduct of exploratory (phase 0) trials in Belgium (FAGG, version 2, January 2012) [online]. Available at http://www.fagg‐afmps.be (last accessed 24 June 2015).
- 34. Marostica E, Sukbuntherng J, Loury D, de Jong J, de Trixhe XW, Vermeulen A, De Nicolao G, O'Brien S, Byrd JC, Advani R, McGreivy J, Poggesi I. Population pharmacokinetic model of ibrutinib, a Bruton tyrosine kinase inhibitor, in patients with B cell malignancies. Cancer Chemother Pharmacol 2015; 75: 111–21. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Determination of plasma concentrations of 13C6‐ibrutinib, Ibrutinib and PCI‐45227
Supporting info item