

Abstract
Glioblastoma is a brain neoplasm with limited 5‐year survival rates. Developments of new treatment regimens that improve patient survival in patients with glioblastoma are needed. It is likely that a number of existing drugs used in other conditions have potential anticancer effects that offer significant survival benefit to glioblastoma patients. Identification of such drugs could provide a novel treatment paradigm.
Keywords: antidepressants, antiepileptic drugs, antihypertensive, beta‐blockers, glioblastoma, statins
Glioblastoma: current knowledge of therapeutics
Glioblastoma (GBM) is the most common and most lethal form of primary brain tumour worldwide 1. Most often, GBM is diagnosed at between 45 and 70 years of age 2. Currently, the standard treatment for patients with GBM involves tumour resection followed by radiotherapy and administration of a chemotherapeutic agent, temozolomide (TMZ). In spite of current treatment regimens, survival remains poor; median survival time ranges from 12 to 16 months 2, 3. To date, no additional treatment strategies that prolong overall survival (OS) further have been identified 4. Developments of new treatment regimens that improve patient survival in patients with GBM are needed. It is likely that a number of existing drugs used to treat other conditions have potential anticancer effects that offer significant survival benefit to GBM patients. Identification of such drugs could provide a novel treatment paradigm.
One significant limitation to the standard treatment protocol is tumour cell resistance to TMZ. O6‐methylguanine methyltransferase (MGMT) is a DNA repair protein which confers cellular resistance to TMZ 5. Spontaneous methylation, and subsequent silencing, of the MGMT gene promoter occurs in up to 50% of patients with GBM and has been associated with a significant increase in OS compared to patients with unmethylated MGMT promoters 6. For patients with unmethylated MGMT gene promoters, who may not respond well to standard treatment, there are currently no alternative treatment options. Furthermore, given that survival in all patients remains poor, more effective regimens are required.
In order to consider pharmacological agents that might have potential in the treatment of GBM, a thorough knowledge of GBM cancer biology is required. In summary, it is well documented that the growth and pathogenesis of GBM is driven by various genetic and epigenetic aberrations 7, 8, 9, 10. A number of recent studies have reported on the heterogeneous nature of GBM 4, 8, 10. Given this, it is probable that within a single tumour there are subpopulations of cells with different genetic alterations and potentially different susceptibilities to chemotherapeutic agents 11. It is unlikely that a ‘single‐hit’ treatment strategy will provide significant benefit to many patients diagnosed with GBM. The administration of several therapeutic agents will allow clinicians to target a range of GBM phenotypes and, as such, therapy involving multiple adjuvants might provide more benefit than standard chemotherapy with TMZ alone.
Two of the main pathological features of GBM include increased cellular proliferation and angiogenesis (Figure 1) 8. Increased cellular proliferation is a hallmark of neoplasia and in the context of GBM probably results from aberrant growth factor signalling, inactivation of tumour suppressor pathways (such as the p53 and retinoblastoma pathways), upregulation of the phosphatidyl inositol 3‐kinase (PI3K) pathway 8 and epidermal growth factor receptor (EGFR) gene mutations 10. In GBM, extensive angiogenesis might be attributed to elevated levels of vascular endothelial growth factor (VEGF) and might also involve fibroblast growth factor (FGF), nerve growth factor (NGF) and angiotensin II 12, 13.
Figure 1.
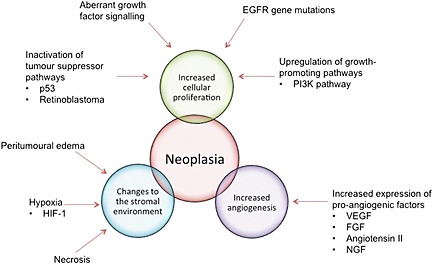
The main pathological features of glioblastoma. EGFR, epidermal growth factor receptor; FGF, fibroblast growth factor; HIF‐1, hypoxia‐inducible factor 1; PI3K, phosphatidyl‐inosotol‐3 kinase; NGF, nerve growth factor; VEGF, vascular endothelial growth factor
Additional pathological features of GBM include changes to the stromal environment such as necrosis, hypoxia and peritumoural oedema (Figure 1) 9, 14. A number of studies have reported that hypoxia might stimulate angiogenesis and increase vascularity through the upregulation of hypoxia‐inducible factor 1 (HIF‐1) 15, 16. Furthermore, hypoxic tumour cells are known to be resistant to radiotherapy, and this might also be attributed to the upregulation of HIF‐1 17, 18, 19. There is evidence in the literature that hypoxia might also confer tumour cell resistance to chemotherapy 20, 21. Several studies have shown that peritumoural oedema and necrosis in GBM have been associated with poor patient survival 9, 14.
At present, there is a pressing need to explore alternate treatment options which could improve OS and quality of life in patients with GBM. Given this, the principal aim of the present review was to identify existing drugs or therapeutic agents already registered or used in previous clinical trials that might impair one or more of the pathological processes of gliomagenesis outlined above (these will be referred to as antineoplastic drugs or antineoplastic agents), and therefore be helpful to consider in a clinical trial development portfolio.
Method
A literature search was conducted using the PubMed database. MeSH search terms and subheadings included glioma; GBM; brain neoplasms, treatment; therapeutics, combined drug therapy; therapeutics, blood–brain barrier; and pharmacology, therapeutic use. The search was restricted to primary research articles published in English. Studies were selected for review based on the inclusion of a drug that crosses the blood–brain barrier (BBB), that is primarily prescribed for noncancer indications and that might have antineoplastic actions. The ability of a drug or its active metabolite to cross the BBB and reach the tumour is paramount in the treatment of brain neoplasms. For the purpose of the present review, the ability of a drug to cross the BBB was determined using clinical pharmacology and therapeutics principles and clinical experience; drugs known to act in the central nervous system (CNS) or to elicit side effects in the CNS were included.
Exploration of the existing literature revealed a number of drug classes that might improve survival in patients with GBM. We restricted the present review to include drugs that are currently marketed, widely used in clinical practice and with favourable side‐effect profiles. This is because there is a short time to clinical use, and there is little point in developing drugs that cannot be used because of a life‐threatening side‐effect profile. In particular, we focused the present review on several agents within the following categories, with the belief that these are the most promising targets for future research: histone deacetylase inhibitors (HDACi), drugs that inhibit neurotransmitters, beta‐adrenoceptor antagonists (beta‐blockers), statins and antihypertensive medications. A total of 45 studies were included in the present review. It is important to note that this was not a comprehensive review of all drugs that might be useful in the treatment of GBM. Additional drug classes that might improve patient survival include antimalarial drugs, antiviral drugs, drugs used in the treatment of diabetes and anti‐inflammatory medications; however, these were not included in the present review. These additional drug classes are reviewed briefly in the conclusions.
Drugs that inhibit neurotransmitters
Psychotropic agents are well known to influence neurotransmission and have more recently been shown to have antiproliferative effects that might be valuable in the treatment of neoplasms 22, 23. Several subclasses of psychotropic drugs, including tricyclic antidepressants (TCAs), selective serotonin reuptake inhibitors (SSRIs), phenothiazines and antiepileptic drugs (AEDs; these will be reviewed later), have been investigated as potential adjuvants for cancer therapy 7, 22, 23, 24. The TCAs act at presynaptic adrenoceptors and serotonin receptors in the CNS to block the reuptake of noradrenaline and serotonin, respectively. SSRIs are more specific and act only on presynaptic serotonin receptors to prevent serotonin reuptake. Phenothiazines are dopamine receptor antagonists traditionally indicated for use in the treatment of psychosis and chemotherapy‐induced emesis.
Recently, TCAs (amitriptyline, imipramine, clomipramine, doxepin and citalopram) and SSRIs (paroxetine, fluoxetine and sertraline) have been reported to reduce tumour cell proliferation in a number of cell lines, including rat C6 glioma, human neuroblastoma and human astrocytoma cell lines 22, 24, 25, 26. Reduced cellular proliferation might occur through several mechanisms. Two recent studies showed an increase in caspase‐3 activity (a cysteine protease involved in the induction of the apoptotic cascade) in response to treatment with TCAs and SSRIs 22, 25. Contrary to this hypothesis, Tzadok et al. 26 demonstrated decreased cell proliferation in response to antidepressant treatment, with an absence of increased caspase‐3 activity, and hypothesized the involvement of the mitogen‐activated protein kinase pathway. A recent in vivo study by Walker et al. 24 investigated TCA use and mortality in patients with glioma or colorectal cancer. In a sample of 2592 patients with glioma and 22 524 patients with colorectal cancer, 4.1–4.2% of patients reported the use of low‐dose TCAs for the period of at least two prescriptions; however, there was no significant reduction in mortality associated with the use of TCAs in patients with glioma or colorectal cancer.
Several studies have reported on the potential antineoplastic activity of dopamine receptor antagonists, including chlorpromazine, thioridazine and perphenazine 23, 26, 27. Tzadok et al. 26 found a significant additive antiproliferative effect following treatment of U87 glioma cells with TMZ and perphenazine. Perphenazine crosses the BBB and has a strong predilection for the blockage of D2 and D3 dopamine receptors. These receptors have been implicated in gliomagenesis through stimulation of neuronal stem cells within the subventricular zone (SVZ) in the lateral ventricles 28. Blockade of D2 and D3 receptors in the SVZ might improve survival in GBM 28. Yde et al. 27 reported increased sensitivity of breast cancer cells to a first‐line therapeutic agent, tamoxifen, with adjuvant chlorpromazine therapy. Furthermore, Sachlos et al. 23 demonstrated that treatment with thioridazine reduced proliferation in acute myeloid leukaemia cell lines, potentially through selective targeting of cancer stem cells. To the best of our knowledge, there is no existing research regarding the efficacy of chlorpromazine and thioridazine as adjuvant anticancer agents in GBM.
AEDs
Epileptic seizures are a common complication of GBM, occurring in 22–60% of patients, and often treatment with an AED such as valproate (VPA), phenytoin, carbamazepine or levetiracetam (LEV) is required to provide symptomatic relief 29. A large number of studies have found a potential benefit of HDACi such as VPA in the treatment of GBM 30, 31, 32 Furthermore, other AEDs have also been hypothesized to offer a survival benefit in patients with glioma 33, 34, 35. HDACi and other AEDs are reviewed below.
HDACi
VPA has a wide spectrum of antiseizure activity and has recently been suggested as a potential adjuvant in cancer treatment 36. It is known to act on gamma‐butyric acid (GABA) levels in the brain, to block voltage‐gated sodium, potassium and calcium channels and to inhibit histone deacetylases 36. Abnormal activation of histone deacetylases can induce neoplasia; deacetylation of histones promotes the condensation of chromatin and repression of the transcription of several genes. HDACi can trigger hyperacetylation of histones, promoting growth arrest and apoptosis 31, 35, 37. Furthermore, inhibition of chromatin condensation might increase the availability of target DNA to alkylating chemotherapeutic agents, and enhance therapy 32, 38.
A number of recent studies have reported the antineoplastic effects of VPA using human glioma cell lines 31, 32, 38. Adjunctive treatment of glioma cell lines with VPA provided an additive effect on inhibition of cellular proliferation following treatment with TMZ. Furthermore, treatment with VPA has been shown to increase tumour cell sensitivity to radiotherapy 31, 32, 38. The mechanisms by which VPA might enhance the cytotoxic effects of TMZ and radiation are unknown. In addition, it has been reported that VPA might increase cellular levels of the MGMT protein 30, 38. In spite of this, VPA is still widely acknowledged to have an antiproliferative effect in glioma cells.
In addition to VPA, some of the newer AEDs, including topiramate (TPM) and the major active metabolite of LEV, 2‐pyrrolidinone‐n‐butyric acid (PBA), might act as HDACi 37. LEV is a newer AED, traditionally indicated for use as monotherapy in the treatment of partial focal seizures. Bobustuc et al. 30 found that treatment with LEV + TMZ significantly reduced cellular proliferation in several GBM cell lines. Furthermore, LEV was reported to have an inhibitory effect on MGMT protein expression in GBM cell lines but not in normal human astrocyte cells. It was hypothesized that increased levels of the p53 protein in cells treated with LEV enhanced p53 binding and inhibition of the MGMT gene promoter, consequently downregulating expression of the MGMT protein and enhancing tumour cell sensitivity to TMZ 30.
TPM is a newer AED, with several mechanisms of action that might contribute to antiseizure effects 39. TPM, in addition to VPA, is classified as a category D drug in pregnancy, owing to an increased risk of fetal malformation 40, 41. Two studies have demonstrated potential antimetastatic and antiangiogenic effects of TPM in mice injected with Lewis lung carcinoma 42, 43. In the earlier study, Ma et al. 42 demonstrated that treatment with TPM for 20 days strongly inhibited tumour metastasis in a dose‐dependent manner. More recently, these authors 43 reported that treatment with TPM for 21 days resulted in a significant reduction of VEGF expression, suggesting that TPM might effectively decrease tumour angiogenesis. It is unknown whether TPM would have similar effects in glioma cells.
Other AEDs
Enzyme‐inducing AEDs (EIAEDs), including carbamazepine and phenytoin, potently induce components of the cytochrome P450 system in the liver and consequently enhance the metabolism of chemotherapeutic agents and other drugs, including steroids that are metabolized by the same system. As such, concomitant treatment with EIAEDs might increase the dosage requirements of chemotherapeutic agents and have an effect on patient outcomes 33, 34, 35, and this is an important consideration in GBM pharmacotherapy.
Several studies have reported on the use of EIAEDs and non‐EIAEDs in patients with GBM 7, 33, 34. Jaeckle et al. 33 investigated the use of AEDs in 620 patients with GBM; 432 patients received treatment with an EIAED, 14 were prescribed a non‐EIAED and 159 did not receive any AED. The median OS in the patients who were prescribed an EIAED was 12.3 months compared to 10.7 months for patients in the other two groups. By contrast, a previous study by Oberndorfer et al. 34 found that, within a retrospective cohort of 168 patients with GBM (of whom 37 were prescribed a non‐EIAED and 43 were prescribed an EIAED), OS was 13.7 months for patients who were prescribed a non‐EIAED compared to 10.8 months in patients prescribed an EIAED. More recently, a study by Weller et al. 35 reported on the use of AEDs in a retrospective cohort of 573 patients with GBM, of whom 110 patients were treated with a non‐EIAED (predominantly VPA) and 277 patients received treatment with an EIAED. These authors found a significant survival benefit for patients treated with VPA compared to those taking other AEDs (including EIAEDs) or no AED 35. A similar retrospective study of 544 patients with primary GBM by Barker et al. 7 reported on the use of AEDs (including phenytoin, LEV, carbamazepine, phenobarbital and VPA) in 403 patients. The median OS of patients in the study was 14 months, irrespective of treatment modality. The median OS in patients treated with VPA was 16.9 months, and in those treated with radiation therapy + TMZ + VPA was 23.9 months, compared to 15.9 months without VPA. However, only a small number (n = 29) of patients were treated with VPA. Recently, there has been a large focus on VPA as a first‐line AED in the treatment of GBM.
Statins
Statins are traditionally prescribed to treat hypercholesterolaemia. More recently, statins have been indicated as potential adjunctive treatments in several cancers, owing to inhibitory effects on cellular signalling pathways involved in proliferation, migration, invasion and induction of apoptosis 44, 45, 46, 47. The primary mechanism by which statins act is inhibition of 3‐hydroxy‐3‐methylglutaryl‐coenzyme A (HMG‐CoA) reductase, preventing the conversion of HMG‐CoA to mevalonate 48. Important downstream products of mevalonate include cholesterol, Ras, Raf and Rho 48, 49. Gabrys et al. 48 discovered that growth in glioma cells might be highly dependent on the mevalonate pathway. As such, this pathway is a significant target for potential antineoplastic therapies.
Several recent in vitro studies have found statins to have proapoptotic and cytotoxic effects on glioma and other cancer cells 50, 51. Tapia‐Perez et al. 50 demonstrated statin‐mediated toxicity in human glioma cell lines U87, U138 and LN405, and in one rat glioma cell line, RGII, treated with simvastatin and lovastatin. This observed cytotoxicity was likely to have been due to stimulation of the proapoptotic protein, Bim. By contrast, Wood et al. 51 found an increase in levels of Bcl 2 (an antiapoptotic protein) in rats exposed to chronic administration of simvastatin (50 mg kg–1 day–1 for 21 days). In humans, the highest daily dose of simvastatin would equate to approximately 0.07 mg kg–1 day–1, much lower than the dosage used in the study by Wood et al. 51.
Additional in vitro studies have reported on the antiproliferative effects of statins on several cancer cell lines 46, 48, 49. Yanae et al. 46 investigated the treatment of rat C6 glioma cells and human‐derived U251MG GBM cells with mevastatin, fluvastatin and simvastatin. In this study, 5 μM of mevastatin and simvastatin, and 2.5 μM of fluvastatin inhibited cell proliferation and induced apoptosis in both cell lines. Yanae et al. 46 stated that the peak plasma concentration of fluvastatin in humans is less than 2 μM. Similarly, Wu et al. 49 found a 35% reduction in cellular proliferation in U87 and U251 human GBM cells treated with simvastatin compared to control cells. Gabrys et al. 48 demonstrated that lovastatin (0.625–50 μM) inhibited proliferation in U87MG and FaDu (hypopharyngeal squamous cell carcinoma) cell lines in a dose‐dependent manner. Mechanisms contributing to the inhibitory effects of statins on cellular proliferation might include inhibition of cell cycle progression as a result of decreased cellular signalling 48, 49. Furthermore, a study by Yongjun et al. 47 reported decreased cell migration in a U87 glioma cell line in response to treatment with atorvastatin, probably due to inhibition of matrix metalloproteinase (MMP) 2).
Antihypertensive drugs
Antihypertensive agents include angiotensin‐converting enzyme inhibitors (ACEIs), angiotensin receptor antagonists/blockers (ARBs) and beta‐blockers (these will be reviewed later) and are typically prescribed to treat hypertension, congestive heart failure and diabetic nephropathy. ACEIs reduce the production of angiotensin II, whereas ARBs selectively inhibit the activation of the angiotensin II type 1 receptor 52. Angiotensin II plays an important role in the regulation of fluid and electrolyte balance and is also involved in cell growth, differentiation, apoptosis and angiogenesis 53, 54. Existing research has hypothesized an antineoplastic role for ACEIs and ARBs, primarily due to the inhibition of angiotensin II 53, 55.
Rivera et al. 55 investigated the effects of treatment with an ARB, losartan, in 12‐week‐old rats implanted with rat C6 glioma cells and found a significant decrease in cell proliferation and a decrease in the number of capillary vessels in tumours resected from rats treated with either 40 mg kg–1 or 80 mg kg–1 once daily compared to controls. A more recent study from the same group reported a significant decrease in tumour volume following 30 days of treatment with either 40 mg kg–1 or 80 mg kg–1 losartan once daily compared to controls 53. Furthermore, Arrieta et al. 53 showed increased apoptosis and decreased levels of the proangiogenic factors VEGF, platelet‐derived growth factor and FGF in rat C6 glioma cell lines treated with losartan compared to controls. A study by Rooprai et al. 56 demonstrated downregulation of MMP‐2 and MMP‐9 in four cell lines (an ependymoma, an oligoastrocytoma, an anaplastic astrocytoma and a GBM cell line) in response to treatment with the ACEi captopril. MMPs are thought to play a role in the degradation and remodelling of the extracellular matrix and might be critically involved in GBM metastasis and invasion; as such, captopril might have potential anti‐invasive effects 57.
A number of retrospective analyses have investigated the impact of antihypertensive treatment on patient outcome in several cancers 12, 52, 58, 59, 60. Cardwell et al. 58 reported on the use of ACEIs and/or ARBs after diagnosis with either breast, colorectal or prostate cancer in over 20 000 patients in the UK. This study found a small, nonsignificant reduction in cancer‐specific mortality in colorectal and prostate cancer patients treated with an ACEI. Overall, there was no significant impact on all‐cause mortality for patients treated with either an ACEI or ARB. Holmes et al. 59 found that treatment with ACEIs, ARBs and/or beta‐blockers had no significant survival benefit in a sample of over 3000 Canadian patients with either breast, colorectal, lung or prostate cancer. Chae et al. 12 investigated the use of antihypertensive agents in 1449 patients with breast cancer. This study found no significant decrease in OS for patients prescribed antihypertensive medications, although treatment with ARBs was associated with a decreased risk of recurrence compared to controls. A smaller study by Engineer et al. 52 showed that combination treatment with a beta‐blocker and either an ACEI or ARB decreased progression and decreased hospitalizations over a 4‐year period in a sample of 262 patients with colorectal cancer compared to patients taking either a beta‐blocker or an ACEI/ARB. One study investigating the effect of treatment with ACEIs/ARBs in GBM demonstrated no significant survival benefit 60. Interestingly, Carpentier et al. 60 reported a significant reduction in preoperative oedema (as visualized on magnetic resonance imaging) in patients treated with either an ARB/ACEI compared to controls, probably due to a reduction in neovascularization that normally enables the development of extensive peritumoural oedema. Furthermore, treatment with an ACEI/ARB resulted in a significant reduction in steroid treatment requirements.
Doxazosin is an apha‐1 adrenoceptor antagonist that has been used to treat hypertension and benign prostatic hyperplasia. A recent study by Petty et al. 61 found that doxazosin inhibited the migration of A172 glioma cells in a dose‐dependent manner. In the same study, mice were implanted with prostate cancer cells and subjected to either treatment with doxazosin for 10 days or to a control treatment. The authors demonstrated a significant reduction in the number and size of lung metastases in treated mice compared to controls. They hypothesized that doxazosin might cause activation of the erythropoietin‐producing hepatocellular A2 family of receptor tyrosine kinases involved in the induction of apoptosis, and inhibition of proliferation and migration.
Beta‐blockers
Beta‐blockers are commonly prescribed in the treatment of hypertension and in prevention of cardiovascular diseases 62, 63. Studies have suggested that these agents might inhibit angiogenesis, invasion and cellular proliferation, as well as increasing apoptosis in several cancer cell lines 64, 65, 66. The most extensively studied beta‐blocker in relation to potential utility as an adjunctive treatment in neoplasia is propranolol, a nonselective beta‐blocker that competes with adrenaline and noradrenaline binding at beta‐1‐ and ‐2 adrenoceptor sites 62, 63. Evidence in the existing literature suggests that blockade of adrenergic stimulation by beta‐blockers might decrease the expression of proangiogenic factors, including VEGF, MMP‐2 and MMP‐9; however, the mechanism by which this occurs is largely unknown 62, 67, 68. Chim et al. 16 hypothesized that decreased levels of hypoxia‐inducible factor 1 associated with propranolol treatment caused a decrease in VEGF levels in haemangioma endothelial cells.
Pasquier et al. 64 investigated the use of propranolol in several cell lines, including human breast cancer, neuroblastoma and U87 GBM cell lines. In this study, treatment with propranolol resulted in decreased proliferation in all cell lines compared to controls. Results were not reported for antiangiogenic effects of propranolol in the U87 GBM cell line. A subsequent study by Pasquier et al. 65 examined the effect of a number of beta‐blockers, including metoprolol, carvedilol, nebivolol, propranolol, butoxamine and atenolol, on two neuroblastoma cell lines. The mixed alpha/beta‐receptor antagonist, carvedilol; the selective beta‐1 adrenoceptor antagonist, nebivolol; and propranolol were reported to have significant antineoplastic effects involving the induction of apoptosis and increased cell cycle arrest compared to controls. Currently, there is little evidence in the literature to support the use of selective or mixed beta‐blockers over propranolol as an adjunct in cancer treatment.
The mechanism by which beta‐adrenoceptor antagonists might promote apoptosis is not yet understood. Kozanoglu et al. 67 reported that treatment with propranolol resulted in increased expression of the proapoptotic gene Bcl‐10, decreased expression of the antiapoptotic gene Bcl‐2 and decreased activation of the nuclear factor‐kappa B (NF‐kappa B) pathway in human U266 multiple myeloma cells. NF‐kappa B is a transcription factor critically involved in many cellular processes, and excessive activation of the NF‐kappa B pathway has been linked to the promotion of cellular proliferation and inhibition of apoptosis 69.
Conclusions
GBM is a relatively rare cancer, with an incidence of 2–4 per 100 000 adults per year worldwide that is unlikely to receive significant research funding or support. Given that drug development is associated with considerable time and financial costs to the pharmaceutical industry, research teams, governments and patients, alternative strategies are required to develop more efficient and timely therapeutic protocols. The ability to use currently marketed drugs in the treatment of GBM provides a financially practicable approach to encourage the development of new therapeutic regimens to improve OS in patients with GBM.
We have demonstrated in the present review that a number of drugs currently in routine use, including antidepressants, AEDs, statins, beta‐blockers and other antihypertensive agents, exhibit promising antineoplastic effects in vitro. We believe that these drug classes offer the most promising survival benefit in GBM. Many of the drugs presented here have been shown to antagonize one or more of the pathological features of GBM. Furthermore, a number of drugs, including VPA, may potentiate the effects of standard radiotherapy and chemotherapy treatments. It is important to note that the drugs presented here do not comprise an exhaustive list of potential therapeutic agents for patients with GBM. Table 1 presents a summary of the indication for use, mechanism of action and potential antineoplastic effects of the afore‐mentioned drugs.
Table 1.
Existing drugs with potential antineoplastic effects as demonstrated through in vitro studies.
| Class Drug | Primary indications for use | Primary mechanism of action | Mechanism of antineoplastic effects | References |
|---|---|---|---|---|
| Tricyclic antidepressants | Major depression | Inhibit reuptake of noradrenaline and serotonin at presynaptic nerve terminals | Reduce cellular proliferation and might induce apoptosis through aberrant MAPK pathway activity or inhibition of mitochondrial activity | Levkovitz et al. 22, Higgins and Pilkington 25, Tzadok et al. 26 |
| Amitriptyline | Neuropathic pain | |||
| Imipramine | ||||
| Migraine prophylaxis | ||||
| Clomipramine | ||||
| Doxepin | ||||
| Citalopram | ||||
| Selective serotonin reuptake inhibitors | Major depression | Inhibit reuptake of serotonin at presynaptic nerve terminals | Reduce cellular proliferation and might induce apoptosis through aberrant MAPK pathway activity or inhibition of mitochondrial activity | Levkovitz et al. 22, Higgins and Pilkington 25, Tzadok et al. 26 |
| Bipolar disorder | ||||
| Anxiety disorders | ||||
| Paroxetine | ||||
| Fluoxetine | ||||
| Sertraline | Bulimia nervosa | |||
| Phenothiazines | Chemotherapy‐induced emesis | Dopamine receptor antagonists | Might decrease cellular proliferation and increase cellular sensitivity to some chemotherapeutic agents | Sachlos et al. 23, Tzadok et al. 26, Yde et al. 27 |
| Chlorpromazine | ||||
| Thioridazine | ||||
| Perphenazine | ||||
| Valproic acid (valproate) | Seizure disorders | Acts on GABA levels in the brain to reduce voltage‐gated sodium, potassium and calcium channels | Acts as a histone deacetylase inhibitor and might contribute to chromatin condensation, growth arrest and apoptosis | Camphausen et al. 31, Chinnaiyan et al. 32, van Nifterik et al. 38 |
| Bipolar disorder | ||||
| Migraine prophylaxis | ||||
| Levetiracetam | Partial focal seizures | Unknown | Might act as a histone deacetylase inhibitor, contributing to chromatin condensation, growth arrest and apoptosis | Bobustuc et al. 30 |
| Adjunct in tonic–clonic and myoclonic seizures | ||||
| Statins | Hypercholesterolaemia | Inhibition of HMG‐CoA reductase, the rate‐limiting enzyme in the cholesterol synthesis pathway | Decreased activity in cell signalling pathways contributes to induction of apoptosis and decreased proliferation | Yanae et al. 46, Yongjun et al. 47, Gabrys et al. 48, Wu et al. 49, Tapia‐Perez et al. 50 |
| Lovastatin | ||||
| Pravastatin | Prevention of cardiovascular disease | |||
| Rosuvastatin | ||||
| Simvastatin | ||||
| Glaucoma | ||||
| Angiotensin‐converting enzyme inhibitors | Hypertension | Decrease production of angiotensin II by inhibition of angiotensin converting‐enzyme | Decreased proliferation and angiogenesis. Might also decrease invasion and migration via reduction of MMP‐2 and MMP‐9 expression | Rooprai et al. 56 |
| Diabetic nephropathy | ||||
| Captopril | ||||
| Angiotensin receptor blockers | Hypertension | Antagonists at angiotensin II type 1 receptors | Decreased angiogenesis as a result of decreased VEGF expression | Arrieta et al. 53, Rivera et al. 55 |
| Diabetic nephropathy | ||||
| Losartan | ||||
| Other antihypertensives | Hypertension | Antagonist at alpha‐1 adrenoceptors | Reduction of cell migration, proliferation and apoptosis through activation of Ephrin A2 receptors | Petty et al. 61 |
| Benign prostatic hyperplasia | ||||
| Doxazosin | ||||
| Beta‐blockers | Hypertension | Antagonists at beta‐1 and/or beta‐2 adrenoceptors | Reduction of angiogenesis through decreased expression of VEGF and MMP‐9. Also decreases cell proliferation through unknown mechanisms | Kozanoglu et al. 67, Pasquier et al. 64 |
| Propranolol * | Prevention of cardiovascular disease | |||
| Butoxamine | ||||
| Metoprolol | ||||
| Nebivolol |
GABA, gamma‐aminobutyric acid; HMG‐CoA, 3‐hydroxy‐3‐methylglutaryl‐coenzyme A; MAPK, mitogen‐activated protein kinase; MMP, matrix metalloproteinase; VEGF, vascular endothelial growth factor.
The majority of studies reporting on anti‐neoplastic effects of beta‐blockers involved only propranolol.
Recent research has identified additional classes of drugs that might have survival benefit in GBM. These drug classes have been excluded from the present review owing to poor side‐effect profiles and/or limited available data. Nonsteroidal anti‐inflammatory drugs – in particular, celecoxib – have been investigated as adjuvants in glioma therapy owing to their ability to enhance radiosensitivity in glioma cell lines; however, celecoxib was recently shown to have no survival benefit in combination with TMZ and thalidomide in a Phase II study 70, 71. Drugs used in the treatment of diabetes, including metformin and pioglitazone, might inhibit glioma cell growth 72, 73. A number of studies have reported that the antimalarial drug, chloroquine, may enhance the cytotoxic effects of TMZ in glioma cell lines 74, 75. Disulfuram, a drug used in the maintenance of alcohol abstinence, has been demonstrated to reverse tumour cell resistance to TMZ 76. Antiviral therapy with acyclovir and valganciclovir has also been hypothesized to offer a survival benefit in GBM 77, 78. Furthermore, it is likely that a number of older drugs that have been withdrawn from use might also exhibit antineoplastic effects, especially in combination with radiotherapy.
As evident in the current literature, the majority of studies reporting on the possible antineoplastic effects of existing therapeutic agents are limited to in vitro investigations, in cell lines with known differences and a lack of heterogeneity in gene expression and activity against human cancers. Further, the concentration of drug used during in vitro experiments often exceeds by several‐fold the in vivo concentration achieved with the therapeutic dosage of a given drug. It is uncertain whether antineoplastic effects reported from in vitro studies would be observed during in vivo trials. The majority of human studies to date have been limited to retrospective analyses. In these cases, researchers are unable to investigate the effects of different dosages of drugs or different phenotypic variables, and research is limited when only a small number of patients are prescribed any given drug.
There have been few studies reporting on the concurrent use of more than one class of drug for the treatment of GBM and cancer cell lines. Multidrug therapy introduces many challenges, including the management of drug interactions and the cumulative risk of additional adverse effects. In the case of GBM, where survival remains extremely poor, it is likely that the therapeutic benefits of multidrug adjunctive therapy and the known safety profile of these older drugs would greatly outweigh any therapeutic risk.
Currently, new therapeutic regimens are required to improve OS in patients with GBM. Given the evidence presented in the present review, future randomized controlled trials incorporating existing agents with potential antineoplastic effects into current therapy could demonstrate an enhancement of the treatment protocol. However, in spite of their limitations, preliminary in vitro and retrospective in vivo studies to determine effective and appropriate dosing, as well as effective drug combinations, are required. Furthermore, a retrospective in vivo study of patients with GBM, correlating OS and progression‐free survival with individual patients' prescription medication, should be carried out to provide more information on the potential survival benefit obtained from adjunctive treatment with existing drugs.
Conflict of Interest/Disclosure
All authors have completed the Unified Competing Interest form at http://www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare: no support from any organization for the submitted work; no financial relationships with any organizations that might have an interest in the submitted work in the previous 3 years; and no other relationships or activities that could appear to have influenced the submitted work.
Contributors
DR‐T wrote the manuscript. JHM was involved in the design of the research, the development of ideas and the preparation of the manuscript. LC was involved in the development of ideas and acted as a scientific adviser. RH was involved in the design of the research, the development of ideas and the preparation of the manuscript, and acted as a scientific adviser.
Rundle‐Thiele, D. , Head, R. , Cosgrove, L. , and Martin, J. H. (2016) Repurposing some older drugs that cross the blood–brain barrier and have potential anticancer activity to provide new treatment options for glioblastoma. Br J Clin Pharmacol, 81: 199–209. doi: 10.1111/bcp.12785.
References
- 1. Reardon DA, Wen PY. Therapeutic advances in the treatment of glioblastoma: rationale and potential role of targeted agents. Oncologist 2006; 11: 152–64. [DOI] [PubMed] [Google Scholar]
- 2. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, Curschmann J, Janzer RC, Ludwin SK, Gorlia T, Allgeier A, Lacombe D, Cairncross JG, Eisenhauer E, Mirimanoff RO; European Organisation for Research and Treatment of Cancer Brain Tumor and Radiotherapy Groups; National Cancer Institute of Canada Clinical Trials Group . Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 2005; 352: 987–96. [DOI] [PubMed] [Google Scholar]
- 3. Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC, Ludwin SK, Allgeier A, Fisher B, Belanger K, Hau P, Brandes AA, Gijtenbeek J, Marosi C, Vecht CJ, Mokhtari K, Wesseling P, Villa S, Eisenhauer E, Gorlia T, Weller M, Lacombe D, Cairncross JG, Mirimanoff RO; European Organisation for Research and Treatment of Cancer Brain Tumor and Radiation Oncology Groups; National Cancer Institute of Canada Clinical Trials Group . Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5‐year analysis of the EORTC‐NCIC trial. Lancet Oncol 2009; 10: 459–66. [DOI] [PubMed] [Google Scholar]
- 4. Kast RE, Boockvar JA, Bruning A, Cappello F, Chang WW, Cvek B, Dou QP, Duenas‐Gonzalez A, Efferth T, Focosi D, Ghaffari SH, Karpel‐Massler G, Ketola K, Khoshnevisan A, Keizman D, Magne N, Marosi C, McDonald K, Munoz M, Paranjpe A, Pourgholami MH, Sardi I, Sella A, Srivenugopal KS, Tuccori M, Wang W, Wirtz CR, Halatsch ME. A conceptually new treatment approach for relapsed glioblastoma: coordinated undermining of survival paths with nine repurposed drugs (CUSP9) by the International Initiative for Accelerated Improvement of Glioblastoma Care. Oncotarget 2013; 4: 502–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Esteller M, Garcia‐Foncillas J, Andion E, Goodman SN, Hidalgo OF, Vanaclocha V, Baylin SB, Herman JG. Inactivation of the DNA‐repair gene MGMT and the clinical response of gliomas to alkylating agents. N Engl J Med 2000; 343: 1350–4. [DOI] [PubMed] [Google Scholar]
- 6. Hegi ME, Diserens AC, Gorlia T, Hamou MF, de Tribolet N, Weller M, Kros JM, Hainfellner JA, Mason W, Mariani L, Bromberg JE, Hau P, Mirimanoff RO, Cairncross JG, Janzer RC, Stupp R. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med 2005; 352: 997–1003. [DOI] [PubMed] [Google Scholar]
- 7. Barker CA, Bishop AJ, Chang M, Beal K, Chan TA. Valproic acid use during radiation therapy for glioblastoma associated with improved survival. Int J Radiat Oncol Biol Phys 2013; 86: 504–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cancer Genome Atlas Research Network . Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008; 455: 1061–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Carrillo JA, Lai A, Nghiemphu PL, Kim HJ, Phillips HS, Kharbanda S, Moftakhar P, Lalaezari S, Yong W, Ellingson BM, Cloughesy TF, Pope WB. Relationship between tumor enhancement, edema, IDH1 mutational status, MGMT promoter methylation, and survival in glioblastoma. Am J Neuroradiol 2012; 33: 1349–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, Zheng S, Chakravarty D, Sanborn JZ, Berman SH, Beroukhim R, Bernard B, Wu CJ, Genovese G, Shmulevich I, Barnholtz‐Sloan J, Zou L, Vegesna R, Shukla SA, Ciriello G, Yung WK, Zhang W, Sougnez C, Mikkelsen T, Aldape K, Bigner DD, Van Meir EG, Prados M, Sloan A, Black KL, Eschbacher J, Finocchiaro G, Friedman W, Andrews DW, Guha A, Iacocca M, O'Neill BP, Foltz G, Myers J, Weisenberger DJ, Penny R, Kucherlapati R, Perou CM, Hayes DN, Gibbs R, Marra M, Mills GB, Lander E, Spellman P, Wilson R, Sander C, Weinstein J, Meyerson M, Gabriel S, Laird PW, Haussler D, Getz G, Chin L; TCGA Research Network . The somatic genomic landscape of glioblastoma. Cell 2013; 155: 462–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Visnyei K, Onodera H, Damoiseaux R, Saigusa K, Petrosyan S, De Vries D, Ferrari D, Saxe J, Panosyan EH, Masterman‐Smith M, Mottahedeh J, Bradley KA, Huang J, Sabatti C, Nakano I, Kornblum HI. A molecular screening approach to identify and characterize inhibitors of glioblastoma stem cells. Mol Cancer Ther 2011; 10: 1818–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chae YK, Brown EN, Lei X, Melhem‐Bertrandt A, Giordano SH, Litton JK, Hortobagyi GN, Gonzalez‐Angulo AM, Chavez‐Macgregor M. Use of ACE inhibitors and angiotensin receptor blockers and primary breast cancer outcomes. J Cancer 2013; 4: 549–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Walsh EM, Kim R, Del Valle L, Weaver M, Sheffield J, Lazarovici P, Marcinkiewicz C. Importance of interaction between nerve growth factor and alpha9beta1 integrin in glial tumor angiogenesis. Neuro Oncol 2012; 14: 890–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lacroix M, Abi‐Said D, Fourney DR, Gokaslan ZL, Shi W, DeMonte F, Lang FF, McCutcheon IE, Hassenbusch SJ, Holland E, Hess K, Michael C, Miller D, Sawaya R. A multivariate analysis of 416 patients with glioblastoma multiforme: prognosis, extent of resection, and survival. J Neurosurg 2001; 95: 190–8. [DOI] [PubMed] [Google Scholar]
- 15. Emara M, Allalunis‐Turner J. Effect of hypoxia on angiogenesis related factors in glioblastoma cells. Oncol Rep 2014; 31: 1947–53. [DOI] [PubMed] [Google Scholar]
- 16. Chim H, Armijo BS, Miller E, Gliniak C, Serret MA, Gosain AK. Propranolol induces regression of hemangioma cells through HIF‐1alpha‐mediated inhibition of VEGF‐A. Ann Surg 2012; 256: 146–56. [DOI] [PubMed] [Google Scholar]
- 17. Spence AM, Muzi M, Swanson KR, O'Sullivan F, Rockhill JK, Rajendran JG, Adamsen TC, Link JM, Swanson PE, Yagle KJ, Rostomily RC, Silbergeld DL, Krohn KA. Regional hypoxia in glioblastoma multiforme quantified with [18F]fluoromisonidazole positron emission tomography before radiotherapy: correlation with time to progression and survival. Clin Cancer Res 2008; 14: 2623–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Marampon F, Gravina GL, Zani BM, Popov VM, Fratticci A, Cerasani M, Di Genova D, Mancini M, Ciccarelli C, Ficorella C, Di Cesare E, Festuccia C. Hypoxia sustains glioblastoma radioresistance through ERKs/DNA‐PKcs/HIF‐1alpha functional interplay. Int J Oncol 2014; 44: 2121–31. [DOI] [PubMed] [Google Scholar]
- 19. Hsieh CH, Lee CH, Liang JA, Yu CY, Shyu WC. Cycling hypoxia increases U87 glioma cell radioresistance via ROS induced higher and long‐term HIF‐1 signal transduction activity. Oncol Rep 2010; 24: 1629–36. [DOI] [PubMed] [Google Scholar]
- 20. Amberger‐Murphy V Hypoxia helps glioma to fight therapy. Curr Cancer Drug Targets 2009; 9: 381–90. [DOI] [PubMed] [Google Scholar]
- 21. Liang BC. Effects of hypoxia on drug resistance phenotype and genotype in human glioma cell lines. J Neurooncol 1996; 29: 149–55. [DOI] [PubMed] [Google Scholar]
- 22. Levkovitz Y, Gil‐Ad I, Zeldich E, Dayag M, Weizman A. Differential induction of apoptosis by antidepressants in glioma and neuroblastoma cell lines: evidence for p‐c‐Jun, cytochrome c, and caspase‐3 involvement. J Mol Neurosci 2005; 27: 29–42. [DOI] [PubMed] [Google Scholar]
- 23. Sachlos E, Risueno RM, Laronde S, Shapovalova Z, Lee JH, Russell J, Malig M, McNicol JD, Fiebig‐Comyn A, Graham M, Levadoux‐Martin M, Lee JB, Giacomelli AO, Hassell JA, Fischer‐Russell D, Trus MR, Foley R, Leber B, Xenocostas A, Brown ED, Collins TJ, Bhatia M. Identification of drugs including a dopamine receptor antagonist that selectively target cancer stem cells. Cell 2012; 149: 1284–97. [DOI] [PubMed] [Google Scholar]
- 24. Walker AJ, Grainge M, Bates TE, Card TR. Survival of glioma and colorectal cancer patients using tricyclic antidepressants post‐diagnosis. Cancer Causes Control 2012; 23: 1959–64. [DOI] [PubMed] [Google Scholar]
- 25. Higgins SC, Pilkington GJ. The in vitro effects of tricyclic drugs and dexamethasone on cellular respiration of malignant glioma. Anticancer Res 2010; 30: 391–7. [PubMed] [Google Scholar]
- 26. Tzadok S, Beery E, Israeli M, Uziel O, Lahav M, Fenig E, Gil‐Ad I, Weizman A, Nordenberg J. In vitro novel combinations of psychotropics and anti‐cancer modalities in U87 human glioblastoma cells. Int J Oncol 2010; 37: 1043–51. [DOI] [PubMed] [Google Scholar]
- 27. Yde CW, Clausen MP, Bennetzen MV, Lykkesfeldt AE, Mouritsen OG, Guerra B. The antipsychotic drug chlorpromazine enhances the cytotoxic effect of tamoxifen in tamoxifen‐sensitive and tamoxifen‐resistant human breast cancer cells. Anticancer Drugs 2009; 20: 723–35. [DOI] [PubMed] [Google Scholar]
- 28. Kast RE, Ellingson BM, Marosi C, Halatsch ME. Glioblastoma treatment using perphenazine to block the subventricular zone's tumor trophic functions. J Neurooncol 2014; 116: 207–12. [DOI] [PubMed] [Google Scholar]
- 29. de Jonge J, Berghauser Pont LM, Idema S, Kloezeman JJ, Noske D, Dirven CM, Lamfers ML. Therapeutic concentrations of anti‐epileptic drugs do not inhibit the activity of the oncolytic adenovirus Delta24‐RGD in malignant glioma. J Gene Med 2013; 15: 134–41. [DOI] [PubMed] [Google Scholar]
- 30. Bobustuc GC, Baker CH, Limaye A, Jenkins WD, Pearl G, Avgeropoulos NG, Konduri SD. Levetiracetam enhances p53‐mediated MGMT inhibition and sensitizes glioblastoma cells to temozolomide. Neuro Oncol 2010; 12: 917–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Camphausen K, Cerna D, Scott T, Sproull M, Burgan WE, Cerra MA, Fine H, Tofilon PJ. Enhancement of in vitro and in vivo tumor cell radiosensitivity by valproic acid. Int J Cancer 2005; 114: 380–6. [DOI] [PubMed] [Google Scholar]
- 32. Chinnaiyan P, Cerna D, Burgan WE, Beam K, Williams ES, Camphausen K, Tofilon PJ. Postradiation sensitization of the histone deacetylase inhibitor valproic acid. Clin Cancer Res 2008; 14: 5410–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jaeckle KA, Ballman K, Furth A, Buckner JC. Correlation of enzyme‐inducing anticonvulsant use with outcome of patients with glioblastoma. Neurology 2009; 73: 1207–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Oberndorfer S, Piribauer M, Marosi C, Lahrmann H, Hitzenberger P, Grisold W. P450 enzyme inducing and non‐enzyme inducing antiepileptics in glioblastoma patients treated with standard chemotherapy. J Neurooncol 2005; 72: 255–60. [DOI] [PubMed] [Google Scholar]
- 35. Weller M, Gorlia T, Cairncross JG, van den Bent MJ, Mason W, Belanger K, Brandes AA, Bogdahn U, Macdonald DR, Forsyth P, Rossetti AO, Lacombe D, Mirimanoff RO, Vecht CJ, Stupp R. Prolonged survival with valproic acid use in the EORTC/NCIC temozolomide trial for glioblastoma. Neurology 2011; 77: 1156–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ghodke‐Puranik Y, Thorn CF, Lamba JK, Leeder JS, Song W, Birnbaum AK, Altman RB, Klein TE. Valproic acid pathway: pharmacokinetics and pharmacodynamics. Pharmacogenet Genomics 2013; 23: 236–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Eyal S, Yagen B, Sobol E, Altschuler Y, Shmuel M, Bialer M. The activity of antiepileptic drugs as histone deacetylase inhibitors. Epilepsia 2004; 45: 737–44. [DOI] [PubMed] [Google Scholar]
- 38. van Nifterik KA, Van den Berg J, Slotman BJ, Lafleur MV, Sminia P, Stalpers LJ. Valproic acid sensitizes human glioma cells for temozolomide and gamma‐radiation. J Neurooncol 2012; 107: 61–7. [DOI] [PubMed] [Google Scholar]
- 39. Gupta PP, Thacker AK, Haider J, Dhawan S, Pandey N, Pandey AK. Assessment of topiramate's efficacy and safety in epilepsy. J Neurosci Rural Pract 2014; 5: 144–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Vajda FJ, Graham J, Roten A, Lander CM, O'Brien TJ, Eadie M. Teratogenicity of the newer antiepileptic drugs – the Australian experience. J Clin Neurosci 2012; 19: 57–9. [DOI] [PubMed] [Google Scholar]
- 41. Veiby G, Daltveit AK, Engelsen BA, Gilhus NE. Fetal growth restriction and birth defects with newer and older antiepileptic drugs during pregnancy. J Neurol 2014; 261: 579–88. [DOI] [PubMed] [Google Scholar]
- 42. Ma B, Xiang Y, Li T, Yu HM, Li XJ. Inhibitory effect of topiramate on Lewis lung carcinoma metastasis and its relation with AQP1 water channel. Acta Pharmacol Sin 2004; 25: 54–60. [PubMed] [Google Scholar]
- 43. Ma B, Pan Y, Song Q, Tie L, Zhang Y, Xiao Y, Zhang J, Han J, Xu Y, Xiang Y, Yu HM, Li XJ. The effect of topiramate on tumor‐related angiogenesis and on the serum proteome of mice bearing Lewis lung carcinoma. Eur J Pharmacol 2011; 663: 9–16. [DOI] [PubMed] [Google Scholar]
- 44. Cemeus C, Zhao TT, Barrett GM, Lorimer IA, Dimitroulakos J. Lovastatin enhances gefitinib activity in glioblastoma cells irrespective of EGFRvIII and PTEN status. J Neurooncol 2008; 90: 9–17. [DOI] [PubMed] [Google Scholar]
- 45. Chan DY, Chen GG, Poon WS, Liu PC. Lovastatin sensitized human glioblastoma cells to TRAIL‐induced apoptosis. J Neurooncol 2008; 86: 273–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yanae M, Tsubaki M, Satou T, Itoh T, Imano M, Yamazoe Y, Nishida S. Statin‐induced apoptosis via the suppression of ERK1/2 and Akt activation by inhibition of the geranylgeranyl‐pyrophosphate biosynthesis in glioblastoma. J Exp Clin Cancer Res 2011; 30: 74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yongjun Y, Shuyun H, Lei C, Xiangrong C, Zhilin Y, Yiquan K. Atorvastatin suppresses glioma invasion and migration by reducing microglial MT1‐MMP expression. J Neuroimmunol 2013; 260: 1–8. [DOI] [PubMed] [Google Scholar]
- 48. Gabrys D, Dorfler A, Yaromina A, Hessel F, Krause M, Oertel R, Baumann M. Effects of lovastatin alone or combined with irradiation on tumor cells in vitro and in vivo . Strahlenther Onkol 2008; 184: 48–53. [DOI] [PubMed] [Google Scholar]
- 49. Wu H, Jiang H, Lu D, Xiong Y, Qu C, Zhou D, Mahmood A, Chopp M. Effect of simvastatin on glioma cell proliferation, migration, and apoptosis. Neurosurgery 2009; 65: 1087–96; discussion 96–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tapia‐Perez JH, Kirches E, Mawrin C, Firsching R, Schneider T. Cytotoxic effect of different statins and thiazolidinediones on malignant glioma cells. Cancer Chemother Pharmacol 2011; 67: 1193–201. [DOI] [PubMed] [Google Scholar]
- 51. Wood WG, Eckert GP, Igbavboa U, Muller WE. Statins and neuroprotection: a prescription to move the field forward. Ann N Y Acad Sci 2010; 1199: 69–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Engineer DR, Burney BO, Hayes TG, Garcia JM. Exposure to ACEI/ARB and beta‐blockers is associated with improved survival and decreased tumor progression and hospitalizations in patients with advanced colon cancer. Transl Oncol 2013; 6: 539–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Arrieta O, Guevara P, Escobar E, Garcia‐Navarrete R, Pineda B, Sotelo J. Blockage of angiotensin II type I receptor decreases the synthesis of growth factors and induces apoptosis in C6 cultured cells and C6 rat glioma. Br J Cancer 2005; 92: 1247–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Juillerat‐Jeanneret L, Celerier J, Chapuis Bernasconi C, Nguyen G, Wostl W, Maerki HP, Janzer RC, Corvol P, Gasc JM. Renin and angiotensinogen expression and functions in growth and apoptosis of human glioblastoma. Br J Cancer 2004; 90: 1059–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Rivera E, Arrieta O, Guevara P, Duarte‐Rojo A, Sotelo J. AT1 receptor is present in glioma cells; its blockage reduces the growth of rat glioma. Br J Cancer 2001; 85: 1396–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Rooprai HK, Kandanearatchi A, Maidment SL, Christidou M, Trillo‐Pazos G, Dexter DT, Rucklidge GJ, Widmer W, Pilkington GJ. Evaluation of the effects of swainsonine, captopril, tangeretin and nobiletin on the biological behaviour of brain tumour cells in vitro . Neuropathol Appl Neurobiol 2001; 27: 29–39. [DOI] [PubMed] [Google Scholar]
- 57. Kast RE, Halatsch ME. Matrix metalloproteinase‐2 and −9 in glioblastoma: a trio of old drugs‐captopril, disulfiram and nelfinavir‐are inhibitors with potential as adjunctive treatments in glioblastoma. Arch Med Res 2012; 43: 243–7. [DOI] [PubMed] [Google Scholar]
- 58. Cardwell CR, Mc Menamin UC, Hicks BM, Hughes C, Cantwell MM, Murray LJ. Drugs affecting the renin–angiotensin system and survival from cancer: a population based study of breast, colorectal and prostate cancer patient cohorts. BMC Med 2014; 12: 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Holmes S, Griffith EJ, Musto G, Minuk GY. Antihypertensive medications and survival in patients with cancer: a population‐based retrospective cohort study. Cancer Epidemiol 2013; 37: 881–5. [DOI] [PubMed] [Google Scholar]
- 60. Carpentier AF, Ferrari D, Bailon O, Ursu R, Banissi C, Dubessy AL, Belin C, Levy C. Steroid‐sparing effects of angiotensin‐II inhibitors in glioblastoma patients. Eur J Neurol 2012; 19: 1337–42. [DOI] [PubMed] [Google Scholar]
- 61. Petty A, Myshkin E, Qin H, Guo H, Miao H, Tochtrop GP, Hsieh JT, Page P, Liu L, Lindner DJ, Acharya C, MacKerell AD Jr, Ficker E, Song J, Wang B. A small molecule agonist of EphA2 receptor tyrosine kinase inhibits tumor cell migration in vitro and prostate cancer metastasis in vivo . PLoS One 2012; 7: e42120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Lamy S, Lachambre MP, Lord‐Dufour S, Beliveau R. Propranolol suppresses angiogenesis in vitro: inhibition of proliferation, migration, and differentiation of endothelial cells. Vascul Pharmacol 2010; 53: 200–8. [DOI] [PubMed] [Google Scholar]
- 63. Larochelle P, Tobe SW, Lacourciere Y. Beta‐blockers in hypertension: studies and meta‐analyses over the years. Can J Cardiol 2014; 30: S16–22. [DOI] [PubMed] [Google Scholar]
- 64. Pasquier E, Ciccolini J, Carre M, Giacometti S, Fanciullino R, Pouchy C, Montero MP, Serdjebi C, Kavallaris M, Andre N. Propranolol potentiates the anti‐angiogenic effects and anti‐tumor efficacy of chemotherapy agents: implication in breast cancer treatment. Oncotarget 2011; 2: 797–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Pasquier E, Street J, Pouchy C, Carre M, Gifford AJ, Murray J, Norris MD, Trahair T, Andre N, Kavallaris M. Beta‐blockers increase response to chemotherapy via direct antitumour and anti‐angiogenic mechanisms in neuroblastoma. Br J Cancer 2013; 108: 2485–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Zhang D, Ma Q, Shen S, Hu H. Inhibition of pancreatic cancer cell proliferation by propranolol occurs through apoptosis induction: the study of beta‐adrenoceptor antagonist's anticancer effect in pancreatic cancer cell. Pancreas 2009; 38: 94–100. [DOI] [PubMed] [Google Scholar]
- 67. Kozanoglu I, Yandim MK, Cincin ZB, Ozdogu H, Cakmakoglu B, Baran Y. New indication for therapeutic potential of an old well‐known drug (propranolol) for multiple myeloma. J Cancer Res Clin Oncol 2013; 139: 327–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Yang EV, Sood AK, Chen M, Li Y, Eubank TD, Marsh CB, Jewell S, Flavahan NA, Morrison C, Yeh PE, Lemeshow S, Glaser R. Norepinephrine up‐regulates the expression of vascular endothelial growth factor, matrix metalloproteinase (MMP)‐2, and MMP‐9 in nasopharyngeal carcinoma tumor cells. Cancer Res 2006; 66: 10357–64. [DOI] [PubMed] [Google Scholar]
- 69. Miller SC, Huang R, Sakamuru S, Shukla SJ, Attene‐Ramos MS, Shinn P, van Leer D, Leister W, Austin CP, Xia M. Identification of known drugs that act as inhibitors of NF‐kappaB signaling and their mechanism of action. Biochem Pharmacol 2010; 79: 1272–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Kang KB, Wang TT, Woon CT, Cheah ES, Moore XL, Zhu C, Wong MC. Enhancement of glioblastoma radioresponse by a selective COX‐2 inhibitor celecoxib: inhibition of tumor angiogenesis with extensive tumor necrosis. Int J Radiat Oncol Biol Phys 2007; 67: 888–96. [DOI] [PubMed] [Google Scholar]
- 71. Kesari S, Schiff D, Henson JW, Muzikansky A, Gigas DC, Doherty L, Batchelor TT, Longtine JA, Ligon KL, Weaver S, Laforme A, Ramakrishna N, Black PM, Drappatz J, Ciampa A, Folkman J, Kieran M, Wen PY. Phase II study of temozolomide, thalidomide, and celecoxib for newly diagnosed glioblastoma in adults. Neuro Oncol 2008; 10: 300–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Sesen J, Dahan P, Scotland SJ, Saland E, Dang VT, Lemarie A, Tyler BM, Brem H, Toulas C, Cohen‐Jonathan Moyal E, Sarry JE, Skuli N. Metformin inhibits growth of human glioblastoma cells and enhances therapeutic response. PLoS One 2015; 10: e0123721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Wan Z, Shi W, Shao B, Shi J, Shen A, Ma Y, Chen J, Lan Q. Peroxisome proliferator‐activated receptor gamma agonist pioglitazone inhibits beta‐catenin‐mediated glioma cell growth and invasion. Mol Cell Biochem 2011; 349: 1–10. [DOI] [PubMed] [Google Scholar]
- 74. Hori YS, Hosoda R, Akiyama Y, Sebori R, Wanibuchi M, Mikami T, Sugino T, Suzuki K, Maruyama M, Tsukamoto M, Mikuni N, Horio Y, Kuno A. Chloroquine potentiates temozolomide cytotoxicity by inhibiting mitochondrial autophagy in glioma cells. J Neurooncol 2015; 122: 11–20. [DOI] [PubMed] [Google Scholar]
- 75. Golden EB, Cho HY, Jahanian A, Hofman FM, Louie SG, Schonthal AH, Chen TC. Chloroquine enhances temozolomide cytotoxicity in malignant gliomas by blocking autophagy. Neurosurg Focus 2014; 37: E12. [DOI] [PubMed] [Google Scholar]
- 76. Paranjpe A, Zhang R, Ali‐Osman F, Bobustuc GC, Srivenugopal KS. Disulfiram is a direct and potent inhibitor of human O6‐methylguanine‐DNA methyltransferase (MGMT) in brain tumor cells and mouse brain and markedly increases the alkylating DNA damage. Carcinogenesis 2014; 35: 692–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Dey M, Ulasov IV, Lesniak MS. Virotherapy against malignant glioma stem cells. Cancer Lett 2010; 289: 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Solomon IH, Ramkissoon SH, Milner DA Jr, Folkerth RD. Cytomegalovirus and glioblastoma: a review of evidence for their association and indications for testing and treatment. J Neuropathol Exp Neurol 2014; 73: 994–8. [DOI] [PubMed] [Google Scholar]