

Abstract
Lysosomal storage diseases are inherited monogenic disorders in which lysosome function is compromised. Although individually very rare, they occur at a collective frequency of approximately one in five thousand live births and usually have catastrophic consequences for health. The lysosomal storage diseases Niemann‐Pick disease type C (NPC) is caused by mutations predominantly in the lysosomal integral membrane protein NPC1 and clinically presents as a progressive neurodegenerative disorder. In this article we review data that demonstrate significant dysregulation of innate immunity in NPC, which occurs both in peripheral organs and the CNS. In particular pro‐inflammatory responses promote disease progression and anti‐inflammatory drugs provide benefit in animal models of the disease and are an attractive target for clinical intervention in this disorder.
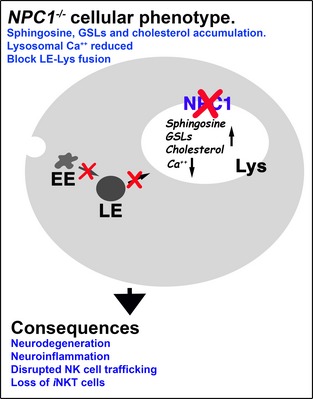
Niemann‐Pick disease type C is a rare, devastating, inherited lysosomal storage disease with a unique cellular phenotype characterized by lysosomal accumulation of sphingosine, various glycosphingolipids and cholesterol and a reduction in lysosomal calcium. In this review we highlight the impact of the disease on innate immune activities in both the central nervous system (CNS) and peripheral tissues and discuss their contributions to pathology and the underlying mechanisms.
Keywords: cytokine, inflammation, lysosomal storage disease, lysosome, microglia, neurodegeneration, Niemann Pick type C
Abbreviations used
- GSLs
glycosphingolipids
- LSDs
lysosomal storage diseases
- NPC
Niemann‐Pick disease type C
Lysosomal storage diseases
Lysosomal storage diseases (LSDs) are a group of metabolic disorders that result from inherited defects in proteins required for normal lysosome function (Ballabio and Gieselmann 2009; Schultz et al. 2011). Affected proteins can include catabolic enzymes, integral membrane proteins, and proteins required for generating specific post‐translational modifications of proteins (Futerman and van Meer 2004; Platt et al. 2012). To date, approximately 70 discrete LSDs have been described that are individually rare; however, collectively they occur at a prevalence of 1: 5000 live births, which may be an underestimate owing to cases being either undiagnosed or misdiagnosed (Cox and Cachon‐Gonzalez 2012).
There are two striking features of LSDs. The first is that disturbance of lysosomal function frequently has severe consequences for the function of cells, organ systems, and the body as a whole, leading to premature death (Platt et al. 2012; Platt 2014). This is perhaps not surprising in light of the accumulating evidence that the lysosome is involved in far more than macromolecule catabolism and re‐cycling. The lysosome is now recognized to be an organelle that has a much broader range of functions including signaling, secretion, and regulation of energy metabolism (Settembre et al. 2013). Second, distinct LSDs that result from the inactivation of different lysosomal proteins often share similar pathologies (Vitner et al. 2010). For example, inappropriate activation of the innate immune system in the form of inflammation is especially frequent in LSDs, but intriguingly it remains an open question as to how exactly lysosomal storage triggers pro‐inflammatory pathways (Platt 2014).
Niemann‐Pick disease type C
Niemann‐Pick disease type C (NPC) is an autosomal recessive condition caused by mutations in either of two independent genes. Mutations in NPC1, an integral transmembrane protein of the limiting membrane of the lysosome (Higgins et al. 1999) accounts for ~ 95% of all clinical cases whereas mutations in NPC2, a soluble cholesterol binding protein are responsible for the remainder (Platt et al. 2014). Therefore, NPC is unlike the majority of LSDs, which are caused by mutations in single genes that encode a lysosomal hydrolase. Patients with mutations in either NPC1 or NPC2 are phenotypically indistinguishable and because loss of activity of either protein is not compensated for by the presence of the other protein, it is probable that they function in a common cell biological pathway (Dixit et al. 2011). For the purpose of this review our discussion will concentrate on the consequences of impaired NPC1 function, as most information is available on NPC1 disease. Clinically, affected individuals are usually diagnosed in childhood although adult onset variants also occur and are under‐diagnosed (Wassif et al. 2015). The course of disease is dominated by progressive neurodegeneration, which presents as cerebellar ataxia, dysphagia, dementia and premature death, typically within the second decade of life (Vanier 2010). Almost three hundred different mutations in NPC1 have been identified to date, but as yet there is neither a clear structure–function relationship that has been established, nor a functional assay for NPC1 (Millat et al. 1999; Ribeiro et al. 2001).
The phenotype of NPC1‐deficient cells
Probably the most prominent feature of NPC cells is the accumulation of free cholesterol in the late endosome/lysosome (Fig. 1). Although this remains the basis for the clinical diagnostic test (Vanier 2010) it is contentious whether it is disease causing or a secondary consequence of the disease. In fact a complex array of lipid species are stored in NPC including glycosphingolipids (GSLs), sphingomyelin, and sphingosine, the latter generated from ceramide catabolism (Rosenbaum and Maxfield 2011). Because storage does not occur as a direct result of impaired catabolic activity NPC is classified as secondary LSD. The function of NPC1 is itself currently unresolved and although the prevailing view is that it is involved in removing cholesterol from the lysosome, evidence that the purified protein can bind the sterol may indicate its requirement as a co‐factor rather than support the hypothesis that NPC1 is responsible for moving cholesterol out of the acidic compartment. Further support for this has been the demonstration that cholesterol can move effectively from the lysosome to mitochondria in NPC1 cells (Kennedy et al. 2014). A combination of experimental data reinforces an alternative model of the pathogenic cascade (Fig. 2). NPC1 has greatest homology with the resistance‐nodulation‐division family of prokaryotic permeases and has been demonstrated to have activity as an efflux pump (Davies et al. 2000). When NPC1 is inactivated in healthy cells the first metabolite to accumulate is sphingosine (which also builds up in tissues in patients and animal models, Rodriguez‐Lafrasse et al. 1994), which requires active transportation out of the lysosome because it is protonated at acidic pH (Lloyd‐Evans et al. 2008). A consequence of the storage of sphingosine is inhibition of the filling of the acidic compartment with calcium. This could be indirect via protein kinase C inhibition (Walter et al. 2009) or a direct effect of sphingosine on the protein that fills the acidic store with calcium, the identity of which remains unknown. Whilst the precise biological roles of calcium release from the lysosome are not fully defined it is known that fusion and trafficking in the endocytic pathway are uniquely dependent upon calcium mobilization from the lysosome (Morgan et al. 2011). Therefore, impaired filling of the lysosome with the cation, as a result of sphingosine accumulation, means insufficient calcium can be released, which in turn leads to a block in endosome/lysosome trafficking and fusion. This is then the cause of the secondary storage of cholesterol, GSLs, and sphingomyelin (Fig. 1). All of these defects, including sphingosine accumulation, the lysosomal calcium deficit, impaired calcium release, and prevention of normal endocytic trafficking have been confirmed independently in multiple studies in NPC cells (Rodriguez‐Lafrasse et al. 1994) (Chen et al. 2010) (Shen et al. 2012; Tamari et al. 2013).
Figure 1.
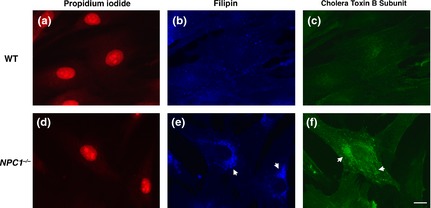
Cholesterol and GM1 ganglioside accumulation are prominent characteristics of the Niemann‐Pick disease type C (NPC) cellular phenotype. WT (a–c) and NPC1 −/− fibroblasts (d–f) stained for nucleus, propidium iodide ‐ red (a and d), cholesterol, filipin—blue (b and e) and GM1 ganglioside, cholera toxin subunit B—green (c and f). Arrows indicate examples of punctate accumulations in NPC1 −/− cells. Scale bar: 10 μm.
Figure 2.
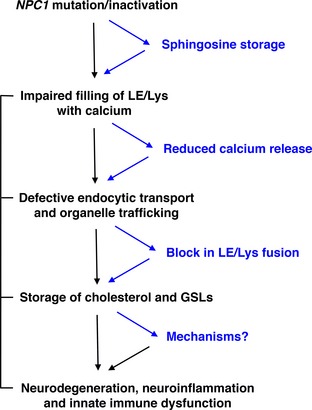
Proposed pathological cascade in Niemann‐Pick disease type C (NPC) cells. Cartoon of proposed pathological process that underlies the NPC cellular phenotype and results in neurodegeneration, neuroinflammation, and dysregulation of innate immune responses.
NPC and innate immune responses
Because neurodegeneration is the main clinical feature of NPC disease, it is unsurprising that a major research focus has been on the effects of loss of NPC1 activity within the CNS. However, because NPC1 is expressed in all cells, its loss is very likely to have widespread consequences. In this review we will discuss the evidence that mutations in NPC1 impact significantly on multiple tissues and cells, in particular within the immune system and result in inflammation and altered innate immune responses.
Animal model of NPC; the Npc1 −/− (BALB/cNctr‐Npc1 m1N /J) mouse
Our understanding of NPC has been advanced enormously by studies of an authentic mouse model, the Npc1 −/− (BALB/cNctr‐Npc1 m1N /J) mouse, which has almost total absence of the protein and displays all the hallmarks of the clinical disease (Loftus et al. 1997). This mutant strain arose spontaneously and has a lifespan in the range of 10–14 weeks and therefore has a course of disease more acute that the vast majority of patients. The mutant mouse has been exploited successfully not only for determining the ontogeny of disease and underlying pathogenic mechanisms but also for the evaluation of experimental therapies. Analyses using these mice have been undertaken at the whole animal, cellular, and molecular levels (Baudry et al. 2003; Smith et al. 2009; Cologna et al. 2012, 2014).
Prior to about 4–5 weeks of age Npc1 −/− mice have no discernible behavioral indication of disease that distinguishes them from wild‐type littermates. First indications of behavioral deficits, such as tremor and ataxic gait, appear by weeks 5–6; by weeks 7–8 defects in motor coordination become more apparent, and by 9–10 weeks ataxia is advanced and accompanied by increased loss in weight and poor coat condition as feeding and drinking becomes difficult (humane end point applied) (Fig. 3) (Smith et al. 2009). Although the BALB/cNctr‐Npc1 m1N /J strain has been the most intensively studied, there are also other authentic mammalian animal models of NPC. Interestingly, loss of Npc1 in mice on the C57BL/6 genetic background results in more acute disease relative to the BALB/cNctr‐Npc1 m1N /J mouse (Parra et al. 2011), suggestive of genetic modifiers. A feline model of NPC has also been characterized (Brown et al. 1994) and successfully used to trial experimental therapies (Stein et al. 2012).
Figure 3.

Details of the progression of disease in Npc1 −/− mice. Npc1 −/− (BALB/cNctr‐Npc1 m1N /J) mice display an acute clinical course, with a lifespan of 10–14 weeks that is characterized by the predictable development of defined symptoms at specific ages.
Neuroinflammation and neurodegeneration in NPC
Pathology within the CNS is a feature of most LSDs, probably because neurons are uniquely vulnerable to perturbation of normal lysosomal activity (Jmoudiak and Futerman 2005; Bellettato and Scarpa 2010; Vitner et al. 2010). Activation of the innate immune system is also a very common feature of neurodegenerative LSDs (Jeyakumar et al. 2003) (Bellettato and Scarpa 2010; Vitner et al. 2010).
Interestingly, microglial activation (as indicated by up‐regulation of CD68 expression) and astrogliosis (Pekny and Nilsson 2005) are apparent from as early as 2 and 4 weeks of age respectively in Npc1 −/− mice (Baudry et al. 2003), prior to any overt behavioral symptoms. Whilst NPC‐activated microglia up‐regulate CD68, in contrast to microglia in the GM1 and GM2 gangliosidoses mice (Jeyakumar et al. 2003), they do not become positive for MHC class II expression, indicating a different activation state, the significance of which is not currently understood (Smith et al. 2009). Neuropathology in NPC develops in a distinct temporal and spatial pattern, with cell loss in both motor and sensory pathways, in addition to the characteristic progressive death of Purkinje cells in the cerebellum (Pressey et al. 2012). Because Npc1 activity is absent throughout the CNS of BALB/cNctr‐Npc1 m1N /J mice an important question has been to what extent does deletion in particular cell types drive disease. Although data points toward cell autonomous neurodegeneration (Lopez and Scott 2013), non‐cell autonomous mechanisms cannot be absolutely excluded because of the complex interdependence of different cell populations within the CNS. For example, the lifespan of Npc1 −/− mice was extended threefold by expression of functional Npc1 in astrocytes (Zhang et al. 2008), greater than fivefold when expressed in neurons and when combined there was an additive effect, but disease symptoms in motor systems still occurred, albeit significantly delayed (Borbon et al. 2012).
Changes in gene expression in the CNS that underlie the pathological cascade have also been documented and one pathway that has been identified is a network of pro‐inflammatory genes (Cologna et al. 2014). Comparable studies performed on post‐mortem patient cerebellum with those from the mutant mouse revealed transcriptional changes and identified complement C3 as the only gene found to be elevated in all samples analyzed (Cologna et al. 2014).
It is still not understood why brain inflammation is triggered in NPC and other LSDs. Clearly, neuroinflammation as a generalized response is independent of the molecular identity of the storage material, because it is universal amongst neurodegenerative LSDs. However, the precise nature of the inflammatory profile is very likely to differ between specific LSDs and is very probably influenced by both the chemical composition of the storage and the underlying mechanistic defect. It is recognized that inflammation is a response to tissue damage or stress and is a protective mechanism that can result in induction of appropriate repair processes (Chovatiya and Medzhitov 2014). However, in chronic conditions the failure of inflammation to resolve, presumably because the pro‐inflammatory stimulus remains, means the destructive phases of the response are significantly extended.
Microglia, the resident myeloid cell populations of the brain are considered to be the most important source of pro‐inflammatory molecules, including cytokines, chemo‐attractants, and reactive oxygen species within the CNS (Gonzalez‐Scarano and Baltuch 1999) (Ransohoff and Perry 2009) as well as providing protective mechanisms (Griffiths et al. 2009; Napoli and Neumann 2010). Several scenarios can be envisaged to explain why microglia are activated in LSDs. It may be as a result of accumulation of storage materials in microglia themselves affecting signaling pathways. On the other hand, as sentinels within the CNS it may be microglial detection of storage in other cell populations, particularly neurons, that is the trigger. Of course, it may not be actual storage, but rather a loss of cellular homeostasis beyond the normal range. Loss of neurons, especially by necrotic death is also very likely to provoke innate immune‐mediated inflammation (Kono and Rock 2008). An important activity of microglia is the phagocytosis of apoptotic cell debris (Napoli and Neumann 2009), but it is not known if this scavenging activity is normal or impaired in NPC and if is the latter, whether it also contributes to the inflammatory profile (Palin et al. 2008). Unfortunately, at this time we have an incomplete understanding of the temporal and spatial details of inflammation and neurodegeneration in NPC.
One of the most important issues that have been resolved is the question of whether neuroinflammation actively promotes pathogenesis. The ability of anti‐inflammatory therapies, such as non‐steroidal drugs (NSAIDs), aspirin and ibuprofen to significantly extend the lifespan, protect against microglial activation and Purkinje cell loss and delay symptom onset in Npc1 −/− mice (Smith et al. 2009) confirmed that neuroinflammation enhances disease and when used in combination with agents that inhibit GSL biosynthesis and modulate calcium they can maximize therapeutic benefit (Williams et al. 2014).
Deficits in the NPC peripheral immune system
Natural killer cells
The immune system in peripheral tissue and the CNS are intimately connected at a number of levels and so the immune system as a whole merits study in these diseases. Natural killer (NK) cells are an immune cell population that, in particular, plays an important role in the killing of virally infected and transformed cells (Vivier et al. 2011). As an effector sub‐group of lymphocyte they express a number of activating and inhibitory receptors, which engage ligands on target cells and when appropriate release cytotoxic products via degranulation (Lanier 2008).
The first lipid to accumulate in the lysosome when NPC1 is inactivated is sphingosine. In healthy cells sphingosine has two potential fates; it can either be recycled back to form ceramide for reutilization in the salvage pathway of GSL biosynthesis or phosphorylated by two specific kinases to yield sphingosine‐1‐phosphate (S1P), a bioactive lipid that has multiple known functions (Maceyka and Spiegel 2014). During inflammation immune cells enter and leave sites of tissue damage by the process of regulated cell migration and trafficking (Butcher and Picker 1996). S1P levels differ significantly between the blood, lymph, and tissues (higher in the first two; lower in the latter) and this gradient has a major role in affecting lymphocyte trafficking as it forms a chemotactic gradient (Cyster and Schwab 2012). Speak Speak et al. (2014) hypothesized that the accumulation of sphingosine in the lysosome in NPC would mean reduced generation of S1P (phosphorylation of sphingosine occurs outside of the lysosome) and result in alteration of the normal systemic/tissue gradient and thereby affect NK cell trafficking. Firstly, the authors confirmed a 10‐fold increase in the sphingosine content of lymph nodes in Npc1 −/− mice and a proportional decrease in conversion to S1P (only a three‐fold increase), suggesting the likelihood of a defect in the gradient between lymphatic fluid and node. Second, as predicted there was a significant increase in the frequency of NK cells in multiple organs as compared to control animals and a corresponding decrease in the circulation. The distribution of NK cells in Npc1 −/− mice is similar to that reported in animals deficient in a specific S1P receptor (Walzer et al. 2007). Comparable differences in frequencies were also measured in blood from NPC patients and interestingly, carrier genotypes also failed to show an age‐related expansion in NK cell numbers. In addition, analysis confirmed an alteration in NK cell development and phenotype (maturation status) as well as frequency in Npc1 −/− animals and patients. Functionally, deficient cells in the mouse were significantly compromised in their ability to kill target cells (cytotoxicity), which correlated with impaired degranulation and reduced calcium release from the lysosome (Speak et al. 2014). These data strongly implicate the requirement for lysosomal calcium in cytotoxic granule (specialized lysosome‐related organelles) secretion by NK cells, which has also been demonstrated for cytotoxic T‐cells degranulation if acidic store calcium release is impaired (Davis et al. 2012). Furthermore, a prediction from this would be that other Npc1 −/− cell types might also have diminished secretion of lysosome‐related organelles.
Invariant natural killer T cells and LSDs
Invariant natural killer T cells (iNKT) cells are a specialized subset of T lymphocytes that bear an invariant TCRα chain and express markers shared with NK cells (Bendelac et al. 2007). They are key effector cells in innate immunity and modulate the activities of the adaptive immune system, especially in host defense against pathogens and cancer (Brennan et al. 2013). Unlike conventional T cells, which respond to antigen presenting cells that express peptides in the context of MHC molecules, iNKTs are activated by GSL ligands presented by the MHC‐related molecule CD1 (CD1a‐e in humans; CD1d in the mouse) (Speak et al. 2008). Importantly, both positive selection of iNKT cells in the thymus and their maintenance in peripheral organs is dependent upon presentation of specific endogenous lipid ligands by CD1 (Mendiratta et al. 1997; Gapin et al. 2001). Because loading of CD1 with lipid molecules occurs in late endocytic/lysosome compartments in the mouse, diseases that alter the distribution, nature or abundance of GSLs (such as LSDs) have the potential to render this process ineffective and thereby affect the frequency and properties of iNKT cells. Consistent with this, it was found that multiple mouse models of LSDs (that store diverse GSL species), including NPC, had decreased numbers of iNKT cells and their maturation was compromised (but T cells specific for peptide ligands were not affected) (Gadola et al. 2006). GSL‐triggered cytokine release was significantly reduced and presentation of endogenous lipids was impaired in all models, but interestingly only Npc1 −/− mice had lower cell surface expression of CD1d (Gadola et al. 2006). However, in contrast to the situation in the mouse, analysis of NPC patients revealed normal frequencies and functions of iNKT cells (Speak et al. 2012). This difference is most likely due to differences in trafficking requirements for iNKT cells between the two species. The evidence is that lipid loading occurs in the early endosome in humans, not in the late endocytic system as it is in the mouse, so is therefore not affected in NPC patients (Chen et al. 2007).
Conclusions
In this review we have highlighted that LSDs are not just diseases of CNS dysfunction but also involve activation of the innate immune system, leading to chronic inflammation that actively contributes to the disease. There are multiple routes of communication between the peripheral immune system and the CNS, including both direct ones mediated by neural circuits (Olofsson et al. 2012) and systemic mechanisms, in the form of circulating cytokines, acute phase proteins and other pro‐inflammatory molecules (Allan and Rothwell 2001). It is well established that the ongoing neurodegenerative processes in the CNS are exacerbated by induction of inflammation at peripheral sites (Perry et al. 2003). Also, neuropathology frequently results in the recruitment of immune cells from peripheral sites into the CNS and should their generation or functioning be ineffective, any neuroprotective benefit their mobilization may offer will be lost. A particularly important reason for exploring the nature of the altered innate immune responses in LSDs is that the vast majority of these disorders are currently without effective treatment and anti‐inflammatories have shown benefit in relevant models and are attractive as adjunct therapeutics. Therefore, targeting the immune system merits clinical evaluation.
Acknowledgments and conflict of interest disclosure
N.P. was funded by NIH (1R21AI102166); A.O.S. by MRC (G0700851); A.C. received funding from the European Union Seventh Framework Program (FP7 2007‐2013) under grant agreement no. 289278 “Sphingonet”; D.A.S. by Niemann‐Pick Research Foundation and NP‐Suisse; I.M.W. by National Niemann‐Pick Foundation; K‐L. W. by MRC (MR/K015338/1); Medical Research Council UK. F.M.P. is a Royal Society Wolfson Research Merit Award holder.
All experiments were conducted in compliance with the ARRIVE guidelines. The authors have no conflict of interest to declare.
References
- Allan S. M. and Rothwell N. J. (2001) Cytokines and acute neurodegeneration. Nat. Rev. Neurosci. 2, 734–744. [DOI] [PubMed] [Google Scholar]
- Ballabio A. and Gieselmann V. (2009) Lysosomal disorders: from storage to cellular damage. Biochim. Biophys. Acta 1793, 684–696. [DOI] [PubMed] [Google Scholar]
- Baudry M., Yao Y., Simmons D., Liu J. and Bi X. (2003) Postnatal development of inflammation in a murine model of Niemann‐Pick type C disease: immunohistochemical observations of microglia and astroglia. Exp. Neurol. 184, 887–903. [DOI] [PubMed] [Google Scholar]
- Bellettato C. M. and Scarpa M. (2010) Pathophysiology of neuropathic lysosomal storage disorders. J. Inherit. Metab. Dis. 33, 347–362. [DOI] [PubMed] [Google Scholar]
- Bendelac A., Savage P. B. and Teyton L. (2007) The biology of NKT cells. Annu. Rev. Immunol. 25, 297–336. [DOI] [PubMed] [Google Scholar]
- Borbon I., Totenhagen J., Fiorenza M. T., Canterini S., Ke W., Trouard T. and Erickson R. P. (2012) Niemann‐Pick C1 mice, a model of “juvenile Alzheimer's disease”, with normal gene expression in neurons and fibrillary astrocytes show long term survival and delayed neurodegeneration. J. Alzheimers Dis. 30, 875–887. [DOI] [PubMed] [Google Scholar]
- Brennan P. J., Brigl M. and Brenner M. B. (2013) Invariant natural killer T cells: an innate activation scheme linked to diverse effector functions. Nat. Rev. Immunol. 13, 101–117. [DOI] [PubMed] [Google Scholar]
- Brown D. E., Thrall M. A., Walkley S. U., Wenger D. A., Mitchell T. W., Smith M. O., Royals K. L., March P. A. and Allison R. W. (1994) Feline Niemann‐Pick disease type C. Am. J. Pathol. 144, 1412–1415. [PMC free article] [PubMed] [Google Scholar]
- Butcher E. C. and Picker L. J. (1996) Lymphocyte homing and homeostasis. Science 272, 60–66. [DOI] [PubMed] [Google Scholar]
- Chen F. W., Li C. and Ioannou Y. A. (2010) Cyclodextrin induces calcium‐dependent lysosomal exocytosis. PLoS ONE 5, e15054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X., Wang X., Keaton J. M., Reddington F., Illarionov P. A., Besra G. S. and Gumperz J. E. (2007) Distinct endosomal trafficking requirements for presentation of autoantigens and exogenous lipids by human CD1d molecules. J. Immunol. 178, 6181–6190. [DOI] [PubMed] [Google Scholar]
- Chovatiya R. and Medzhitov R. (2014) Stress, inflammation, and defense of homeostasis. Mol. Cell 54, 281–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cologna S. M., Cluzeau C. V., Yanjanin N. M. et al (2014) Human and mouse neuroinflammation markers in Niemann‐Pick disease, type C1. J. Inherit. Metab. Dis. 37, 83–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cologna S. M., Jiang X. S., Backlund P. S. et al (2012) Quantitative proteomic analysis of Niemann‐Pick disease, type C1 cerebellum identifies protein biomarkers and provides pathological insight. PLoS ONE 7, e47845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox T. M. and Cachon‐Gonzalez M. B. (2012) The cellular pathology of lysosomal diseases. J. Pathol. 226, 241–254. [DOI] [PubMed] [Google Scholar]
- Cyster J. G. and Schwab S. R. (2012) Sphingosine‐1‐phosphate and lymphocyte egress from lymphoid organs. Annu. Rev. Immunol. 30, 69–94. [DOI] [PubMed] [Google Scholar]
- Davies J. P., Chen F. W. and Ioannou Y. A. (2000) Transmembrane molecular pump activity of Niemann‐Pick C1 protein. Science 290, 2295–2298. [DOI] [PubMed] [Google Scholar]
- Davis L. C., Morgan A. J., Chen J. L. et al (2012) NAADP activates two‐pore channels on T cell cytolytic granules to stimulate exocytosis and killing. Curr. Biol. 22, 2331–2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixit S. S., Jadot M., Sohar I., Sleat D. E., Stock A. M. and Lobel P. (2011) Loss of Niemann‐Pick C1 or C2 protein results in similar biochemical changes suggesting that these proteins function in a common lysosomal pathway. PLoS ONE 6, e23677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Futerman A. H. and van Meer G. (2004) The cell biology of lysosomal storage disorders. Nat. Rev. Mol. Cell Biol. 5, 554–565. [DOI] [PubMed] [Google Scholar]
- Gadola S. D., Silk J. D., Jeans A. et al (2006) Impaired selection of invariant natural killer T cells in diverse mouse models of glycosphingolipid lysosomal storage diseases. J. Exp. Med. 203, 2293–2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gapin L., Matsuda J. L., Surh C. D. and Kronenberg M. (2001) NKT cells derive from double‐positive thymocytes that are positively selected by CD1d. Nat. Immunol. 2, 971–978. [DOI] [PubMed] [Google Scholar]
- Gonzalez‐Scarano F. and Baltuch G. (1999) Microglia as mediators of inflammatory and degenerative diseases. Annu. Rev. Neurosci. 22, 219–240. [DOI] [PubMed] [Google Scholar]
- Griffiths M. R., Gasque P. and Neal J. W. (2009) The multiple roles of the innate immune system in the regulation of apoptosis and inflammation in the brain. J. Neuropathol. Exp. Neurol. 68, 217–226. [DOI] [PubMed] [Google Scholar]
- Higgins M. E., Davies J. P., Chen F. W. and Ioannou Y. A. (1999) Niemann‐Pick C1 is a late endosome‐resident protein that transiently associates with lysosomes and the trans‐Golgi network. Mol. Genet. Metab. 68, 1–13. [DOI] [PubMed] [Google Scholar]
- Jeyakumar M., Thomas R., Elliot‐Smith E. et al (2003) Central nervous system inflammation is a hallmark of pathogenesis in mouse models of GM1 and GM2 gangliosidosis. Brain 126, 974–987. [DOI] [PubMed] [Google Scholar]
- Jmoudiak M. and Futerman A. H. (2005) Gaucher disease: pathological mechanisms and modern management. Br. J. Haematol. 129, 178–188. [DOI] [PubMed] [Google Scholar]
- Kennedy B. E., Madreiter C. T., Vishnu N., Malli R., Graier W. F. and Karten B. (2014) Adaptations of energy metabolism associated with increased levels of mitochondrial cholesterol in Niemann‐Pick type C1‐deficient cells. J. Biol. Chem. 289, 16278–16289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kono H. and Rock K. L. (2008) How dying cells alert the immune system to danger. Nat. Rev. Immunol. 8, 279–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanier L. L. (2008) Evolutionary struggles between NK cells and viruses. Nat. Rev. Immunol. 8, 259–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd‐Evans E., Morgan A. J., He X. et al (2008) Niemann‐Pick disease type C1 is a sphingosine storage disease that causes deregulation of lysosomal calcium. Nat. Med. 14, 1247–1255. [DOI] [PubMed] [Google Scholar]
- Loftus S. K., Morris J. A., Carstea E. D. et al (1997) Murine model of Niemann‐Pick C disease: mutation in a cholesterol homeostasis gene. Science 277, 232–235. [DOI] [PubMed] [Google Scholar]
- Lopez M. E. and Scott M. P. (2013) Genetic dissection of a cell‐autonomous neurodegenerative disorder: lessons learned from mouse models of Niemann‐Pick disease type C. Dis. Models Mech. 6, 1089–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maceyka M. and Spiegel S. (2014) Sphingolipid metabolites in inflammatory disease. Nature 510, 58–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendiratta S. K., Martin W. D., Hong S., Boesteanu A., Joyce S. and Van Kaer L. (1997) CD1d1 mutant mice are deficient in natural T cells that promptly produce IL‐4. Immunity 6, 469–477. [DOI] [PubMed] [Google Scholar]
- Millat G., Marcais C., Rafi M. A., Yamamoto T., Morris J. A., Pentchev P. G., Ohno K., Wenger D. A. and Vanier M. T. (1999) Niemann‐Pick C1 disease: the I1061T substitution is a frequent mutant allele in patients of Western European descent and correlates with a classic juvenile phenotype. Am. J. Hum. Genet. 65, 1321–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan A. J., Platt F. M., Lloyd‐Evans E. and Galione A. (2011) Molecular mechanisms of endolysosomal Ca2+ signalling in health and disease. Biochem. J. 439, 349–374. [DOI] [PubMed] [Google Scholar]
- Napoli I. and Neumann H. (2009) Microglial clearance function in health and disease. Neuroscience 158, 1030–1038. [DOI] [PubMed] [Google Scholar]
- Napoli I. and Neumann H. (2010) Protective effects of microglia in multiple sclerosis. Exp. Neurol. 225, 24–28. [DOI] [PubMed] [Google Scholar]
- Olofsson P. S., Rosas‐Ballina M., Levine Y. A. and Tracey K. J. (2012) Rethinking inflammation: neural circuits in the regulation of immunity. Immunol. Rev. 248, 188–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palin K., Cunningham C., Forse P., Perry V. H. and Platt N. (2008) Systemic inflammation switches the inflammatory cytokine profile in CNS Wallerian degeneration. Neurobiol. Dis. 30, 19–29. [DOI] [PubMed] [Google Scholar]
- Parra J., Klein A. D., Castro J., Morales M. G., Mosqueira M., Valencia I., Cortes V., Rigotti A. and Zanlungo S. (2011) Npc1 deficiency in the C57BL/6J genetic background enhances Niemann‐Pick disease type C spleen pathology. Biochem. Biophys. Res. Commun. 413, 400–406. [DOI] [PubMed] [Google Scholar]
- Pekny M. and Nilsson M. (2005) Astrocyte activation and reactive gliosis. Glia 50, 427–434. [DOI] [PubMed] [Google Scholar]
- Perry V. H., Newman T. A. and Cunningham C. (2003) The impact of systemic infection on the progression of neurodegenerative disease. Nat. Rev. Neurosci. 4, 103–112. [DOI] [PubMed] [Google Scholar]
- Platt F. M. (2014) Sphingolipid lysosomal storage disorders. Nature 510, 68–75. [DOI] [PubMed] [Google Scholar]
- Platt F. M., Boland B. and van der Spoel A. C. (2012) The cell biology of disease: lysosomal storage disorders: the cellular impact of lysosomal dysfunction. J. Cell Biol. 199, 723–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platt F. M., Wassif C., Colaco A., Dardis A., Lloyd‐Evans E., Bembi B. and Porter F. D. (2014) Disorders of cholesterol metabolism and their unanticipated convergent mechanisms of disease. Annu. Rev. Genomics Hum. Genet. 15, 173–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pressey S. N., Smith D. A., Wong A. M., Platt F. M. and Cooper J. D. (2012) Early glial activation, synaptic changes and axonal pathology in the thalamocortical system of Niemann‐Pick type C1 mice. Neurobiol. Dis. 45, 1086–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ransohoff R. M. and Perry V. H. (2009) Microglial physiology: unique stimuli, specialized responses. Annu. Rev. Immunol. 27, 119–145. [DOI] [PubMed] [Google Scholar]
- Ribeiro I., Marcao A., Amaral O., Sa Miranda M. C., Vanier M. T. and Millat G. (2001) Niemann‐Pick type C disease: NPC1 mutations associated with severe and mild cellular cholesterol trafficking alterations. Hum. Genet. 109, 24–32. [DOI] [PubMed] [Google Scholar]
- Rodriguez‐Lafrasse C., Rousson R., Pentchev P. G., Louisot P. and Vanier M. T. (1994) Free sphingoid bases in tissues from patients with type C Niemann‐Pick disease and other lysosomal storage disorders. Biochim. Biophys. Acta 1226, 138–144. [DOI] [PubMed] [Google Scholar]
- Rosenbaum A. I. and Maxfield F. R. (2011) Niemann‐Pick type C disease: molecular mechanisms and potential therapeutic approaches. J. Neurochem. 116, 789–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz M. L., Tecedor L., Chang M. and Davidson B. L. (2011) Clarifying lysosomal storage diseases. Trends Neurosci. 34, 401–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Settembre C., Fraldi A., Medina D. L. and Ballabio A. (2013) Signals from the lysosome: a control centre for cellular clearance and energy metabolism. Nat. Rev. Mol. Cell Biol. 14, 283–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen D., Wang X., Li X. et al (2012) Lipid storage disorders block lysosomal trafficking by inhibiting a TRP channel and lysosomal calcium release. Nat. Commun. 3, 731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith D., Wallom K. L., Williams I. M., Jeyakumar M. and Platt F. M. (2009) Beneficial effects of anti‐inflammatory therapy in a mouse model of Niemann‐Pick disease type C1. Neurobiol. Dis. 36, 242–251. [DOI] [PubMed] [Google Scholar]
- Speak A. O., Cerundolo V. and Platt F. M. (2008) CD1d presentation of glycolipids. Immunol. Cell Biol. 86, 588–597. [DOI] [PubMed] [Google Scholar]
- Speak A. O., Platt N., Salio M. et al (2012) Invariant natural killer T cells are not affected by lysosomal storage in patients with Niemann‐Pick disease type C. Eur. J. Immunol. 42, 1886–1892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speak A. O., Te Vruchte D., Davis L. C. et al (2014) Altered distribution and function of natural killer cells in murine and human Niemann‐Pick disease type C1. Blood 123, 51–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein V. M., Crooks A., Ding W. et al (2012) Miglustat improves purkinje cell survival and alters microglial phenotype in feline Niemann‐Pick disease type C. J. Neuropathol. Exp. Neurol. 71, 434–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamari F., Chen F. W., Li C., Chaudhari J. and Ioannou Y. A. (2013) PKC activation in Niemann pick C1 cells restores subcellular cholesterol transport. PLoS ONE 8, e74169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanier M. T. (2010) Niemann‐Pick disease type C. Orphanet J. Rare Dis. 5, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitner E. B., Platt F. M. and Futerman A. H. (2010) Common and uncommon pathogenic cascades in lysosomal storage diseases. J. Biol. Chem. 285, 20423–20427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vivier E., Raulet D. H., Moretta A., Caligiuri M. A., Zitvogel L., Lanier L. L., Yokoyama W. M. and Ugolini S. (2011) Innate or adaptive immunity? The example of natural killer cells. Science 331, 44–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter M., Chen F. W., Tamari F., Wang R. and Ioannou Y. A. (2009) Endosomal lipid accumulation in NPC1 leads to inhibition of PKC, hypophosphorylation of vimentin and Rab9 entrapment. Biol. Cell 101, 141–152. [DOI] [PubMed] [Google Scholar]
- Walzer T., Chiossone L., Chaix J. et al (2007) Natural killer cell trafficking in vivo requires a dedicated sphingosine 1‐phosphate receptor. Nat. Immunol. 8, 1337–1344. [DOI] [PubMed] [Google Scholar]
- Wassif C. A., Cross J. L., Iben J. et al (2015) High incidence of unrecognized visceral/neurological late‐onset Niemann‐Pick disease, type C prediced by analysis of massively parallel sequencing data sets. Genet. Med. doi:1038/gim.2015.25 [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams I. M., Wallom K. L., Smith D. A., Al Eisa N., Smith C. and Platt F. M. (2014) Improved neuroprotection using miglustat, curcumin and ibuprofen as a triple combination therapy in Niemann‐Pick disease type C1 mice. Neurobiol. Dis. 67, 9–17. [DOI] [PubMed] [Google Scholar]
- Zhang M., Strnatka D., Donohue C., Hallows J. L., Vincent I. and Erickson R. P. (2008) Astrocyte‐only Npc1 reduces neuronal cholesterol and triples life span of Npc1‐/‐ mice. J. Neurosci. Res. 86, 2848–2856. [DOI] [PMC free article] [PubMed] [Google Scholar]