

ABSTRACT
All higher organisms negotiate a truce with their commensal microbes and battle pathogenic microbes on a daily basis. Much attention has been given to the role of the innate immune system in controlling intestinal microbes and to the strategies used by intestinal microbes to overcome the host immune response. However, it is becoming increasingly clear that the metabolisms of intestinal microbes and their hosts are linked and that this interaction is equally important for host health and well-being. For instance, an individual's array of commensal microbes can influence their predisposition to chronic metabolic diseases such as diabetes and obesity. A better understanding of host–microbe metabolic interactions is important in defining the molecular bases of these disorders and could potentially lead to new therapeutic avenues. Key advances in this area have been made using Drosophila melanogaster. Here, we review studies that have explored the impact of both commensal and pathogenic intestinal microbes on Drosophila carbohydrate and lipid metabolism. These studies have helped to elucidate the metabolites produced by intestinal microbes, the intestinal receptors that sense these metabolites, and the signaling pathways through which these metabolites manipulate host metabolism. Furthermore, they suggest that targeting microbial metabolism could represent an effective therapeutic strategy for human metabolic diseases and intestinal infection.
KEY WORDS: Drosophila melanogaster, Commensal, Metabolism, Microbiota, Pathogen
Drosophila Collection: Drosophila melanogaster is an ideal host in which to dissect the impact of intestinal microbes on host metabolism.
Introduction
Shortly after a sterile gestation and birth, our intestines become colonized by non-pathogenic intestinal microbes that we term our commensal microbiota. These are not a random assortment of organisms but rather a diverse community of microbes that coexist and, under ideal circumstances, maintain a mutualistic, symbiotic relationship with us. Periodically, this community is disrupted by infection and/or antibiotic treatment. Invasion of the intestine by pathogens might edge out members of the commensal microbiota by competing for certain intestinal niches, by creating conditions within the intestine that do not favor commensal growth, or by activating a non-specific innate immune response. Antibiotic treatments, which few of us escape during our lifetimes, are intended to target a particular pathogen, but invariably result in collateral damage within the intestinal microbiota owing to lack of specificity (Fouhy et al., 2012; Relman, 2012; Monira et al., 2013). In the absence of transplantation of intestinal microbiota harvested from a healthy host, restoration of equilibrium within the disrupted microbial community can be delayed by weeks or even months (Reid et al., 2011; Relman, 2012; Nobel et al., 2015; Stewardson et al., 2015).
Derangements of our intestinal microbiota can impact growth and development in childhood and contribute to the pathophysiology of chronic metabolic diseases such as diabetes and obesity (Subramanian et al., 2014; Miele et al., 2015). Although the complex and changing conditions that determine the fluctuations of the intestinal microbial community over a lifetime are poorly understood, mounting evidence suggests that the ability to sculpt the intestinal microbial community could lead to new therapeutic modalities for the prevention of chronic metabolic diseases and intestinal infection. To understand host–microbe interactions in mammals, investigators must consider the complex, interdependent metabolic pathways of the host and the trillions of microbes residing in the intestine. In contrast, the simpler microbiota and signaling systems of the fruit fly Drosophila melanogaster as well as the genetic tools available for use in this model organism present the investigator with a unique opportunity to test hypotheses regarding the impact of commensal and pathogenic intestinal microbes on host metabolism in a more controlled and targeted fashion.
Here, we will review what has been learned from the Drosophila model about manipulation of host metabolism by both the commensal microbiota of the intestine and intestinal pathogens. Specifically, we will discuss the contributions made by Drosophila researchers in elucidating the key metabolites secreted by intestinal commensals and pathogens, host sensing of these metabolites, and the endocrine signals released from the host intestine in response to these metabolites.
After comparing the anatomy and metabolic regulatory pathways of the Drosophila and mammalian intestines, we will review in turn studies elucidating the impact of commensal bacteria, bacterial pathogens and viruses of the intestine on host metabolism. Taken together, these studies show that intestinal microbes manipulate host metabolism and, furthermore, that the metabolisms of the host and its intestinal inhabitants are intricately intertwined. Small changes in microbial metabolism can result in large fluctuations in host metabolic homeostasis. We propose that this knowledge can be exploited in the design of prebiotic and probiotic therapies to treat metabolic disease and mitigate the metabolic derangements caused by intestinal infections.
Comparison of the Drosophila and mammalian intestines and their associated metabolic signaling pathways
Intestinal structures and cell types
In order to extrapolate from studies of flies to mammals in a thoughtful way, the parallels and differences between the intestines of these two organisms must first be considered. The mammalian intestinal epithelium consists of enterocytes, goblet cells, enteroendocrine cells, Paneth cells and stem cells. These cells are arranged to form protrusions and invaginations termed villi and crypts, respectively (Fig. 1A). Ubiquitously distributed goblet cells are dedicated to the synthesis of the intestinal mucus, a proteoglycan that covers and protects the intestinal epithelium (Birchenough et al., 2015). Enterocytes are principally responsible for absorption of nutrients. To aid with this, their luminal face is lined with small cellular protrusions known as microvilli, which increase surface area.
Fig. 1.
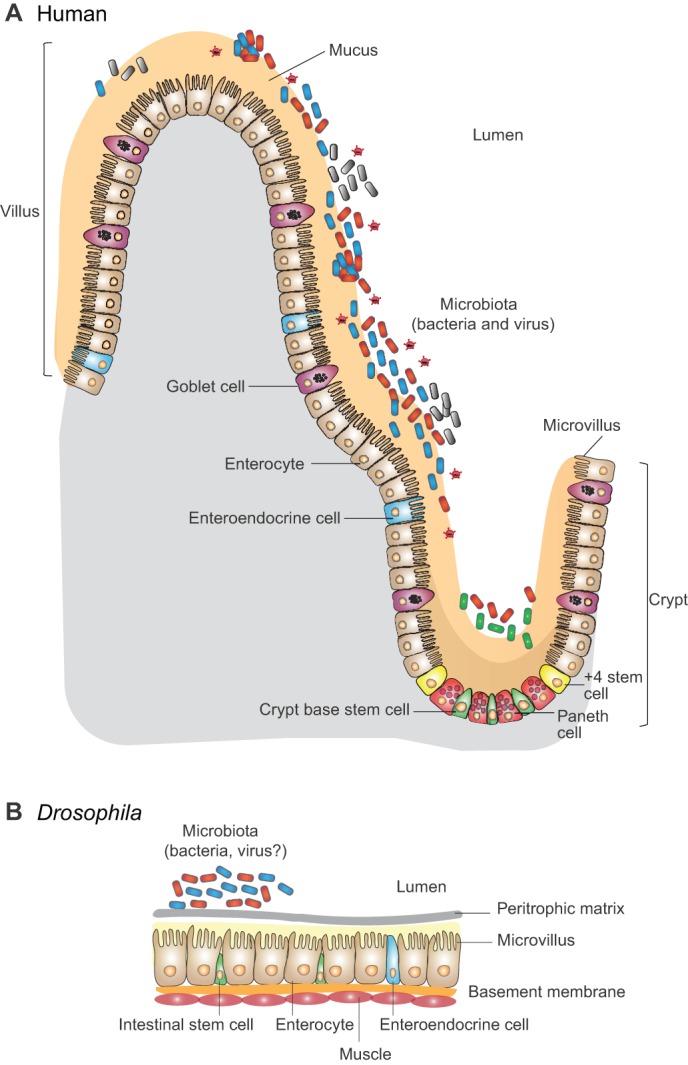
A comparison of the mammalian and Drosophila melanogaster intestines. (A) The contour of the mammalian intestinal epithelium consists of peaks and valleys termed villi (singular: villus) and crypts, respectively. Several cell types with distinct functions are found within the epithelium. Enterocytes, whose surface area is maximized by numerous protrusions known as microvilli, are principally responsible for nutrient absorption. Goblet cells, which are distributed throughout the epithelium, secrete the protective mucus layer composed of polysaccharides and proteins that covers the epithelial surface. Located in crypts, enteroendocrine cells secrete small bioactive peptides in response to signals from nutrients and commensal bacteria in the intestinal lumen. Paneth cells, which are found at the crypt base, secrete antimicrobial peptides and create a stem cell niche. Two stem cell populations are found in the mammalian intestine. Stem cells positioned at the crypt base divide at a constant rate to replenish the epithelium. Division of stem cells located at the +4 position is activated by intestinal insult or infection. The resident commensal microbiota is found within and on top of the mucus layer. (B) The Drosophila intestinal epithelium lacks villi and crypts and consists of only three cell types: enterocytes, enteroendocrine cells and stem cells. The peritrophic membrane (or matrix), a structure analogous to intestinal mucus, covers the epithelial surface. The intestinal lumen is colonized by a much less diverse microbiota.
Nutrients in the mammalian intestinal lumen are sensed by enteroendocrine cells through active transport across the cell membrane or activation of G-protein coupled receptors (GPCRs) on the cell surface (Gribble and Reimann, 2016). Interestingly, some of these GPCRs also function as taste (gustatory) receptors in the mouth (Jang et al., 2007; Dotson et al., 2008; Kokrashvili et al., 2009). For this reason, enteroendocrine cells have been described as the taste organs of the gastrointestinal tract (Sternini et al., 2008; Breer et al., 2012). Activation of these sensors on enteroendocrine cells stimulates the release of small peptide hormones from cytoplasmic vesicles into the local extracellular milieu and systemic circulation. These peptides modulate carbohydrate metabolism, lipid metabolism, intestinal peristalsis and satiety (Drucker, 2001; Naslund and Hellstrom, 2007; Mellitzer and Gradwohl, 2011). Therefore, enteroendocrine cells coordinate local and systemic metabolic responses with the intestinal contents.
The intestinal epithelium is maintained by active and quiescent populations of stem cells (Umar, 2010). The former, positioned at the base of crypts, continually renews the epithelium by producing cells that migrate up to the villus tips and are eventually sloughed off. A second, quiescent, population of stem cells, which is positioned four cells away from the crypt base, divides in response to intestinal insult. Paneth cells, which neighbor the active population of stem cells within the crypt, play a role in maintaining the stem cell niche and also secrete antibacterial peptides and enzymes (Sato et al., 2011; Clevers and Bevins, 2013).
Unlike the mammalian intestinal epithelium, the luminal surface of the Drosophila intestine is unconvoluted and comprises only three cell types: enterocytes, enteroendocrine cells and stem cells (Fig. 1B). Only one type of intestinal stem cell has been identified in Drosophila (Micchelli and Perrimon, 2006; Ohlstein and Spradling, 2006). These cells replenish all intestinal cell types that are lost as a result of normal senescence or acute intestinal injury (Amcheslavsky et al., 2009; Apidianakis et al., 2009; Buchon et al., 2009; Ren et al., 2010). The Drosophila epithelium is covered by the peritrophic matrix, a structure that is analogous to intestinal mucus; this matrix consists of chitin (a polymer of N-acetylglucosamine), and proteins such as peritrophins and drosocrystallin (Lehane, 1997; Kuraishi et al., 2011; Moussian, 2013; Shibata et al., 2015). Enterocytes carry out digestive, absorptive and innate immune functions of the intestine (Marianes and Spradling, 2013; Dutta et al., 2015). In contrast to the wide body of published research elucidating the function of mammalian enteroendocrine cells, Drosophila enteroendocrine cells remain relatively unexplored. However, these cells also express gustatory receptors on their cell surface (Park and Kwon, 2011). In addition, they harbor vesicles filled with small peptides that regulate lipid metabolism, carbohydrate metabolism and gut peristalsis (Veenstra, 2009; Song et al., 2014; Vanderveken and O'Donnell, 2014; Kohyama-Koganeya et al., 2015); therefore, they possess all the components required to fulfill the same function as mammalian enteroendocrine cells in coordinating a systemic response to nutrients and metabolites in the gut lumen.
Control of systemic carbohydrate mobilization and storage by the mammalian and fly intestinal epithelia
Appropriate carbohydrate utilization and storage is important for the maintenance of metabolic homeostasis in all animals and, therefore, is tightly controlled by the endocrine system. In response to ingestion of nutrients, both mammals and Drosophila release small peptide hormones termed insulin or insulin-like peptides from specialized cells (Barbieri et al., 2003; Nassel et al., 2013). Insulin receptors on adipose tissue and muscle cells sense these peptides and activate the insulin/insulin-like growth factor signaling (IIS) pathways, which inhibit gluconeogenesis and glycogenolysis and promote glycogen and triglyceride storage (Khan and Pessin, 2002; Teleman, 2010; Padmanabha and Baker, 2014). In mammals, low levels of carbohydrates result in secretion of glucagon, an endocrine peptide that activates glycogen catabolism (Marroqui et al., 2014). In Drosophila, adipokinetic hormone seems to fulfill the role of glucagon (Bednarova et al., 2013).
In mammals, the commensal microbiota plays a role in the regulation of systemic glucagon and insulin secretion through its production of short-chain fatty acids (SCFAs), whose aliphatic tails contain fewer than six carbon atoms (Shen et al., 2013; Everard and Cani, 2014). Microbial metabolism within the intestine, which results in excretion of acetate [two carbon atoms (C2)], propionate (C3) and butyrate (C4), accounts for 95% of the body's SCFAs. These bacterial metabolites not only serve as nutrition for enterocytes but are also sensed by the GPCRs GPR109A, FFAR2 and FFAR3 on intestinal cells (Fig. 2). GPR109A is expressed in colonic enterocytes, where activation by butyrate suppresses inflammation. Both FFAR2 and FFAR3 are expressed in enteroendocrine L-cells, which secrete regulatory peptides such as protein YY (PYY) and glucagon-like peptide 1 (GLP-1) in response to SCFAs. GLP-1 inhibits glucagon release and promotes insulin secretion, resulting in lowering of blood glucose, whereas PYY reduces appetite (Kasubuchi et al., 2015; Psichas et al., 2015). Thus, bacteria communicate the status of their own metabolism to enteroendocrine cells through the metabolites they produce. The enteroendocrine cells, in turn, appropriately adjust host food intake and metabolism.
Fig. 2.

Signaling pathways through which gut microbiota modulate host carbohydrate and lipid metabolism. Consumed nutrients are metabolized by the gut microbiota to produce bioactive metabolites that are sensed by the mammalian and possibly the Drosophila epithelium. Short-chain fatty acids (SCFAs), the product of bacterial carbohydrate fermentation, and other bacterial metabolites are taken up by enterocytes (note that the villi are not shown in this representation) and converted into metabolically active molecules such as acetyl-CoA, or sensed by specific G-protein-coupled receptors (GPCRs) expressed on the surfaces of enterocytes and enteroendocrine cells. This, in turn, triggers release of enteroendocrine peptides into the systemic circulation and activates signaling cascades that modulate host carbohydrate and lipid utilization both in the intestine and systemically. Dietary triacylglycerides (TAGs) are hydrolyzed into monoacylglycerides (MAGs) and free fatty acids (FFAs) before being absorbed by enterocytes. These lipids accumulate within the leaflets of the endoplasmic reticulum (ER) membrane and are then packaged either into lipid droplets for storage or into lipoprotein particles for transport to other tissues.
Although the regulatory cascade is not as well defined in flies, all indications are that a similar process is at work. The regulatory peptide IMPL2, which is synthesized and secreted by enteroendocrine and possibly other cell types, blocks insulin signaling (Garbe et al., 1993; Marianes and Spradling, 2013; Sarraf-Zadeh et al., 2013; Dutta et al., 2015). It seems to function by directly binding to and inhibiting the function of the Drosophila insulin-like peptides (Dilps), which activate the insulin receptor (Alic et al., 2011). Furthermore, evidence suggests that intestinal acetate increases signaling through the insulin pathway by repressing transcription of IMPL2 (Shin et al., 2011; Hang et al., 2014; Kwon et al., 2015). Taken together, these data suggest that a receptor for acetate is present in the Drosophila intestine, possibly on the surface of enteroendocrine cells, and that, similarly to the mammalian system, activation of this receptor modulates insulin signaling.
Lipid uptake and mobilization in the intestine
In mammals, continued ingestion of lipid-laden foods correlates with the development of diabetes and obesity (Moran-Ramos et al., 2012; de Souza et al., 2015), and modulation of the metabolic response to dietary lipids through manipulation of the intestinal microbiota has been proposed as a therapeutic option for these diseases (Cani et al., 2008; Serino et al., 2014). An in-depth understanding of how intestinal microbiota modulate lipid uptake is key to the development of such therapies.
The dietary lipids of mammals are principally composed of triacylglycerols (TAGs). These lipids are emulsified by bile and then enzymatically degraded by pancreatic lipase, yielding free fatty acids (FFAs) and monoacylglycerols (MAGs) in the intestinal lumen (Fig. 2) (Mansbach and Gorelick, 2007). Through as-yet poorly defined pathways, these lipid products are taken up by enterocytes by mechanisms that include both passive diffusion and active transport. The fatty acid transport proteins (FATPs), which are hypothesized to function as acyl-CoA synthases and/or transporters, the fatty acid translocase FAT (CD36) and the plasma-membrane-associated fatty-acid-binding protein FABpm have all been implicated in fatty acid uptake from the intestinal lumen (Mansbach and Gorelick, 2007). Once imported, fatty acids are transported to the endoplasmic reticulum, where they are reassembled into TAGs, which accumulate between the leaflets of the endoplasmic reticulum membrane. These collections of TAGs eventually bud off to form lipid-storage droplets or are packaged into lipoprotein particles, known as chylomicrons, within the ER lumen and released into the lymph (Buttet et al., 2014). In addition to proteins, chylomicrons contain large amounts of TAG as well as cholesterol and fat-soluble vitamins (Iqbal and Hussain, 2009). They are surrounded by a phospholipid monolayer that is principally composed of phosphatidylcholine. Chylomicron-associated proteins as well as the particles themselves are synthesized by enterocytes expressly for transport of dietary fat.
Many aspects of the lipid uptake mechanisms of the Drosophila intestine remain largely unexplored. A biliary system is not present in the fly intestine and, although an emulsifying substance might be secreted by enterocytes, none has been identified to date. In fact, one might argue that, because TAG is not abundant in the natural food sources of Drosophila (such as rotting fruit), an emulsifying agent analogous to bile is not essential for lipid absorption in this organism. Luminal digestion of TAG is carried out by the Magro protein, a homolog of the mammalian gastric lipase (Sieber and Thummel, 2009, 2012). Absorption of fatty acids from the intestinal lumen has not been studied. However, homologs of FATP, CD36 and FABPpm are all present in the Drosophila genome (Adams et al., 2000). Absorbed dietary lipids are presumably then trafficked to the endoplasmic reticulum, where they are either retained in lipid droplets or packaged for transport through the hemolymph to the specialized Drosophila adipose tissue known as the fat body for storage, or to other organs for catabolism. Tachykinin, an enteroendocrine-cell-derived regulatory peptide, activates mobilization of lipids from the intestine to the hemolymph (Song et al., 2014). Although the precise mechanism by which Tachykinin acts has not been elucidated, alterations in the transcriptional profile of genes involved in lipid metabolism seem to play a role.
Three lipoproteins – lipophorin (Lpp), the lipid transfer particle (LTP) and Crossveinless D (Cv-D) – have been implicated in systemic lipid transport in Drosophila (Palm et al., 2012). Lpp is responsible for the transport of 95% of the lipids carried in the hemolymph, whereas LTP and Cv-D contribute to a much lesser extent. Knockdown of both LTP and Lpp leads to accumulation of lipid droplets in the intestine, demonstrating that these proteins are required for mobilization of intestinal lipid stores and dietary fat (Palm et al., 2012). By facilitating recruitment of Lpp to the Lpp receptor, LTP, in particular, seems to be crucial for the transfer of lipids from the gut to Lpp and for the uptake of lipids by distant tissues (Rodriguez-Vazquez et al., 2015).
Whereas the mammalian proteins dedicated to the transport of dietary lipids are synthesized by enterocytes, Lpp and LTP are synthesized in the fat body and are transported to the intestine. Furthermore, whereas chylomicrons principally contain TAG and phosphatidylcholine, the principal lipids associated with lipophorin in the hemolymph are diacylglycerol and phosphatidylethanolamine (Panakova et al., 2005; Palm et al., 2012). However, the evolutionary and physiological significance of these small differences in lipid transport has not been investigated.
Commensal microbiota and host metabolism: impact on development and health
A universal feature of organisms with open digestive tracts is colonization of the gastrointestinal tract by a characteristic commensal microbiota. This microbiota, which thrives on the nutrients produced by digestion of the host's diet and intestinal secretions, is shaped by host-specific selective pressures such as the intestinal environment, food preference and eating habits (Ni et al., 2015). In turn, the microbiota manipulates host metabolism by altering nutrient availability, generating essential nutrients, and excreting metabolites that serve as a form of interspecies communication (Fig. 3). As touched upon above, this complex interaction plays a key role in childhood growth and development, and in adult metabolic homeostasis (Ukhanova et al., 2012; Ahmed et al., 2014).
Fig. 3.

Pathogens and non-pathogens modulate intestinal metabolism differently. The metabolites produced by commensal (non-pathogenic) bacteria play a key role in maintaining gut homeostasis, and bacteriophages trim and tailor the bacterial population. The peritropic membrane comprises chitin and proteins. By secreting proteases and chitinases, bacterial pathogens can digest, and thus weaken, the peritrophic barrier, allowing these bacteria to invade the intestinal epithelium. Alternatively, a non-invasive pathogen might interrupt signaling between commensals and the host intestine by consuming commensal metabolites or producing virulence factors that mute host signaling pathways. If intestinal lipid metabolism is dysregulated, the resulting lipid droplets within enterocytes can provide a platform for replication of viruses that exploit these organelles, thus promoting viral superinfection.
Because of its less-complex and more-tractable microbiota in comparison to mammals, Drosophila melanogaster provides a useful model in which to study the governing principles of the host's metabolic interaction with its microbiota. Whereas humans are exposed to the diverse microbiotas that populate a wide array of plant and animal food sources, wild Drosophila have a more limited diet of overripe fruits and vegetables, decomposing plants, and fungi. Accordingly, the intestinal microbiota of the wild fly consists of five to 30 taxa (Wong et al., 2011; Broderick and Lemaitre, 2012), as compared with the greater than 500 taxa in the human intestinal microbiota (The Human Microbiome Project Consortium, 2012). The laboratory fly's microbiota is further limited by its artificial environment. The intestinal microbiome of a captive fly consists principally of the microbes expelled by its predecessors into the fly medium (Blum et al., 2013). In spite of this, some bacterial species, such as those belonging to the genera Acetobacteraceae and Lactobacillaceae, are found in both laboratory-raised and wild Drosophila (Cox and Gilmore, 2007). Of course, it is possible that the genomes of the Acetobacteraceae and Lactobacillaceae found in wild and laboratory-raised flies have diverged in metabolically important ways owing to the distinct selective pressures of the laboratory and natural environments.
The simplicity of the laboratory-raised fly microbiome affords experimental tractability. Acetobacter and Lactobacillus species, which predominate in the intestines of laboratory-raised flies, are readily cultured and easily eliminated to generate axenic, or ‘germ-free’, animals. Furthermore, comparative studies of development, nutrient allocation and metabolic signaling in conventional and axenic flies suggest that the intestinal microbiota impacts the physiology of flies and mammals in parallel ways.
The microbiome and development
In the developing world, the microbiota has been shown to influence risk of malnutrition, growth retardation and cognitive delay (Smith et al., 2013; Ahmed et al., 2014; Kane et al., 2015). However, the particular microbes involved and their specific functions in these pathophysiological processes are often difficult to pinpoint. Similar to mammals, the Drosophila microbiota is important for normal development. Because the Drosophila genome is genetically tractable and less redundant than mammalian genomes, the mechanism by which the microbiome impacts development is better understood. Drosophila are generally raised on nutrient media containing a source of sugar and protein as well as yeast or yeast extract. Yeast is a rich source of lipids and B vitamins as well as proteins and carbohydrates. A decrease in the yeast content of Drosophila medium has been shown to cause developmental delay due to a reduction in signaling through the target of rapamycin (TOR) pathway, a highly conserved pathway also found in mammals (Layalle et al., 2008; Storelli et al., 2011; Wong et al., 2014). A similar developmental delay was found in axenic flies, and this developmental delay could be rescued not only by supplementation with yeast or B vitamins (Shin et al., 2011; Wong et al., 2014) but also by re-association with Acetobacter pomorum (Shin et al., 2011) or Lactobacillus plantarum (Storelli et al., 2011). Re-colonization with either of these commensal organisms activated insulin signaling, a process that is central to Drosophila development (Nassel et al., 2015).
Highlighting the power of the fly model, Shin and colleagues went on to identify the A. pomorum protein pyrroloquinoline quinone-dependent alcohol dehydrogenase (PQQ-ADH) as being required to rescue development, by generating a library of A. pomorum transposon mutants and re-associating each mutant singly with an axenic fly population (Shin et al., 2011). Finally, Shin determined that acetate, the product of PQQ-ADH, was the microbial signal that was essential for activation of insulin signaling and normal Drosophila development. In another example of the power of the Drosophila model, Erkosar et al. recently showed that the beneficial effect of L. plantarum on host development is mediated by its activation of host intestinal proteases (Erkosar et al., 2015). This effect could be replicated by intestinal overexpression of one of these proteases in germ-free larvae and negated by infection of L. plantarum-monocolonized flies with a pathogen.
Because screens of individual bacterial mutants in mice are labor intensive and costly, technologies such as signature-tagged mutagenesis (STM) and transposon sequencing (Tn-seq) have been developed to assess sub-libraries of 100 or more mutants simultaneously (Shea et al., 1996; van Opijnen et al., 2009). Importantly, using these high-throughput approaches, the presence of a mutant carrying a mutation in a gene such as PQQ-ADH, which eliminates excretion of the bioactive metabolite acetate, would be detected neither by the host nor the investigator. Thus, the approach taken by Shin, namely screening of bacterial mutants singly, is the most powerful approach for identification of bacterial genes responsible for the production of active microbial metabolites in the host intestine. Such an approach is accessible to most investigators only when undertaken in a model invertebrate host such as D. melanogaster, highlighting the potential of this model to accelerate investigations of the role of the microbiota in normal development and developmental disorders. In line with the scope of this article, below we focus specifically on insights into the role of the intestinal microbiota in the development of obesity that have been gleaned from studies of the fly intestinal microbiome.
The microbiota and obesity
In mammals, metabolites secreted by the microbiota modulate adult metabolism as well as juvenile development. For instance, SCFAs regulate appetite, insulin signaling and adipogenesis through specific GPCRs located on enteroendocrine cells, sympathetic neurons and adipose tissue (Ichimura et al., 2009, 2014; Hara et al., 2014). Not surprisingly, therefore, the intestinal microbiota can direct host storage of lipids in adipose tissue, leading to obesity. For instance, specific antibiotic-driven perturbations of the mouse microbiota during development predispose these mice to obesity (Cox et al., 2014). By contrast, when fed a high-fat diet, germ-free mice are less prone to obesity as compared with their conventionally raised counterparts (Rabot et al., 2010). However, in the mouse, the specific microbe or microbes responsible for this effect have not been identified. Experimental manipulation of the Drosophila microbiota also suggests a role in nutrient allocation and metabolism. Investigators in the Douglas and Lee labs showed that germ-free flies given access to standard fly medium develop hyperglycemia and hyperlipidemia (Shin et al., 2011; Ridley et al., 2012; Newell and Douglas, 2014; Wong et al., 2014). Recolonization with single bacterial species reversed hyperglycemia, whereas multiple bacterial partners were required to reverse hyperlipidemia (Newell and Douglas, 2014). These studies demonstrate the power of the Drosophila model as a platform on which to dissect the effects of distinct intestinal microbes on host metabolism and to experiment with the design of an intestinal microbiota that maximizes metabolic health and minimizes obesity risk.
In humans, genetic background is known to contribute to the development of diabetes and obesity (Bouchard, 1997; Ridaura et al., 2013; Basile et al., 2014). Because the intestinal microbiota also plays a role in the development of these diseases, the role of host genetics in shaping the intestinal microbiota remains a top research priority. In humans, accumulating evidence suggests that the genetic background of the host alters the intestinal microbiota and this, in turn, modulates host metabolism (Goodrich et al., 2014; Zhang et al., 2015). In one study, mice of three different genetic backgrounds with varying susceptibility to obesity and diabetes were bred in a common environment (Ussar et al., 2015). Metabolic phenotypes were found to be the product of not only the environment, but also the host genetic background. However, precise host genes were not implicated. The power and ease of Drosophila genetics make this an ideal model for such studies. In a genome-wide association study (GWAS) using the Drosophila Genetic Reference Panel (DGRP), Dobson and colleagues demonstrated that host genetic polymorphisms greatly influence microbiota-dependent nutritional phenotypes (Dobson et al., 2015). These studies suggested that a single gene mutation could, in some cases, reverse a microbiota-dependent nutritional effect. However, correlations of nutritional phenotypes with host genotype were evaluated only by testing loss-of-function mutants. Although additional genetic experiments are required to solidify these associations, this work paves the way for future mechanistic studies exploring the impact of host genotype on the host–microbiota metabolic interaction.
The intestinal microbiome affects our well-being by ensuring normal development, adequate nutrition, and appropriate carbohydrate and lipid metabolism throughout life. Therefore, the ability to maintain a microbiome ideally matched to the host would greatly improve human health. Because of the genetic tools available to Drosophila researchers and the obvious parallels to the mammalian system, this is an ideal host in which to explore the genetic barriers to the maintenance of the designer microbiome.
Bacterial pathogens and host metabolism
Anorexia, malaise and diarrhea are all symptoms of intestinal infection. In the developing world, multiple intestinal infections in rapid succession are an important cause of malnutrition, wasting and a general failure to thrive in children under five (Kotloff et al., 2013). Although the rapid transit of nutrients through the intestine that defines diarrhea is bound to be responsible for some of this, research in Drosophila has elegantly revealed pathogen impacts on host metabolism that extend beyond decreased intestinal transit time.
Mycobacterium marinum, a mycobacterial species that causes skin infections associated with abrasions acquired during water exposure, is sometimes used as a model for Mycobacterium tuberculosis (Deng et al., 2011). Early work by the Schneider laboratory showed that, when injected into the Drosophila hemolymph, this extra-intestinal pathogen caused wasting through dysregulation of insulin signaling (Dionne et al., 2006). The ultimate result was a decrease in glycogen and triglyceride stores along with an elevation in systemic glucose levels, suggesting that systemic infections might cause insulin resistance in Drosophila as they do in mammals (Gheorghita et al., 2015). The study by Dionne et al. set the stage for subsequent explorations of the impact of diarrheal pathogens on Drosophila metabolism.
It has been shown using flies that systemic infection with the intestinal pathogens Salmonella typhimurium and Listeria monocytogenes, but not the common intestinal inhabitant Enterococcus faecalis, results in anorexia (Ayres and Schneider, 2009). Development of anorexia, in turn, impacts expression of antimicrobial peptides and susceptibility to infection. Thus, the host metabolic state can alter interactions with invading pathogens by modulating the innate immune response. Interestingly, this group also reported that anorexia was induced by decreased expression of the gustatory receptor Gr28b (Ayres and Schneider, 2009), which is highly expressed in enteroendocrine cells (Buchon et al., 2013; Marianes and Spradling, 2013). This gustatory receptor, as well as others expressed on the surface of intestinal cells, provides a mechanism whereby the products of pathogenic intestinal microbes can activate signaling pathways that alter host satiety and susceptibility to infection.
The group led by David Schneider subsequently employed metabolomic studies to demonstrate that L. monocytogenes infection decreased glycogen and triglyceride stores as well as the glucose concentration in the hemolymph (Chambers et al., 2012). The group also noted that levels of the anti-oxidant uric acid were decreased. Although these changes are presumably the result of infection-induced anorexia and other bacterial impacts on the host, this study did not conclusively identify these changes in host metabolism as components of a pathogen virulence program, a host innate immune response or a specific host–pathogen interaction pathway.
Because of the speed and affordability of genetics, the comprehensive mutant and transgenic RNA interference (RNAi) lines, and the eminently accessible and extensive databases, the Drosophila model is ideally suited to rapid dissection of host–pathogen interactions (Ni et al., 2008, 2011; Cook et al., 2010; dos Santos et al., 2015). Using these tools, subsequent studies have thus far revealed three distinct pathways co-opted by intestinal pathogens to modulate host metabolism. Pseudomonas entomophila, a bacterium originally isolated from flies, causes a lethal infection when ingested (Liehl et al., 2006). By contrast, Pectobacterium carotovorum strain 15 (Ecc15) induces a strong innate immune response when ingested, but does not kill the fly (Basset et al., 2000). Chakrabarti and colleagues compared the transcriptomes of flies infected with these two bacteria and found that a number of stress response genes were selectively activated in P. entomophila infection (Chakrabarti et al., 2012). In addition, whereas transcription of antimicrobial peptides was greatly activated by infection, expression of a diptericin-lacZ reporter fusion could not be detected, leading the authors to conclude that translation but not transcription was inhibited by P. entomophila. The mechanism of translation inhibition was determined to be the result of phosphorylation of elf2α by the GCN2 kinase and inhibition of the TOR pathway, which is known to activate protein translation. The P. entomophila pore-forming toxin, monalysin, was found to be at least partially responsible for this block in translation. Although a connection between host metabolism and protein translation in the intestine was not explored in this study, we hypothesize that a burst of transcription and subsequent translation is likely part of the intestinal response to the ingestion of dietary nutrients. When this is blocked by an intestinal pathogen, the host metabolic response might be altered such that intestinal nutrients are not optimally utilized. Therefore, a block in protein translation could be a cause of wasting in particular intestinal infections.
The human intestine has evolved to detect and respond to the metabolic waste products of its commensal bacterial inhabitants, and the importance of the host response to these bacterial metabolites in human health and metabolic disease is just beginning to be appreciated (Canfora et al., 2015). Parallel symbiotic interactions between Drosophila and its commensal intestinal microbiota have been identified, although the cell types and receptors that detect these bacterial metabolites have not yet been identified (Shin et al., 2011). We hypothesize that intestinal pathogens contribute to and catabolize metabolites within the host intestine uniquely such that the host response to pathogens is distinct from its response to commensals. However, this aspect of the host–pathogen interaction remains poorly understood and is an area in which Drosophila researchers have made and are poised to make seminal contributions.
In Drosophila, unique metabolites of intestinal pathogens have been reported to activate the host intestinal innate immune system. In particular, Lee et al. determined that the metabolite uracil secreted by Ecc15 is an activator of intestinal transcription of the Drosophila gene encoding the reactive-oxygen-generating protein dual oxidase (Duox) (Lee et al., 2013). Based on the signaling pathway mediating duox activation, they proposed the existence of a GPCR that responds to uracil (Lee et al., 2015). Furthermore, they reported that this metabolite is also secreted by the intestinal pathogens Vibrio fluvialis, Shigella sonnei, Pseudomonas aeruginosa and Serratia marcescens but not by the commensal organism Commensalibacter intestini when cultured in minimal medium. Notably, Klebsiella pneumoniae, which is a normal inhabitant of the human intestine, also produced uracil in culture. Because essential metabolic pathways of bacteria are highly conserved, we propose that commensal bacteria and pathogens could differ primarily in how these metabolic pathways are regulated within the intestinal environment, resulting in differences in the repertoire of metabolites produced. Although the Lee et al. study did not specifically study the effect of Duox activation on host metabolism, we predict that the resulting activation of a non-specific intestinal innate immune response, which decreases the number of pathogenic and commensal bacteria, could result in disruption of host metabolic homeostasis.
Another example of the interaction of host and pathogen metabolisms was discovered by Hang et al. (2014). In this case, acetate, a metabolite normally supplied to the host by the commensal microbiota, was consumed by the intestinal pathogen Vibrio cholerae, leading to a decrease in insulin signaling, depletion of lipid stores in the fat body and the appearance of large lipid droplets in enterocytes. This metabolic phenomenon is likely due to the observed transcriptional activation of intestinal IMPL2, an inhibitor of insulin signaling (Honegger et al., 2008). Interestingly, overexpression of IMPL2 has recently been implicated in organ-wasting phenotypes (Figueroa-Clarevega and Bilder, 2015; Kwon et al., 2015). This presents an additional mechanism by which intestinal infection could lead to host wasting.
Manipulation of host metabolism by bacterial pathogens could have the added effect of predisposing the host to viral infection. Recently, the Cherry laboratory has demonstrated that insulin signaling protects D. melanogaster against infection by certain viruses, through activation of the MAPK signaling pathway and phosphorylation of ERK (Xu et al., 2012, 2013). This supports the hypothesis that, by suppressing insulin signaling, bacterial infection of the intestine might predispose the host to viral superinfection.
The metabolites of commensal intestinal bacteria are sensed by enterocytes, enteroendocrine cells, and possibly cells of other types in the intestine. The host responds to these bacterial signals by adjusting carbohydrate and lipid metabolism. Wasting in the setting of intestinal infection, which has been documented in both humans and Drosophila, is likely to be partially the result of pathogen interference with these ‘conversations’ between the host and its intestinal microbiota. Studies of this phenomenon in Drosophila have revealed a variety of mechanisms by which pathogens interrupt this communication (Fig. 3). Intestinal pathogens might secrete toxins that block the host translational response to bacterial signals. They might activate the innate immune response leading to a shift in the commensal population and, therefore, the bacterial metabolites produced by this population. Finally, they might silence the communication by consuming the metabolites secreted by the commensal population. The benefit to the pathogen of silencing the host–commensal communication has not yet been explored. However, these studies suggest the hypothesis that disruption of intestinal nutrient transport and metabolism leaves more dietary nutrients in the intestinal lumen. These nutrients are available to the luminal pathogen to support its growth and replication. In other words, the host wastes while the intestinal pathogen feasts.
Intestinal viruses and host metabolism
Researchers are just beginning to mine the virome of the healthy and diseased mammalian intestine (Minot et al., 2013; Norman et al., 2015). This virome includes both enteric viruses that target eukaryotic cells and viruses also known as bacteriophage, which target intestinal bacteria. Owing to the requirement for a specific host receptor, bacteriophage cause lysis in a very narrow range of bacterial hosts (Buckling and Brockhurst, 2012; Diaz-Munoz and Koskella, 2014). Therefore, it has been hypothesized that enteric bacteriophage exclude particular microbes from the intestinal milieu, thus shaping the commensal bacterial population (Mills et al., 2013). Because the intestinal microbiota plays a role in regulating host metabolism, it seems inevitable that relationships between the enteric virome and host metabolism will also be established. A similar multi-species interaction is likely to be at play in the Drosophila intestine, supporting its use as a model to explore these relationships and their importance in the maintenance of human health.
Many viruses that are pathogenic in humans, including dengue and hepatitis C, use lipid droplets as a platform for viral replication (Saka and Valdivia, 2012). In the case of hepatitis C, infection is associated with an increase in the number of lipid droplets in hepatocytes (Filipe and McLauchlan, 2015). Although the mechanism by which lipid droplets multiply in response to infection has not been elucidated, the virus might use this to amplify its own replication.
A similar virulence mechanism is observed in D. melanogaster infection by the flock house virus (FHV), a natural insect pathogen. Infection of Drosophila cells with FHV increased transcription of genes encoding lipid metabolism proteins such as the CTP:phosphocholine cytidylyltransferases, Cct1 and Cct2 (Castorena et al., 2010). These proteins, which are found in association with the surface of lipid droplets, are required for the synthesis of phosphatidylcholine from diacylglycerol (Moessinger et al., 2014). RNAi inhibition of Cct1 and Cct2 expression resulted in decreased viral replication, suggesting that FHV might also use lipid droplets as a platform for replication. Although additional studies are required, these examples suggest that use of the lipid droplet surface as a replication platform is a conserved virulence mechanism in viruses that infect both mammals and Drosophila.
Summary and future directions
Although the cast of commensal and pathogenic microbes in the Drosophila and mammalian intestines are distinct, the literature reviewed here strongly suggests that the pathways by which these microbes interact with their host intestines are highly conserved. In both, the relevant intestinal structure holds bacteria at a distance from the epithelium, provides a surface for attachment, and serves as a source of nutrition for intestinal bacteria (Ritchie et al., 2010; Kuraishi et al., 2011; Purdy and Watnick, 2011; Hang et al., 2014; Faderl et al., 2015; Tailford et al., 2015). Furthermore, the intestinal epithelium senses and responds to nutrients and microbial metabolites by altering intestinal motility and host metabolism (Drucker, 2001; LaJeunesse et al., 2010; Vanderveken and O'Donnell, 2014).
One important aspect in which these two epithelia differ is in their topology (Fig. 1). Whereas the intestinal epithelium of the fly is flat, the villi and crypts of the mammalian epithelium create invaginations in the epithelial surface that might not be accessible to all intestinal bacteria. Although the factors that enable crypt colonization have not been defined, this ability is characteristic of particular intestinal pathogens (Iimura et al., 2005; Olivier et al., 2007). Because the antimicrobial-peptide-secreting Paneth cells reside at the crypt base, we hypothesize that the high concentration of antimicrobial peptides at the crypt base shapes both the crypt microbiota and susceptibility to pathogen colonization (Shim et al., 2010). Furthermore, because stem cells and enteroendocrine cells reside towards the bottom of these crypts, access of both commensal and pathogenic bacteria to these spaces might impact their influence on renewal of the epithelial surface and host metabolism. Therefore, although the cell types and intestinal signaling pathways that control host metabolism are similar, the proximity of the microbiota to the analogous mammalian host cell must be considered before extrapolating from Drosophila to mammals.
The differences in the microbiota of mammals and flies must also be considered when drawing parallels between these two organisms. Whereas the principal members of the fly microbiota are Lactobacilli, which are Gram-positive facultative anaerobic rods of the Firmicutes phylum, and Acetobacter species, which are Gram-negative aerobic rods of the Proteobacteria phylum, a much more diverse microbiota is found in the mammalian gastrointestinal tract. However, because each microbiota is exquisitely matched to its host intestine, considerable differences in microbiota are found even when using a mammal, such as the mouse, to model the human gastrointestinal tract (Nguyen et al., 2015). These differences in microbiota do not invalidate the use of non-human models. As we have highlighted in our discussion, studies with model organisms suggest that, although the participants in the conversation are very different, the metabolic dialog itself is quite similar.
In mice and humans, many of the intestinal receptors that sense the byproducts of microbial metabolism have been identified. In Drosophila, these receptors are also likely to exist. However, not one has been identified to date. This represents a great gap in our understanding of the interaction of Drosophila with its intestinal microbiota, and one that can be rapidly filled given the genetic tools available for this model. We believe that this is one area where future research efforts should be focused.
Microbes that reside within our intestines can both promote and impede optimal nutrient utilization by sharing and catabolizing the nutrients we consume. We share our nutrients with commensal microbes, and they, in return, catabolize indigestible foods, delivering to us digestible byproducts (Hooper et al., 2002; Rakoff-Nahoum et al., 2014). To streamline this process, the eukaryotic intestine has evolved pathways, such as those housed by enteroendocrine cells, to sense and respond to the metabolites produced by symbiotic enteric microbes (Cani et al., 2013; Galland, 2014). Although it is not surprising that this tenuous metabolic equilibrium between the eukaryotic host and its intestinal inhabitants is easily perturbed, it is essential that we understand and learn to control such perturbations because they might contribute greatly to the obesity and diabetes epidemics that are rampant in the developed world. Because of the ease with which the Drosophila model can be designed and manipulated, the accessibility of host genetic tools, and the simplicity of the fly genome and microbiota, this organism provides an ideal system in which to investigate the principles of the host interaction with its enteric microbial population.
Footnotes
This article is part of a subject collection on Spotlight on Drosophila: Translational Impact. See related articles in this collection at http://dmm.biologists.org/collection/drosophila-disease-model.
Competing interests
The authors declare no competing or financial interests.
Funding
This work was supported by NIH grant R21AI109436 to P.I.W.
References
- Adams M. D., Celniker S. E., Holt R. A., Evans C. A., Gocayne J. D., Amanatides P. G., Scherer S. E., Li P. W., Hoskins R. A., Galle R. F., et al. (2000). The genome sequence of Drosophila melanogaster. Science 287, 2185-2195. 10.1126/science.287.5461.2185 [DOI] [PubMed] [Google Scholar]
- Ahmed T., Auble D., Berkley J. A., Black R., Ahern P. P., Hossain M., Hsieh A., Ireen S., Arabi M. and Gordon J. I. (2014). An evolving perspective about the origins of childhood undernutrition and nutritional interventions that includes the gut microbiome. Ann. N. Y. Acad. Sci. 1332, 22-38. 10.1111/nyas.12487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alic N., Hoddinott M. P., Vinti G. and Partridge L. (2011). Lifespan extension by increased expression of the Drosophila homologue of the IGFBP7 tumour suppressor. Aging Cell 10, 137-147. 10.1111/j.1474-9726.2010.00653.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amcheslavsky A., Jiang J. and Ip Y. T. (2009). Tissue damage-induced intestinal stem cell division in Drosophila. Cell Stem Cell 4, 49-61. 10.1016/j.stem.2008.10.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apidianakis Y., Pitsouli C., Perrimon N. and Rahme L. (2009). Synergy between bacterial infection and genetic predisposition in intestinal dysplasia. Proc. Natl. Acad. Sci. USA 106, 20883-20888. 10.1073/pnas.0911797106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayres J. S. and Schneider D. S. (2009). The role of anorexia in resistance and tolerance to infections in Drosophila. PLoS Biol. 7, e1000150 10.1371/journal.pbio.1000150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbieri M., Bonafè M., Franceschi C. and Paolisso G. (2003). Insulin/IGF-I-signaling pathway: an evolutionarily conserved mechanism of longevity from yeast to humans. Am. J. Physiol. Endocrinol. Metab. 285, E1064-E1071. 10.1152/ajpendo.00296.2003 [DOI] [PubMed] [Google Scholar]
- Basile K. J., Johnson M. E., Xia Q. and Grant S. F. A. (2014). Genetic susceptibility to type 2 diabetes and obesity: follow-up of findings from genome-wide association studies. Int. J. Endocrinol. 2014, 769671 10.1155/2014/769671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basset A., Khush R. S., Braun A., Gardan L., Boccard F., Hoffmann J. A. and Lemaitre B. (2000). The phytopathogenic bacteria Erwinia carotovora infects Drosophila and activates an immune response. Proc. Natl. Acad. Sci. USA 97, 3376-3381. 10.1073/pnas.97.7.3376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bednarova A., Kodrik D. and Krishnan N. (2013). Unique roles of glucagon and glucagon-like peptides: parallels in understanding the functions of adipokinetic hormones in stress responses in insects. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 164, 91-100. 10.1016/j.cbpa.2012.10.012 [DOI] [PubMed] [Google Scholar]
- Birchenough G. M. H., Johansson M. E. V., Gustafsson J. K., Bergström J. H. and Hansson G. C. (2015). New developments in goblet cell mucus secretion and function. Mucosal Immunol. 8, 712-719. 10.1038/mi.2015.32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blum J. E., Fischer C. N., Miles J. and Handelsman J. (2013). Frequent replenishment sustains the beneficial microbiome of Drosophila melanogaster. Mbio 4, e00860-e00813 10.1128/mbio.00860-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchard C. (1997). Genetics of human obesity: recent results from linkage studies. J. Nutr. 127, 1887S-1890S. [DOI] [PubMed] [Google Scholar]
- Breer H., Eberle J., Frick C., Haid D. and Widmayer P. (2012). Gastrointestinal chemosensation: chemosensory cells in the alimentary tract. Histochem. Cell Biol. 138, 13-24. 10.1007/s00418-012-0954-z [DOI] [PubMed] [Google Scholar]
- Broderick N. A. and Lemaitre B. (2012). Gut-associated microbes of Drosophila melanogaster. Gut Microbes 3, 307-321. 10.4161/gmic.19896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchon N., Broderick N. A., Chakrabarti S. and Lemaitre B. (2009). Invasive and indigenous microbiota impact intestinal stem cell activity through multiple pathways in Drosophila. Genes Dev. 23, 2333-2344. 10.1101/gad.1827009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchon N., Osman D., David F. P. A., Fang H. Y., Boquete J.-P., Deplancke B. and Lemaitre B. (2013). Morphological and molecular characterization of adult midgut compartmentalization in Drosophila. Cell Rep. 3, 1725-1738. 10.1016/j.celrep.2013.04.001 [DOI] [PubMed] [Google Scholar]
- Buckling A. and Brockhurst M. (2012). Bacteria-virus coevolution. Adv. Exp. Med. Biol. 751, 347-370. 10.1007/978-1-4614-3567-9_16 [DOI] [PubMed] [Google Scholar]
- Buttet M., Traynard V., Tran T. T., Besnard P., Poirier H. and Niot I. (2014). From fatty-acid sensing to chylomicron synthesis: role of intestinal lipid-binding proteins. Biochimie 96, 37-47. 10.1016/j.biochi.2013.08.011 [DOI] [PubMed] [Google Scholar]
- Canfora E. E., Jocken J. W. and Blaak E. E. (2015). Short-chain fatty acids in control of body weight and insulin sensitivity. Nat. Rev. Endocrinol. 11, 577-591. 10.1038/nrendo.2015.128 [DOI] [PubMed] [Google Scholar]
- Cani P. D., Delzenne N. M., Amar J. and Burcelin R. (2008). Role of gut microflora in the development of obesity and insulin resistance following high-fat diet feeding. Pathol. Biol. 56, 305-309. 10.1016/j.patbio.2007.09.008 [DOI] [PubMed] [Google Scholar]
- Cani P. D., Everard A. and Duparc T. (2013). Gut microbiota, enteroendocrine functions and metabolism. Curr. Opin. Pharmacol. 13, 935-940. 10.1016/j.coph.2013.09.008 [DOI] [PubMed] [Google Scholar]
- Castorena K. M., Stapleford K. A. and Miller D. J. (2010). Complementary transcriptomic, lipidomic, and targeted functional genetic analyses in cultured Drosophila cells highlight the role of glycerophospholipid metabolism in Flock House virus RNA replication. BMC Genomics 11, 183 10.1186/1471-2164-11-183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarti S., Liehl P., Buchon N. and Lemaitre B. (2012). Infection-induced host translational blockage inhibits immune responses and epithelial renewal in the Drosophila gut. Cell Host Microbe 12, 60-70. 10.1016/j.chom.2012.06.001 [DOI] [PubMed] [Google Scholar]
- Chambers M. C., Song K. H. and Schneider D. S. (2012). Listeria monocytogenes infection causes metabolic shifts in Drosophila melanogaster. PLoS ONE 7, e50679 10.1371/journal.pone.0050679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clevers H. C. and Bevins C. L. (2013). Paneth cells: maestros of the small intestinal crypts. Annu. Rev. Physiol. 75, 289-311. 10.1146/annurev-physiol-030212-183744 [DOI] [PubMed] [Google Scholar]
- Cook K. R., Parks A. L., Jacobus L. M., Kaufman T. C. and Matthews K. A. (2010). New research resources at the Bloomington Drosophila Stock Center. Fly 4, 88-91. 10.4161/fly.4.1.11230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox C. R. and Gilmore M. S. (2007). Native microbial colonization of Drosophila melanogaster and its use as a model of Enterococcus faecalis pathogenesis. Infect. Immun. 75, 1565-1576. 10.1128/IAI.01496-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox L. M., Yamanishi S., Sohn J., Alekseyenko A. V., Leung J. M., Cho I., Kim S. G., Li H., Gao Z., Mahana D. et al. (2014). Altering the intestinal microbiota during a critical developmental window has lasting metabolic consequences. Cell 158, 705-721. 10.1016/j.cell.2014.05.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Souza R. J., Mente A., Maroleanu A., Cozma A. I., Ha V., Kishibe T., Uleryk E., Budylowski P., Schünemann H., Beyene J. et al. (2015). Intake of saturated and trans unsaturated fatty acids and risk of all cause mortality, cardiovascular disease, and type 2 diabetes: systematic review and meta-analysis of observational studies. BMJ 351, h3978 10.1136/bmj.h3978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng W., Tang X., Hou M., Li C. and Xie J. (2011). New insights into the pathogenesis of tuberculosis revealed by Mycobacterium marinum: the zebrafish model from the systems biology perspective. Crit. Rev. Eukaryot. Gene Expr. 21, 337-346. 10.1615/CritRevEukarGeneExpr.v21.i4.40 [DOI] [PubMed] [Google Scholar]
- Diaz-Munoz S. L. and Koskella B. (2014). Bacteria-phage interactions in natural environments. Adv. Appl. Microbiol. 89, 135-183. 10.1016/B978-0-12-800259-9.00004-4 [DOI] [PubMed] [Google Scholar]
- Dionne M. S., Pham L. N., Shirasu-Hiza M. and Schneider D. S. (2006). Akt and FOXO dysregulation contribute to infection-induced wasting in Drosophila. Curr. Biol. 16, 1977-1985. 10.1016/j.cub.2006.08.052 [DOI] [PubMed] [Google Scholar]
- Dobson A. J., Chaston J. M., Newell P. D., Donahue L., Hermann S. L., Sannino D. R., Westmiller S., Wong A. C.-N., Clark A. G., Lazzaro B. P. et al. (2015). Host genetic determinants of microbiota-dependent nutrition revealed by genome-wide analysis of Drosophila melanogaster. Nat. Commun. 6, 6312 10.1038/ncomms7312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- dos Santos G., Schroeder A. J., Goodman J. L., Strelets V. B., Crosby M. A., Thurmond J., Emmert D. B. and Gelbart W. M. (2015). FlyBase: introduction of the Drosophila melanogaster Release 6 reference genome assembly and large-scale migration of genome annotations. Nucleic Acids Res. 43, D690-D697. 10.1093/nar/gku1099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dotson C. D., Zhang L., Xu H., Shin Y.-K., Vigues S., Ott S. H., Elson A. E. T., Choi H. J., Shaw H., Egan J. M. et al. (2008). Bitter taste receptors influence glucose homeostasis. PLoS ONE 3, e3974 10.1371/journal.pone.0003974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drucker D. J. (2001). Minireview: the glucagon-like peptides. Endocrinology 142, 521-527. 10.1210/endo.142.2.7983 [DOI] [PubMed] [Google Scholar]
- Dutta D., Dobson A. J., Houtz P. L., Glässer C., Revah J., Korzelius J., Patel P. H., Edgar B. A. and Buchon N. (2015). Regional cell-specific transcriptome mapping reveals regulatory complexity in the adult Drosophila midgut. Cell Rep. 12, 346-358. 10.1016/j.celrep.2015.06.009 [DOI] [PubMed] [Google Scholar]
- Erkosar B., Storelli G., Mitchell M., Bozonnet L., Bozonnet N. and Leulier F. (2015). Pathogen virulence impedes mutualist-mediated enhancement of host juvenile growth via inhibition of protein digestion. Cell Host Microbe 18, 445-455. 10.1016/j.chom.2015.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everard A. and Cani P. D. (2014). Gut microbiota and GLP-1. Rev. Endocr. Metab. Disord. 15, 189-196. 10.1007/s11154-014-9288-6 [DOI] [PubMed] [Google Scholar]
- Faderl M., Noti M., Corazza N. and Mueller C. (2015). Keeping bugs in check: the mucus layer as a critical component in maintaining intestinal homeostasis. IUBMB Life 67, 275-285. 10.1002/iub.1374 [DOI] [PubMed] [Google Scholar]
- Figueroa-Clarevega A. and Bilder D. (2015). Malignant Drosophila tumors interrupt insulin signaling to induce cachexia-like wasting. Dev. Cell 33, 47-55. 10.1016/j.devcel.2015.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filipe A. and McLauchlan J. (2015). Hepatitis C virus and lipid droplets: finding a niche. Trends Mol. Med. 21, 34-42. 10.1016/j.molmed.2014.11.003 [DOI] [PubMed] [Google Scholar]
- Fouhy F., Guinane C. M., Hussey S., Wall R., Ryan C. A., Dempsey E. M., Murphy B., Ross R. P., Fitzgerald G. F., Stanton C. et al. (2012). High-throughput sequencing reveals the incomplete, short-term recovery of infant gut microbiota following parenteral antibiotic treatment with ampicillin and gentamicin. Antimicrob. Agents Chemother. 56, 5811-5820. 10.1128/AAC.00789-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galland L. (2014). The gut microbiome and the brain. J. Med. Food 17, 1261-1272. 10.1089/jmf.2014.7000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garbe J. C., Yang E. and Fristrom J. W. (1993). IMP-L2: an essential secreted immunoglobulin family member implicated in neural and ectodermal development in Drosophila. Development 119, 1237-1250. [DOI] [PubMed] [Google Scholar]
- Gheorghita V., Barbu A. E., Gheorghiu M. L. and Caruntu F. A. (2015). Endocrine dysfunction in sepsis: a beneficial or deleterious host response? Germs 5, 17-25. 10.11599/germs.2015.1067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodrich J. K., Waters J. L., Poole A. C., Sutter J. L., Koren O., Blekhman R., Beaumont M., Van Treuren W., Knight R., Bell J. T. et al. (2014). Human genetics shape the gut microbiome. Cell 159, 789-799. 10.1016/j.cell.2014.09.053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gribble F. M. and Reimann F. (2016). Enteroendocrine Cells: chemosensors in the intestinal epithelium. Annu. Rev. Physiol. 78, 277-299. 10.1146/annurev-physiol-021115-105439 [DOI] [PubMed] [Google Scholar]
- Hang S., Purdy A. E., Robins W. P., Wang Z., Mandal M., Chang S., Mekalanos J. J. and Watnick P. I. (2014). The acetate switch of an intestinal pathogen disrupts host insulin signaling and lipid metabolism. Cell Host Microbe 16, 592-604. 10.1016/j.chom.2014.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara T., Kashihara D., Ichimura A., Kimura I., Tsujimoto G. and Hirasawa A. (2014). Role of free fatty acid receptors in the regulation of energy metabolism. Biochim. Biophy. Acta 1841, 1292-1300. 10.1016/j.bbalip.2014.06.002 [DOI] [PubMed] [Google Scholar]
- Honegger B., Galic M., Köhler K., Wittwer F., Brogiolo W., Hafen E. and Stocker H. (2008). Imp-L2, a putative homolog of vertebrate IGF-binding protein 7, counteracts insulin signaling in Drosophila and is essential for starvation resistance. J. Biol. 7, 10 10.1186/jbiol72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooper L. V., Midtvedt T. and Gordon J. I. (2002). How host-microbial interactions shape the nutrient environment of the mammalian intestine. Annu. Rev. Nutr. 22, 283-307. 10.1146/annurev.nutr.22.011602.092259 [DOI] [PubMed] [Google Scholar]
- Ichimura A., Hirasawa A., Hara T. and Tsujimoto G. (2009). Free fatty acid receptors act as nutrient sensors to regulate energy homeostasis. Prostaglandins Other Lipid Mediat. 89, 82-88. 10.1016/j.prostaglandins.2009.05.003 [DOI] [PubMed] [Google Scholar]
- Ichimura A., Hasegawa S., Kasubuchi M. and Kimura I. (2014). Free fatty acid receptors as therapeutic targets for the treatment of diabetes. Front. Pharmacol. 5, 236 10.3389/fphar.2014.00236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iimura M., Gallo R. L., Hase K., Miyamoto Y., Eckmann L. and Kagnoff M. F. (2005). Cathelicidin mediates innate intestinal defense against colonization with epithelial adherent bacterial pathogens. J. Immunol. 174, 4901-4907. 10.4049/jimmunol.174.8.4901 [DOI] [PubMed] [Google Scholar]
- Iqbal J. and Hussain M. M. (2009). Intestinal lipid absorption. Am. J. Physiol. Endocrinol. Metab. 296, E1183-E1194. 10.1152/ajpendo.90899.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang H.-J., Kokrashvili Z., Theodorakis M. J., Carlson O. D., Kim B.-J., Zhou J., Kim H. H., Xu X., Chan S. L., Juhaszova M. et al. (2007). Gut-expressed gustducin and taste receptors regulate secretion of glucagon-like peptide-1. Proc. Natl. Acad. Sci. USA 104, 15069-15074. 10.1073/pnas.0706890104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane A. V., Dinh D. M. and Ward H. D. (2015). Childhood malnutrition and the intestinal microbiome. Pediatr. Res. 77, 256-262. 10.1038/pr.2014.179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasubuchi M., Hasegawa S., Hiramatsu T., Ichimura A. and Kimura I. (2015). Dietary gut microbial metabolites, short-chain fatty acids, and host metabolic regulation. Nutrients 7, 2839-2849. 10.3390/nu7042839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan A. and Pessin J. (2002). Insulin regulation of glucose uptake: a complex interplay of intracellular signalling pathways. Diabetologia 45, 1475-1483. 10.1007/s00125-002-0974-7 [DOI] [PubMed] [Google Scholar]
- Kohyama-Koganeya A., Kurosawa M. and Hirabayashi Y. (2015). Differential effects of tissue-specific deletion of BOSS on feeding behaviors and energy metabolism. PLoS ONE 10, e0133083 10.1371/journal.pone.0133083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokrashvili Z., Mosinger B. and Margolskee R. F. (2009). Taste signaling elements expressed in gut enteroendocrine cells regulate nutrient-responsive secretion of gut hormones. Am. J. Clin. Nutr. 90, 822S-825S. 10.3945/ajcn.2009.27462T [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotloff K. L., Nataro J. P., Blackwelder W. C., Nasrin D., Farag T. H., Panchalingam S., Wu Y., Sow S. O., Sur D., Breiman R. F. et al. (2013). Burden and aetiology of diarrhoeal disease in infants and young children in developing countries (the Global Enteric Multicenter Study, GEMS): a prospective, case-control study. Lancet 382, 209-222. 10.1016/S0140-6736(13)60844-2 [DOI] [PubMed] [Google Scholar]
- Kuraishi T., Binggeli O., Opota O., Buchon N. and Lemaitre B. (2011). Genetic evidence for a protective role of the peritrophic matrix against intestinal bacterial infection in Drosophila melanogaster. Proc. Natl. Acad. Sci. USA 108, 15966-15971. 10.1073/pnas.1105994108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon Y., Song W., Droujinine I. A., Hu Y., Asara J. M. and Perrimon N. (2015). Systemic organ wasting induced by localized expression of the secreted insulin/IGF antagonist ImpL2. Dev. Cell 33, 36-46. 10.1016/j.devcel.2015.02.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaJeunesse D. R., Johnson B., Presnell J. S., Catignas K. and Zapotoczny G. (2010). Peristalsis in the junction region of the Drosophila larval midgut is modulated by DH31 expressing enteroendocrine cells. BMC Physiol. 10, 14 10.1186/1472-6793-10-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Layalle S., Arquier N. and Léopold P. (2008). The TOR pathway couples nutrition and developmental timing in Drosophila. Dev. Cell 15, 568-577. 10.1016/j.devcel.2008.08.003 [DOI] [PubMed] [Google Scholar]
- Lee K.-A., Kim S.-H., Kim E.-K., Ha E.-M., You H., Kim B., Kim M.-J., Kwon Y., Ryu J.-H. and Lee W.-J. (2013). Bacterial-derived uracil as a modulator of mucosal immunity and gut-microbe homeostasis in Drosophila. Cell 153, 797-811. 10.1016/j.cell.2013.04.009 [DOI] [PubMed] [Google Scholar]
- Lee K.-A., Kim B., Bhin J., Kim D. H., You H., Kim E.-K., Kim S.-H., Ryu J.-H., Hwang D. and Lee W.-J. (2015). Bacterial uracil modulates Drosophila DUOX-dependent gut immunity via Hedgehog-induced signaling endosomes. Cell Host Microbe 17, 191-204. 10.1016/j.chom.2014.12.012 [DOI] [PubMed] [Google Scholar]
- Lehane M. J. (1997). Peritrophic matrix structure and function. Annu. Rev. Entomol. 42, 525-550. 10.1146/annurev.ento.42.1.525 [DOI] [PubMed] [Google Scholar]
- Liehl P., Blight M., Vodovar N., Boccard F. and Lemaitre B. (2006). Prevalence of local immune response against oral infection in a Drosophila/Pseudomonas infection model. PLoS Pathogens 2, e56 10.1371/journal.ppat.0020056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansbach C. M. II and Gorelick F. (2007). Development and physiological regulation of intestinal lipid absorption. II. Dietary lipid absorption, complex lipid synthesis, and the intracellular packaging and secretion of chylomicrons. Am. J. Physiol. Gastrointest. Liver Physiol. 293, G645-G650. 10.1152/ajpgi.00299.2007 [DOI] [PubMed] [Google Scholar]
- Marianes A. and Spradling A. C. (2013). Physiological and stem cell compartmentalization within the Drosophila midgut. Elife 2, e00886 10.7554/eLife.00886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marroqui L., Alonso-Magdalena P., Merino B., Fuentes E., Nadal A. and Quesada I. (2014). Nutrient regulation of glucagon secretion: involvement in metabolism and diabetes. Nutr. Res. Rev. 27, 48-62. 10.1017/S0954422414000031 [DOI] [PubMed] [Google Scholar]
- Mellitzer G. and Gradwohl G. (2011). Enteroendocrine cells and lipid absorption. Curr. Opin. Lipidol. 22, 171-175. 10.1097/MOL.0b013e32834622a2 [DOI] [PubMed] [Google Scholar]
- Micchelli C. A. and Perrimon N. (2006). Evidence that stem cells reside in the adult Drosophila midgut epithelium. Nature 439, 475-479. 10.1038/nature04371 [DOI] [PubMed] [Google Scholar]
- Miele L., Giorgio V., Alberelli M. A., De Candia E., Gasbarrini A. and Grieco A. (2015). Impact of gut microbiota on obesity, diabetes, and cardiovascular disease risk. Curr. Cardiol. Rep. 17, 120 10.1007/s11886-015-0671-z [DOI] [PubMed] [Google Scholar]
- Mills S., Shanahan F., Stanton C., Hill C., Coffey A. and Ross R. P. (2013). Movers and shakers: influence of bacteriophages in shaping the mammalian gut microbiota. Gut Microbes 4, 4-16. 10.4161/gmic.22371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minot S., Bryson A., Chehoud C., Wu G. D., Lewis J. D. and Bushman F. D. (2013). Rapid evolution of the human gut virome. Proc. Natl. Acad. Sci. USA 110, 12450-12455. 10.1073/pnas.1300833110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moessinger C., Klizaite K., Steinhagen A., Phillippou-Massier J., Shevchenko A., Hoch M., Ejsing C. S. and Thiele C. (2014). Two different pathways of phosphatidylcholine synthesis, the Kennedy pathway and Lands cycle, differentially regulate triacylglycerol storage. BMC Cell Biol. 15, 43 10.1186/s12860-014-0043-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monira S., Nakamura S., Gotoh K., Izutsu K., Watanabe H., Alam N. H., Nakaya T., Horii T., Ali S. I., Iida T. et al. (2013). Metagenomic profile of gut microbiota in children during cholera and recovery. Gut Pathogens 5, 1 10.1186/1757-4749-5-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran-Ramos S., Tovar A. R. and Torres N. (2012). Diet: friend or foe of enteroendocrine cells--how it interacts with enteroendocrine cells. Adv. Nutr. 3, 8-20. 10.3945/an.111.000976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moussian B. (2013). The apical plasma membrane of chitin-synthesizing epithelia. Insect Sci. 20, 139-146. 10.1111/j.1744-7917.2012.01549.x [DOI] [PubMed] [Google Scholar]
- Naslund E. and Hellstrom P. M. (2007). Appetite signaling: from gut peptides and enteric nerves to brain. Physiol. Behav. 92, 256-262. 10.1016/j.physbeh.2007.05.017 [DOI] [PubMed] [Google Scholar]
- Nassel D. R., Kubrak O. I., Liu Y., Luo J. and Lushchak O. V. (2013). Factors that regulate insulin producing cells and their output in Drosophila. Front. Physiol. 4, 252 10.3389/fphys.2013.00252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nassel D. R., Liu Y. and Luo J. (2015). Insulin/IGF signaling and its regulation in Drosophila. Gen. Comp. Endocrinol. 221, 255-266. 10.1016/j.ygcen.2014.11.021 [DOI] [PubMed] [Google Scholar]
- Newell P. D. and Douglas A. E. (2014). Interspecies interactions determine the impact of the gut microbiota on nutrient allocation in Drosophila melanogaster. Appl. Environ. Microbiol. 80, 788-796. 10.1128/AEM.02742-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen T. L. A., Vieira-Silva S., Liston A. and Raes J. (2015). How informative is the mouse for human gut microbiota research? Dis. Model. Mech. 8, 1-16. 10.1242/dmm.017400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni J.-Q., Markstein M., Binari R., Pfeiffer B., Liu L.-P., Villalta C., Booker M., Perkins L. and Perrimon N. (2008). Vector and parameters for targeted transgenic RNA interference in Drosophila melanogaster. Nat. Methods 5, 49-51. 10.1038/nmeth1146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni J.-Q., Zhou R., Czech B., Liu L.-P., Holderbaum L., Yang-Zhou D., Shim H.-S., Tao R., Handler D., Karpowicz P. et al. (2011). A genome-scale shRNA resource for transgenic RNAi in Drosophila. Nat. Methods 8, 405-407. 10.1038/nmeth.1592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni Y., Li J. and Panagiotou G. (2015). A molecular-level landscape of diet-gut microbiome interactions: toward dietary interventions targeting bacterial genes. Mbio 6, e01263-15 10.1128/mBio.01263-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobel Y. R., Cox L. M., Kirigin F. F., Bokulich N. A., Yamanishi S., Teitler I., Chung J., Sohn J., Barber C. M., Goldfarb D. S. et al. (2015). Metabolic and metagenomic outcomes from early-life pulsed antibiotic treatment. Nat. Commun. 6, 7486 10.1038/ncomms8486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norman J. M., Handley S. A., Baldridge M. T., Droit L., Liu C. Y., Keller B. C., Kambal A., Monaco C. L., Zhao G., Fleshner P. et al. (2015). Disease-specific alterations in the enteric virome in inflammatory bowel disease. Cell 160, 447-460. 10.1016/j.cell.2015.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohlstein B. and Spradling A. (2006). The adult Drosophila posterior midgut is maintained by pluripotent stem cells. Nature 439, 470-474. 10.1038/nature04333 [DOI] [PubMed] [Google Scholar]
- Olivier V., Salzman N. H. and Satchell K. J. F. (2007). Prolonged colonization of mice by Vibrio cholerae El Tor O1 depends on accessory toxins. Infect. Immun. 75, 5043-5051. 10.1128/IAI.00508-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padmanabha D. and Baker K. D. (2014). Drosophila gains traction as a repurposed tool to investigate metabolism. Trends Endocrinol. Metab. 25, 518-527. 10.1016/j.tem.2014.03.011 [DOI] [PubMed] [Google Scholar]
- Palm W., Sampaio J. L., Brankatschk M., Carvalho M., Mahmoud A., Shevchenko A. and Eaton S. (2012). Lipoproteins in Drosophila melanogaster--assembly, function, and influence on tissue lipid composition. PLoS Genet. 8, e1002828 10.1371/journal.pgen.1002828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panakova D., Sprong H., Marois E., Thiele C. and Eaton S. (2005). Lipoprotein particles are required for Hedgehog and Wingless signalling. Nature 435, 58-65. 10.1038/nature03504 [DOI] [PubMed] [Google Scholar]
- Park J.-H. and Kwon J. Y. (2011). Heterogeneous expression of Drosophila gustatory receptors in enteroendocrine cells. PLoS ONE 6, e29022 10.1371/journal.pone.0029022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Psichas A., Sleeth M. L., Murphy K. G., Brooks L., Bewick G. A., Hanyaloglu A. C., Ghatei M. A., Bloom S. R. and Frost G. (2015). The short chain fatty acid propionate stimulates GLP-1 and PYY secretion via free fatty acid receptor 2 in rodents. Int. J. Obes. 39, 424-429. 10.1038/ijo.2014.153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purdy A. E. and Watnick P. I. (2011). Spatially selective colonization of the arthropod intestine through activation of Vibrio cholerae biofilm formation. Proc. Natl. Acad. Sci. USA 108, 19737-19742. 10.1073/pnas.1111530108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabot S., Membrez M., Bruneau A., Gerard P., Harach T., Moser M., Raymond F., Mansourian R. and Chou C. J. (2010). Germ-free C57BL/6J mice are resistant to high-fat-diet-induced insulin resistance and have altered cholesterol metabolism. FASEB J. 24, 4948-4959. 10.1096/fj.10-164921 [DOI] [PubMed] [Google Scholar]
- Rakoff-Nahoum S., Coyne M. J. and Comstock L. E. (2014). An ecological network of polysaccharide utilization among human intestinal symbionts. Curr. Biol. 24, 40-49. 10.1016/j.cub.2013.10.077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid G., Younes J. A., Van der Mei H. C., Gloor G. B., Knight R. and Busscher H. J. (2011). Microbiota restoration: natural and supplemented recovery of human microbial communities. Nat. Rev. Microbiol. 9, 27-38. 10.1038/nrmicro2473 [DOI] [PubMed] [Google Scholar]
- Relman D. A. (2012). The human microbiome: ecosystem resilience and health. Nutr. Rev. 70 Suppl. 1, S2-S9. 10.1111/j.1753-4887.2012.00489.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren F., Wang B., Yue T., Yun E.-Y., Ip Y. T. and Jiang J. (2010). Hippo signaling regulates Drosophila intestine stem cell proliferation through multiple pathways. Proc. Natl. Acad. Sci. USA 107, 21064-21069. 10.1073/pnas.1012759107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridaura V. K., Faith J. J., Rey F. E., Cheng J., Duncan A. E., Kau A. L., Griffin N. W., Lombard V., Henrissat B., Bain J. R. et al. (2013). Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science 341, 1241214 10.1126/science.1241214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridley E. V., Wong A. C.-N., Westmiller S. and Douglas A. E. (2012). Impact of the resident microbiota on the nutritional phenotype of Drosophila melanogaster. PLoS ONE 7, e36765 10.1371/journal.pone.0036765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie J. M., Rui H., Bronson R. T. and Waldor M. K. (2010). Back to the future: studying cholera pathogenesis using infant rabbits. Mbio 1, e00047-10 10.1128/mBio.00047-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Vazquez M., Vaquero D., Parra-Peralbo E., Mejia-Morales J. E. and Culi J. (2015). Drosophila lipophorin receptors recruit the lipoprotein LTP to the plasma membrane to mediate lipid uptake. PLoS Genet. 11, e1005356 10.1371/journal.pgen.1005356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saka H. A. and Valdivia R. (2012). Emerging roles for lipid droplets in immunity and host-pathogen interactions. Annu. Rev. Cell Dev. Biol. 28, 411-437. 10.1146/annurev-cellbio-092910-153958 [DOI] [PubMed] [Google Scholar]
- Sarraf-Zadeh L., Christen S., Sauer U., Cognigni P., Miguel-Aliaga I., Stocker H., Köhler K. and Hafen E. (2013). Local requirement of the Drosophila insulin binding protein imp-L2 in coordinating developmental progression with nutritional conditions. Dev. Biol. 381, 97-106. 10.1016/j.ydbio.2013.06.008 [DOI] [PubMed] [Google Scholar]
- Sato T., van Es J. H., Snippert H. J., Stange D. E., Vries R. G., van den Born M., Barker N., Shroyer N. F., van de Wetering M. and Clevers H. (2011). Paneth cells constitute the niche for Lgr5 stem cells in intestinal crypts. Nature 469, 415-418. 10.1038/nature09637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serino M., Blasco-Baque V., Nicolas S. and Burcelin R. (2014). Managing the manager: gut microbes, stem cells and metabolism. Diabet. Metab. 40, 186-190. 10.1016/j.diabet.2013.12.004 [DOI] [PubMed] [Google Scholar]
- Shea J. E., Hensel M., Gleeson C. and Holden D. W. (1996). Identification of a virulence locus encoding a second type III secretion system in Salmonella typhimurium. Proc. Natl. Acad. Sci. USA 93, 2593-2597. 10.1073/pnas.93.6.2593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen J., Obin M. S. and Zhao L. (2013). The gut microbiota, obesity and insulin resistance. Mol. Aspects Med. 34, 39-58. 10.1016/j.mam.2012.11.001 [DOI] [PubMed] [Google Scholar]
- Shibata T., Maki K., Hadano J., Fujikawa T., Kitazaki K., Koshiba T. and Kawabata S.-I. (2015). Crosslinking of a peritrophic matrix protein protects gut epithelia from bacterial exotoxins. PLoS Pathog. 11, e1005244 10.1371/journal.ppat.1005244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shim D.-H., Ryu S. and Kweon M.-N. (2010). Defensins play a crucial role in protecting mice against oral Shigella flexneri infection. Biochem. Biophys. Res. Commun. 401, 554-560. 10.1016/j.bbrc.2010.09.100 [DOI] [PubMed] [Google Scholar]
- Shin S. C., Kim S.-H., You H., Kim B., Kim A. C., Lee K.-A., Yoon J.-H., Ryu J.-H. and Lee W.-J. (2011). Drosophila microbiome modulates host developmental and metabolic homeostasis via insulin signaling. Science 334, 670-674. 10.1126/science.1212782 [DOI] [PubMed] [Google Scholar]
- Sieber M. H. and Thummel C. S. (2009). The DHR96 nuclear receptor controls triacylglycerol homeostasis in Drosophila. Cell Metab. 10, 481-490. 10.1016/j.cmet.2009.10.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieber M. H. and Thummel C. S. (2012). Coordination of triacylglycerol and cholesterol homeostasis by DHR96 and the Drosophila LipA homolog magro. Cell Metab. 15, 122-127. 10.1016/j.cmet.2011.11.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith M. I., Yatsunenko T., Manary M. J., Trehan I., Mkakosya R., Cheng J., Kau A. L., Rich S. S., Concannon P., Mychaleckyj J. C. et al. (2013). Gut microbiomes of Malawian twin pairs discordant for kwashiorkor. Science 339, 548-554. 10.1126/science.1229000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song W., Veenstra J. A. and Perrimon N. (2014). Control of lipid metabolism by tachykinin in Drosophila. Cell Rep. 9, 40-47. 10.1016/j.celrep.2014.08.060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sternini C., Anselmi L. and Rozengurt E. (2008). Enteroendocrine cells: a site of ‘taste’ in gastrointestinal chemosensing. Curr. Opin. Endocr. Diabet. Obes. 15, 73-78. 10.1097/MED.0b013e3282f43a73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewardson A. J., Gaia N., Francois P., Malhotra-Kumar S., Delemont C., Martinez de Tejada B., Schrenzel J., Harbarth S., Lazarevic V., The SATURN WP1 and WP3 Study Groups (2015). Collateral damage from oral ciprofloxacin versus nitrofurantoin in outpatients with urinary tract infections: a culture-free analysis of gut microbiota. Clin. Microbiol. Infect. 21, 344.e341-344.e311 10.1016/j.cmi.2014.11.016 [DOI] [PubMed] [Google Scholar]
- Storelli G., Defaye A., Erkosar B., Hols P., Royet J. and Leulier F. (2011). Lactobacillus plantarum promotes Drosophila systemic growth by modulating hormonal signals through TOR-dependent nutrient sensing. Cell Metab. 14, 403-414. 10.1016/j.cmet.2011.07.012 [DOI] [PubMed] [Google Scholar]
- Subramanian S., Huq S., Yatsunenko T., Haque R., Mahfuz M., Alam M. A., Benezra A., DeStefano J., Meier M. F., Muegge B. D. et al. (2014). Persistent gut microbiota immaturity in malnourished Bangladeshi children. Nature 510, 417-421. 10.1038/nature13421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tailford L. E., Crost E. H., Kavanaugh D. and Juge N. (2015). Mucin glycan foraging in the human gut microbiome. Front. Genet. 6, 81 10.3389/fgene.2015.00081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teleman A. A. (2010). Molecular mechanisms of metabolic regulation by insulin in Drosophila. Biochem. J. 425, 13-26. 10.1042/BJ20091181 [DOI] [PubMed] [Google Scholar]
- The Human Microbiome Project Consortium. (2012). Structure, function and diversity of the healthy human microbiome. Nature 486, 207-214. 10.1038/nature11234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ukhanova M., Culpepper T., Baer D., Gordon D., Kanahori S., Valentine J., Neu J., Sun Y., Wang X. and Mai V. (2012). Gut microbiota correlates with energy gain from dietary fibre and appears to be associated with acute and chronic intestinal diseases. Clin. Microbiol. Infect. 18 Suppl. 4, 62-66. 10.1111/j.1469-0691.2012.03859.x [DOI] [PubMed] [Google Scholar]
- Umar S. (2010). Intestinal stem cells. Curr. Gastroenterol. Rep. 12, 340-348. 10.1007/s11894-010-0130-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ussar S., Griffin N. W., Bezy O., Fujisaka S., Vienberg S., Softic S., Deng L., Bry L., Gordon J. I. and Kahn C. R. (2015). Interactions between gut microbiota, host genetics and diet modulate the predisposition to obesity and metabolic syndrome. Cell Metab. 22, 516-530. 10.1016/j.cmet.2015.07.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Opijnen T., Bodi K. L. and Camilli A. (2009). Tn-seq: high-throughput parallel sequencing for fitness and genetic interaction studies in microorganisms. Nat. Methods 6, 767-772. 10.1038/nmeth.1377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderveken M. and O'Donnell M. J. (2014). Effects of diuretic hormone 31, drosokinin, and allatostatin A on transepithelial K(+) transport and contraction frequency in the midgut and hindgut of larval Drosophila melanogaster. Arch. Insect. Biochem. Physiol. 85, 76-93. 10.1002/arch.21144 [DOI] [PubMed] [Google Scholar]
- Veenstra J. A. (2009). Peptidergic paracrine and endocrine cells in the midgut of the fruit fly maggot. Cell Tissue Res. 336, 309-323. 10.1007/s00441-009-0769-y [DOI] [PubMed] [Google Scholar]
- Wong C. N. A., Ng P. and Douglas A. E. (2011). Low-diversity bacterial community in the gut of the fruitfly Drosophila melanogaster. Environ. Microbiol. 13, 1889-1900. 10.1111/j.1462-2920.2011.02511.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong A. C.-N., Dobson A. J. and Douglas A. E. (2014). Gut microbiota dictates the metabolic response of Drosophila to diet. J. Exp. Biol. 217, 1894-1901. 10.1242/jeb.101725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J., Grant G., Sabin L. R., Gordesky-Gold B., Yasunaga A., Tudor M. and Cherry S. (2012). Transcriptional pausing controls a rapid antiviral innate immune response in Drosophila. Cell Host Microbe 12, 531-543. 10.1016/j.chom.2012.08.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J., Hopkins K., Sabin L., Yasunaga A., Subramanian H., Lamborn I., Gordesky-Gold B. and Cherry S. (2013). ERK signaling couples nutrient status to antiviral defense in the insect gut. Proc. Natl. Acad. Sci. USA 110, 15025-15030. 10.1073/pnas.1303193110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C., Yin A., Li H., Wang R., Wu G., Shen J., Zhang M., Wang L., Hou Y., Ouyang H. et al. (2015). Dietary modulation of gut microbiota contributes to alleviation of both genetic and simple obesity in children. EBioMedicine 2, 966-982. 10.1016/j.ebiom.2015.07.007 [DOI] [PMC free article] [PubMed] [Google Scholar]