

Abstract
We developed the densest single-nucleotide polymorphism (SNP)-based linkage genetic map to date for the genus Quercus. An 8k gene-based SNP array was used to genotype more than 1,000 full-sibs from two intraspecific and two interspecific full-sib families of Quercus petraea and Quercus robur. A high degree of collinearity was observed between the eight parental maps of the two species. A composite map was then established with 4,261 SNP markers spanning 742 cM over the 12 linkage groups (LGs) of the oak genome. Nine genomic regions from six LGs displayed highly significant distortions of segregation. Two main hypotheses concerning the mechanisms underlying segregation distortion are discussed: genetic load vs. reproductive barriers. Our findings suggest a predominance of pre-zygotic to post-zygotic barriers.
Keywords: high-density linkage map, SNP, segregation distortion, reproductive barriers, Quercus
1. Introduction
`The genus Quercus is a major forest tree genus with 300–600 species spread throughout the world (http://www.mobot.org/MOBOT/research/APweb/orders/fagalesweb.htm#Fagales). The European white oak species complex—including Quercus robur (Qr), pedunculate oak and Quercus petraea (Qp), sessile oak, in particular—is an exceptional model for studies of the genetics of speciation in sympatry.1–4 These two main oak species have similar distribution areas, but display morphological differences for many traits, including leaf shape, acorn peduncle length, and hairiness.5 They also have different ecophysiological requirements: Qr tolerates waterlogging and light exposure more effectively than Qp,6 whereas Qp has a higher water use efficiency and is more shade-tolerant, consistent with its role as a post-pioneer species.7–9 These two sympatric species are interfertile, forming natural hybrids in mixed stands and displaying only partial reproductive isolation (RI).3,10
Low-density genetic linkage maps in oaks have been constructed with various genotyping techniques: random amplified polmorphic DNA, amplified fragment length polymorphism and simple sequence repeat (SSR).11–13 Progress has recently been made, with the development of a single-nucleotide polymorphism (SNP) assay for Qr and Qp.14 Using this resource, we were able in this present study to construct high-density gene-based maps with a large number of SNP markers, providing a detailed scan of segregation distortion (SD) within the genome. SD, corresponding to deviation from the expected Mendelian proportion of individuals in a given genotypic class within a segregating population, has been widely documented in plant species, within the framework of genetic mapping studies, e.g. in barley,15 clementine,16 eucalyptus,17 maize,18 rice,19 monkeyflower,20 poplar21 and tomato.22 The detection of SD depends heavily on the type of cross. In most cases, SD is stronger and, therefore, easier to detect, in interspecific crosses than in intraspecific crosses.23 Some of these studies showed that distortion spreads over linked markers (and even over large genetic distances in some cases), creating SD regions (SDR). SD may result from three underlying causes. Gametic incompatibility (one of the many types of prezygotic barrier)24 and/or reduced hybrid viability (one of the many forms of post-zygotic selection)25,26 may be involved. Indeed, various developmental processes in the pollen, embryo and seedling offer opportunities for differential selection at the gamete (pollen tube competition, pollen tube growth) and/or zygote (embryo competition, ovule abortion) level. The accumulation of deleterious mutations is an alternative mechanism that may give rise to SD. Genetic load has often been advocated as a source of SD, particularly in trees, which form large populations likely to have accumulated large number of deleterious mutations.27,28 SD may also arise due to asymmetric allelic inheritance in heterozygotes (i.e. meiotic drive),29 but this process is not considered here as it relates to hybrid fertility rather than hybrid viability.
In this context, the first objective of this study was to establish a high-density gene-based genetic map of oak by genotyping more than 1,000 full-sibs from two intraspecific and two interspecific full-sib families of Q. petraea and Q. robur. A single composite map was constructed by merging the eight parental maps. This map includes 4,261 gene-based markers and is the densest linkage map ever produced for oak. The second objective of our study was to carry out a detailed analysis of the SDRs based on the patterns of SD observed for intraspecific and interspecific crosses. The SDRs identified on the eight linkage maps were compared to investigate the causes of the observed SDRs that span 6 of the 12 chromosomes. More precisely, we describe the extent of SD along linkage groups (LGs) and depict the distribution of SDRs, compare the consistency of SDRs across different genetic backgrounds and draw inferences about the potential sources of the widespread SD in the oak genome.
2. Materials and methods
2.1. Plant material and DNA extraction
Six parental trees, three Q. robur (3P, A4, 11P) and three Q. petraea (QS28, QS21, QS29), were used to generate four full-sib families (F1) (Fig. 1):
One Q. robur intraspecific family (3P × A4) referred to as P1.
One Q. petraea intraspecific family (QS28f × QS21) referred to as P4.
Two interspecific families (11P1 × QS28m, 11P2 × QS29) referred to as P2 and P3.
Figure 1.
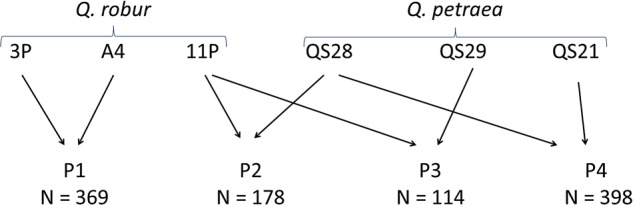
Description of the four pedigrees (3P, A4, 11P, QS28, QS29 and QS21 referred to the name of the parents used for the controlled crosses. P1, P2, P3 and P4 correspond to the name of the pedigree; n represents the number of offspring in each pedigree).
Crosses are denoted ‘female parent × male parent’ below. The same parental tree of Q. petraea (QS28) was used as the male parent in one cross (QS28m) and the female parent (QS28f) in another cross. Furthermore, the Q. robur parent 11P was used as a female parent in two different crosses and is therefore named 11P1 and 11P2.
Quercus robur had to be used as the female parent in all interspecific crosses, due to the strong asymmetry of hybridization success3,30 between these two species. The sizes of the progenies of the full-sib crosses varied from 398 (QS28f × QS21) to 114 (11P2 × QS29) individuals. Intraspecific and interspecific controlled crosses were implemented over the years since 1993. The offspring were installed in stool beds at the nursery of the INRA Forestry Research Station Pierroton (latitude 44.44°N, longitude 0.46°W) in South West of France, before subsequent vegetative propagation of the offspring. Bud and leaf material was collected on the stool beds and DNA was extracted as described by Bodénès et al.13
2.2. SNP genotyping
SNP genotyping was carried out on the four mapping populations (1,059 samples in total), with the Illumina® Infinium iSelect Custom Genotyping Array (Illumina Inc., San Diego, CA, USA), according to the manufacturer's standard protocol, using 200 ng of genomic DNA per sample. The selected SNPs correspond to two different data sets, the first one was obtained from the resequencing of six trees used as parents of the four controlled crosses and the second one was obtained from the oak Unigene established by Ueno et al.31 In total, 7,913 SNPs were selected for array design. We deliberately selected a limited number of markers (from 1 to 3) per contig, to ensure a broad distribution of SNPs across the genome.14
2.3. Linkage map construction
For each cross, two data sets were generated, each containing the meiotic segregation information from one of the parents. Markers segregating in a 1:1 ratio (testcross configuration) were mapped with JoinMap 4.0 (Kyazma, Wageningen, NL).32 For each population, the minimum logarithm of odds (LOD) threshold for grouping was determined by identifying grouping tree branches with stable marker numbers over increasing consecutive LOD values for a total number of groups of 12 which corresponds to the number of chromosome. The minimum LOD threshold for most of the linkage maps was 5 and increased to 6 for P4m map or 7 for P2m map. The two parental linkage maps for each mapping population were constructed with the maximum likelihood (ML) algorithm, with a minimum LOD of 5 and the default parameters (recombination frequency of 0.4 and a maximum threshold value of 1 for the jump). Recombination frequencies were converted to map distances in cM with the Kosambi's mapping function. The maps shown were plotted with MapChart 2.033 or using an R script. LG numbering was based on SSR markers, as in the study by Bodénès et al.13 The cosegregation of SSRs and SNPs (data not shown) made it possible to identify homologous LGs unambiguously.
2.4. Composite map construction
We constructed a composite map from the eight parental maps, with the LPmerge software developed by Endelman and Plomion,34 which is available as an R package https://cran.r-project.org/web/packages/LPmerge/index.html. This approach is based on the integration of linkage map data rather than observed recombination between markers, with linear programming used to minimize the mean absolute error between the composite map and the linkage map for each population as efficiently as possible. For assessment of the goodness-of-fit for the composite map, LPmerge computes a root-mean-square error (RMSE) per LG by comparing the position (in cM) of all markers on the composite map with that on the component maps (http://w3.pierroton.inra.fr/cgi-bin/cmap/viewer?ref_map_set_acc=51&ref_map_accs=-1). We calculated this metric for different maximum interval sizes (k in the algorithm), ranging from 1 to 8. The value of k minimizing the mean RMSE per LG was selected for construction of the composite map (Supplementary File S1). Intercross markers (segregating in a 1:2:1 ratio) were added as accessory markers in a second step taking the rate of recombination between these loci and the closest linked test-cross framework marker (FM) into account.
2.5. SD analysis
We tested each marker for significant deviation from the expected Mendelian genotype frequencies (χ2 with 1 degree of freedom for codominant markers, α= 0.05 calculated with JoinMap software) to detect SD. Assuming that each LG corresponds to one chromosome (n= 12 in Quercus) and that each chromosome contains at least two independent regions (the mean length of LGs was 66 cM, this paper), we expected there to be at least 24 independent genomic regions. A threshold of at least 0.05/24 ≈ 0.002 would therefore be required to obtain a genome wide error rate of α = 0.05. However, we applied a more stringent threshold (α = 0.001) and only considered distorted regions with more than three tightly linked distorted loci, to decrease the false-positive rate and to ensure that only biologically meaningful SDRs were detected. Markers displaying SD were conserved and integrated into the map. We investigated the patterns of distortion on the eight parental maps, by plotting the χ2 value of each marker along the 12 LG of the composite map obtained with the LPmerge programme (Fig. 2).
Figure 2.

Distribution of SD for the 12 LGs for the 8 parental maps, α = 0.001. The vertical line corresponds to a type I error of 0.001. The x-axis corresponds to the χ2 value and the y-axis corresponds to the map length of the LG in cM. The black vertical line corresponds to the χ2 threshold value, orange line corresponds to P1f, yellow line: P1m, light green: P2f, dark green: P2m, pink: P3f, violet: P3m, light blue: P4f and dark blue: P4m.
3. Results
3.1. SNP genotyping
The 7,913 SNPs were submitted to Illumina for Oligo Pool All (OPA) design for use in the Infinium assay. From the initial set of 7,913 SNPs, 903 (11.4%) did not pass Illumina production quality control and were eliminated. The genotyping data, across the four mapping populations, for the 7,010 SNPs retained were analyzed with Illumina GenomeStudio software, which clusters and calls data automatically, making it possible to visualize the data directly.14 For each SNP, the representation of the genotyping data included three main clusters, corresponding to the AA homozygote, the AB heterozygote and the BB homozygote. We obtained four different segregating configurations in the F1 mapping populations: AB×AA (heterozygous in the female parent), AA×AB (heterozygous in the male parent) (Fig. 3a), AB×AB (both parents heterozygous) (Fig. 3b) and AA×AA, BB×BB or AA×BB (both parents homozygous, not informative for genetic mapping). We inspected all the SNPs by eye on the Illumina clusters, making use of the distribution of the segregating full-sibs relative to the parental positions. Observations were carried out individually for each mapping pedigree. The results for the four mapping populations were merged, and 5,726 SNPs were retained (Table 1). We discarded SNPs that did not yield well defined, clearly separated clusters (i.e. for which the genotype could not be called unambiguously). We optimized the positions of the segregating loci, by mapping as FMs only the SNPs segregating in a testcross configuration (1:1 ratio). Intercross markers (1:2:1 ratio) are less informative for linkage analysis.35 They were therefore excluded from construction of the framework map.
Figure 3.

(a) AA×AB configuration [one parent is homozygous, the other, heterozygous, two classes of genotypes are observed among the progeny, AA (in red, left part) and AB (violet part, middle part)]. (b) AB×AB configuration [both parents are heterozygous, three classes of genotypes are observed among the progeny, AA (in red, left part) AB (in violet, middle part) and BB (in blue, right part)]. This figure is available in black and white in print and in colour at DNA Research online.
Table 1.
Number of polymorphic (P), monomorphic (M) and failed (NA) SNPs in the four pedigrees, P1, P2, P3 and P4
| P1 | P2 | P3 | P4 | All pedigrees | |
|---|---|---|---|---|---|
| P | 3,515 | 3,734 | 3,399 | 3,418 | 5,726 |
| M | 2,012 | 2,120 | 2,488 | 2,222 | 637 |
| NA | 1,483 | 1,156 | 1,123 | 1,370 | 647 |
3.2. Construction of eight parental maps and one composite map
From the 7,010 SNPs passing Illumina production quality control testing, 6,363 SNPs were ‘scorable’ and were used to genotype the four mapping populations.14 The number of SNPs mapped differed between parental trees, ranging from 1,421 SNPs for P3f to 889 for P4m (Table 2). The 12 expected LGs were retrieved for all the parental trees. The size of the genetic maps varied from 684 cM (P4f) to 840 cM (P4m) (Supplementary File S2). The number of markers per LG varied from 259 (LG2 for P4f) to 40 (LG4 for P1m) (Supplementary File S2). The LGs were all of similar size (mean of 62 ± 11 cM, suggesting the presence of at least one chiasma per chromosome), except for LG2, which was ∼1.5 times longer than the other LGs. Alignment of the eight parental maps obtained for the two species (Q. robur and Q. petraea) revealed a high degree of collinearity between the maps, making it possible to construct a composite map with LPmerge software composed of 4,261 FMs and 129 accessory markers (provided by intercross markers). We noticed that markers at the end of some LGs (LG3, LG4, LG5, LG7, LG8 and LG11) were around 5–14 cM distant from adjacent markers (Supplementary File S3). These markers were found to be present in only one (LG3, LG4, LG7, LG8) or two (for LG5 and LG11) contributing maps and distorted the merged map distances calculated by LPmerge. Therefore, these markers were moved at the position of their nearest adjacent marker calculated by JoinMap on the parental map. This URL allows us to compare LGs from different parental linkage maps: http://w3.pierroton.inra.fr/cgi-bin/cmap/map_details?ref_pmap_set_acc=47;ref_map_accs=1251;comparative_maps=1%3dmap_acc%3d1287;highlight=%22s_1BHPHQ_1326%22. The 12 composite LGs constructed from testcross markers covered 742 cM in total, with individual LG lengths of 51 cM (LG1) to 93 cM (LG2) and a mean density of 1 SNP marker per 0.2 cM (Table 3, Fig. 4).
Table 2.
Genotyping results for each parental map (testcross markers only) of the four pedigrees (P1–P4), f and m referred to the female and male maps
| P1f | P1m | P2f | P2m | P3f | P3m | P4f | P4m | |
|---|---|---|---|---|---|---|---|---|
| Number of individuals | 369 | 369 | 178 | 178 | 114 | 114 | 398 | 398 |
| Individuals with >10% missing data | 45 | 45 | 0 | 0 | 0 | 0 | 48 | 48 |
| Number of individuals considered | 324 | 324 | 178 | 178 | 114 | 114 | 350 | 350 |
| Total number of SNPs | 1,303 | 1,272 | 1,401 | 1,375 | 1,622 | 1,030 | 1,556 | 969 |
| SNPs same contig, different LGs | 0 | 1 | 0 | 2 | 2 | 1 | 2 | 0 |
| SNPs same contig, same map position (replicated SNP) | 131 | 133 | 125 | 142 | 178 | 101 | 159 | 73 |
| Analysed SNPs | 1,172 | 1,138 | 1,276 | 1,231 | 1,442 | 928 | 1,395 | 896 |
| Ungrouped SNPs | 1 | 0 | 10 | 26 | 21 | 22 | 1 | 6 |
| Excluded SNPs | 1 | 2 | 37 | 0 | 0 | 1 | 1 | 1 |
| Mapped SNPs | 1,170 | 1,136 | 1,229 | 1,205 | 1,421 | 905 | 1,393 | 889 |
Table 3.
Features of the composite map constructed with testcross markers. k is a parameter of LPmerge software, varying from 1 to 8, corresponding to the maximum size of the interval
| LG | Number of loci | k var/min RMSE | LG size (in cM) | SNP/cM |
|---|---|---|---|---|
| LG1 | 310 | 1 | 51 | 6.1 |
| LG2 | 712 | 8 | 93 | 7.7 |
| LG3 | 302 | 3 | 63 | 4.8 |
| LG4 | 229 | 3 | 63 | 3.6 |
| LG5 | 300 | 1 | 64 | 4.7 |
| LG6 | 406 | 5 | 63 | 6.4 |
| LG7 | 329 | 3 | 56 | 5.9 |
| LG8 | 437 | 4 | 67 | 6.5 |
| LG9 | 299 | 3 | 58 | 5.2 |
| LG10 | 289 | 5 | 58 | 5,0 |
| LG11 | 298 | 1 | 54 | 5.6 |
| LG12 | 350 | 1 | 53 | 6.6 |
| Total | 4,261 | 742 | ||
| Mean | 355 | 62 | 5.7 |
The composite map with the lowest RMSE (obtained with various values of k) was retained.
Figure 4.

Composite linkage map for the 12 LGs and localization of the 9 segregation distorted regions (SDRs). The SDRs are represented by a vertical colour trait along the LG2, in orange: P1f, LG4, in dark green:, P2m, LG6, left to right, in light blue: P4f, dark blue: P4m, orange: P1f, LG8, in dark green: P2m, LG10, in yellow: P1m, LG11, in dark green: P2m, in violet: P3m). The LG size in cM is given in the LG name. This figure is available in black and white in print and in colour at DNA Research online.
3.3. Segregation distortion
The genome-wide patterns of SD for the eight parental maps are presented in Fig. 2. Overall, 0 (P2f, P3f) to 15% (P2m) of SNPs, depending on the mapping population and the parental tree, displayed significant SD (α = 0.001) (Table 4). SD was non-randomly distributed along LGs: nine SDRs were identified on the eight parental maps. These SDRs were unevenly distributed on six LGs. Three types of SDR were observed: (i) SDRs at the ends of LGs (LG2, LG6, LG10), (ii) SDRs in the middle of the LG (LG4, LG8) and (iii) SDRs encompassing the whole LG (LG11) (Figs 2 and 4). The most significant distortions were observed on LG8 (P2m, 96% of markers displayed SD), LG11 (P2m and P3m, 60–88% of loci displaying SD, respectively), LG4 (P2m, 44% of loci displaying SD), LG6 (P1f, P4f and P4m, 20 to 32% of loci displaying SD) and LG10 (P1m, 40% of loci displaying SD) (Table 5). We found that 79% of the 359 loci displaying SD belonged to the male parent (χ² test, P-value of 9 × 10−5) and 62% belonged to the two interspecific crosses (χ² test, P-value of 1 × 10−3).
Table 4.
Number of loci displaying SD for the eight parental maps, α = 0.001
| Pedigree name | Cross type | Name of the parents | Mapping population size | SNP genotyped | Distorded loci α = 0.001 | LG (number of distorted loci) | Percentage of distorted SNPs |
|---|---|---|---|---|---|---|---|
| P1 | Intraspecific | P1f | 324 | 1,170 | 38 | LG2 (11); LG6 (27) | 3 |
| 3P×A4 | F1 | P1m | 1,136 | 25 | LG7 (4); LG10 (23) | 2 | |
| P2 | Interspecific | P2f | 178 | 1,229 | 0 | 0 | |
| 11P×QS28 | F1 | P2m | 1,205 | 177 | LG4 (34); LG8 (95); LG11 (48) | 15 | |
| P3 | Interspecific | P3f | 114 | 1,421 | 0 | 0 | |
| 11P×QS29 | F1 | P3m | 905 | 47 | LG11 (47) | 5 | |
| P4 | Intraspecific | P4f | 350 | 1,393 | 38 | LG6 (38) | 3 |
| QS28×QS21 | F1 | P4m | 889 | 34 | LG6 (34) | 4 |
LG for linkage group.
Table 5.
Number and coverage of SDRs over the LGs, α = 0.001
| Parental genotype | LG | Number of distorted SNP loci | Proportion of distorted loci (%) | SDR (in cM) | LG size (in cM) | Proportion of distorted LG (%) |
|---|---|---|---|---|---|---|
| P1f | LG2 | 11 | 6 | 1 | 89 | 1 |
| LG6 | 28 | 28 | 10 | 52 | 20 | |
| P1m | LG10 | 23 | 28 | 24 | 59 | 40 |
| P2f | — | — | — | — | — | — |
| P2m | LG4 | 34 | 61 | 31 | 70 | 44 |
| LG8 | 95 | 91 | 55 | 58 | 96 | |
| LG11 | 48 | 64 | 34 | 57 | 60 | |
| P3f | — | — | — | — | — | — |
| P3m | LG11 | 47 | 87 | 34 | 38 | 88 |
| P4f | LG6 | 38 | 30 | 17 | 52 | 32 |
| P4m | LG6 | 34 | 33 | 15 | 71 | 21 |
4. Discussion
4.1. Construction of a high-density gene-based linkage map and application in genetic studies in oak and beyond
We report here the development and validation of the first high-throughput Illumina SNP genotyping assay for oaks, with a success rate of 88.6%, i.e. 7,010 of the 7,913 SNPs were successfully genotyped. Overall, 82% of the SNPs successfully genotyped were polymorphic in at least one of the four pedigrees and 63% were mapped as FMs on the parental genetic maps.
The use of multiple segregating populations of diverse genetic backgrounds made it possible to map a larger number of markers and to achieve greater genome coverage. This has already been illustrated in watermelon, for example, in which the genotyping of four mapping populations increased the proportion of mapped loci from the most polymorphic pedigree by 29%.36 In this study, the gain in terms of the number of newly segregating test-cross markers from the pedigree with the largest number of SNPs (2,472 in P2) with respect to the other three pedigrees was remarkably high, reaching +1,756 SNPs (i.e. +41% newly mapped markers).
The development of array-based genotyping technologies has made it possible to generate high-density linkage maps rapidly, but the integration of independent maps containing thousands of loci remains a real challenge.37–39 Two main approaches have been developed for the construction of combined maps. The first involves pooling genotypic data and minimizing the sum of recombination frequencies, as proposed in the ML method implemented in JoinMap (e.g. as used in triticale by Alheit et al.37 and in sunflower by Bowers et al.).38 However, this method is time-consuming and may be not appropriate when thousands of markers are used, due to the computational time required. The second approach involves the direct integration of independent linkage maps, as proposed in MergeMap40,41 and LPmerge34 and recently used in pine,39,42 barley43 and wheat.44 Using this second approach, we merged eight linkage maps each containing 889–1,421 markers, to obtain a composite map including 4,261 loci corresponding to 4,239 different contigs of the oak UniGene database45 and covering 742 cM on the 12 LGs. This map is considerably denser than a previously published linkage map based on 397 EST and genomic SSRs.13
Framework maps were constructed exclusively from markers with a testcross configuration. All intercross markers were excluded from construction of the framework map because they provide little information about linkage.35 We added markers segregating in the intercross configuration at a later stage, as accessory markers. The eight parental maps displayed remarkably high degrees of collinearity and no chromosomal rearrangements were observed, providing support for our approach of merging maps to construct a unified composite map for the genus Quercus, including both species maps. In the framework of the oak genome sequencing project,46 this genomic resource will be crucial for the assembly of genome scaffolds into chromosomal pseudomolecules. High-density sequence-based linkage maps have been used to anchor and orient scaffolds in many plant species, including Cicer arietinum (chickpea),47 Rubus (raspberry)48 and Eucalyptus.49 The oak composite linkage map also provides a framework for genome-wide analysis at the centimorgan scale, for genomic scans of species/population divergence,50 studies of the evolutionary relationships between related species,16,51 the detection of recombination hot and cold spots,52 studies of the extent of long-distance linkage disequilibrium and genetic diversity,39,53 the detection and positional characterization of QTLs through co-localization with gene-based markers (e.g.54–56), and the identification of chromosomal rearrangements.57 This unified linkage map for the genus Quercus will also be useful for analyses of synteny and collinearity within the Fagaceae at a much higher resolution than previously reported (e.g. between Quercus and Castanea),13,14,58 and for extending such analyses to other Eurosids, providing insight into genome evolution59 and a framework for the transfer of genetic information between species.
4.2. Patterns of SD identify gametic incompatibility as a major RI barrier in oak
Deviation from the ratios expected for Mendelian inheritance reveal disturbances in the transmission of genetic information from one generation to the next, generating interesting hypotheses about the mechanisms underlying RI for exploration in further studies. Overall, our results revealed that SD was widespread in the oak genome. Regardless of the species or cross, half of the 12 LGs displayed SD, for 6–91% of markers, depending on the LG. We observed large differences in SD values between intra- and interspecific crosses, and between male and female parents.
These SDRs may result from chromosome loss or rearrangements, genetic load or pre- or post-zygotic selection, and interpretation may differ between intra- and interspecific crosses.
Peculiar life history traits of trees may raise their genetic load and result in substantial SD. Oaks, like most forest trees species, form very large populations, and generally outcross through wind pollination, resulting in high levels of gene flow.60,61 These characteristics lead to the accumulation of deleterious mutations and the build-up of a large genetic load.27 The accumulation of recessive deleterious mutations in fitness genes (pollen fertility, anther receptivity, seed fertility) or loci closely linked to fitness genes decreases the viability of plants with homozygous at these loci.62 Genetic load may be partially purged at early embryonic stages, by the death or sterility of hybrids carrying homozygous recessive deleterious mutations. Genetic load varies considerably between plant species and may be a major source of SDRs.17 For the intraspecific pedigree P4, we observed SDRs on LG6 that were conserved between the two parental maps. This could indicate the presence of lethal or sublethal genes compromising seedling survival in both parental genotypes.48
Gametic incompatibility and/or reduced hybrid viability can also contribute to SD. In this study, the proportion of loci displaying SD differed between the eight linkage maps (0–14.5%). The frequency of SD was higher on three LGs: LG6 (28% of loci displaying SD distributed over 16 cM, for three parents), LG 8 (26.5%, 55 cM, one parent) and LG11 (26.5%, 33 cM, two parents). In several previous studies (eucalyptus,63 rice19 and monkeyflower),20 clusters of distorted loci were found to extend over all or most of the LG. In rice, for example, distortion gradually decreases with increasing distance from the markers displaying the highest levels of SD, located at or near previously reported gametophytic gene loci or sterility loci. In our study, 79% of the loci presenting SD were of paternal origin whatever the type of cross (intra- or inter-specific). LG11 in the cross with QS29 as the male parent (P3m) provides an extreme example, with an SDR encompassing 88% of the SNPs on this LG, as previously reported in a study with far fewer EST–SSR markers and a much smaller number of offspring.13 These figures may reflect strong pollen incompatibilities between the parents of the different crosses, as previously reported by Abadie et al.64 Indeed, the results obtained for one genotype (QS28), used as either the male or the female parent in controlled crosses, strongly support the observed trend: the number of markers displaying SD was five times higher when this genotype was used as the male parent (P2m) than when it was used as the female parent (P4f). A confounding effect of the type of cross may also have contributed to the observed pattern (see next paragraph) because QS28 was used as the male in the interspecific cross and as the female in the intraspecific cross. Male gametophytic selection has been identified as the phenomenon most frequently causing skewed segregation, due to selective influences of the gynoecium, resulting in genetic incompatibility.19
Interestingly, 62% of the loci displaying SD were derived from interspecific crosses and were only detected in the male LGs. SD is frequently observed in interspecific crosses, and has been attributed to biological factors, such as pollen–pistil incompatibility, hybrid viability, sterility due to gametophytic competition, negative epistatic interactions between alleles or positive introgression.17,21 Pre-reproductive barriers play a major role in the directionality of introgression: genetically based pollen discrimination is a major barrier, as it greatly increases assortative mating within species and the parental species fidelity of hybrids.65 Alternatively, natural selection may play a key role by acting against unfit genetic combinations. Lepais and Gerber66 observed lower levels of mating success for interspecific crosses compared with intraspecific crosses or backcross mating events, in a mixed stand of four European white oak species (including Qr and Qp). They clearly showed that the different species contributed unequally to reproduction success through differences in pollen efficacy. Most pure-bred plants reproduce preferentially with conspecific individuals. These findings were confirmed by another independent hybridization study conducted in a mixed stand of four European white oak species, Qr, Qp, Quercus pubescens and Quercus frainetto.1 This previous study highlighted the importance of selection against hybrids, resulting in the maintenance of four distinct parental gene pools in sympatry. Based on these observations, we suggest that gametic incompatibility could lead to SD in chromosomal regions on either the female or the male map, whereas reduced hybrid variability would be likely to cause SD in the corresponding regions of both parental maps. Most of the observed SDRs were identified in only one parent, generally the male. Thus, a large fraction of the SDRs were sex-specific, suggesting that gametic selection plays an important role in shaping SDRs in oak.
5. Conclusion
Our study demonstrates the relevance of Illumina technology for SNP genotyping for multipedigree studies in oaks. The high rate of successful SNP development for the six different parental genotypes used in the four controlled crosses provided us with a very large set of mappable SNPs, which was essential for comparative mapping and construction of the oak high-density composite linkage map.
We established a high-density composite linkage map based on more than 4,261 SNP loci suitable for use as a reference gene-based map for the genus Quercus and for the Fagaceae in general. Genomic resources are available for very few species within the Fagaceae family and the map developed here could be useful for studies of species from the same genus or for related genera belonging to this family.
Finally, we identified regions of SD potentially related to RI or genetic load. Further studies assessing seed abortion and the viability of young hybrid seedlings during juvenile development are required to shed light on the causal mechanisms underlying RI. Additional investigations of the co-localization between SDRs, QTLs for adaptive traits67,68 and species divergence hot spots should also be carried out. The recent availability of a whole-genome sequence for Quercus46 will finally help to identify genes located in and underlying SDRs and the genes involved in RI.
Authors' contributions
C.B. performed the genetic analysis, E.C. extracted the DNA, E.C. and C.B. carried out the genotyping analysis, F.E. updated the databases, C.B. and C.P. wrote the manuscript. C.B., C.P. and A.K. conceived and designed the project. All authors read and approved the final manuscript.
Supplementary Data
Supplementary Data are available at www.dnaresearch.oxfordjournals.org.
Funding
The study was funded by the European Commission, as part of the FP5 OAKFLOW project (Intra and interspecific geneflow in oaks as mechanisms promoting genetic diversity and adaptive potential, N°QLK5-2000-00960), the FP6 program (FP6-2004-GLOBAL-3, Network of Excellence EVOLTREE ‘Evolution of Trees as Drivers of Terrestrial Biodiversity’, No. 016322). Funding to pay the Open Access publication charges for this article was provided by Treepeace (European Research Council Advanced Grant FP7-339728).
Web portal
Quercus portal:
https://w3.pierroton.inra.fr/QuercusPortal/
CMap Comparative Map Viewer for the composite LGs:
http://w3.pierroton.inra.fr/cgi-bin/cmap/viewer?ref_map_set_acc=51&ref_map_accs=-1
CMap Comparative Map Viewer for different parental LGs:
Supplementary Material
Acknowledgements
We thank Benjamin Dencausse and Guy Roussel for technical support for the creation and monitoring of the controlled crosses and the team of the INRA experimental unit for assistance with field work. We thank Cyril Firmat, Jérôme Bartholomé and Hélène Lagraulet for their help for the development of R scripts.
References
- 1.Curtu A., Gailing O., Finkeldey R.. 2007, Evidence for hybridization and introgression within a species-rich oak (Quercus spp.) community, BMC Evol. Biol., 7, 218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Guichoux E., Garnier-Géré P., Lagache L., Lang T., Boury C., Petit R.J.. 2013, Outlier loci highlight the direction of introgression in oaks, Mol. Ecol., 22, 450–62. [DOI] [PubMed] [Google Scholar]
- 3.Lepais O., Roussel G., Hubert F., Kremer A., Gerber S.. 2013, Strength and variability of postmating reproductive isolating barriers between four European white oak species, Tree Genet. Genomics, 9, 841–53. [Google Scholar]
- 4.Goicoechea P.G., Herrán A., Durand J., Bodénès C., Plomion C., Kremer A.. 2015, A linkage disequilibrium perspective on the genetic mosaic of speciation in two hybridizing Mediterranean white oaks, Heredity, 114, 373–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kremer A., Dupouey J.L., Deans J.D. et al. . 2002, Leaf morphological differentiation between Quercus robur and Quercus petraea is stable across western European mixed oak stands, Ann. For. Sci., 59, 777–87. [Google Scholar]
- 6.Parelle J., Brendel O., Bodénès C. et al. . 2006, Differences in morphological and physiological responses to water-logging between two sympatric oak species (Quercus petraea [Matt.] Liebl., Quercus robur L.), Ann. Sci. For., 63, 849–59. [Google Scholar]
- 7.Ducousso A., Bodénès C., Petit R.J., Kremer A.. 1996, Le point sur les chênes blancs européens, Forêt Entreprise, 112, 49–56. [Google Scholar]
- 8.Ponton S., Dupouey J.L., Bréda N., Feuillat F., Bodénès C., Dreyer E.. 2001, Carbon isotope discrimination and wood anatomy variations in mixed stands of Quercus robur and Quercus petraea , Plant Cell Environment, 24, 861–8. [Google Scholar]
- 9.Petit R.J., Bodénès C., Ducousso A., Roussel G., Kremer A.. 2003, Hybridization as a mechanism of invasion in oaks, New Phytol., 161, 151–64. [Google Scholar]
- 10.Bacilieri R., Ducousso A., Petit R.J., Kremer A.. 1996, Mating system and asymetric hybridization in a mixed stand of European oaks, Evolution, 50, 900–8. [DOI] [PubMed] [Google Scholar]
- 11.Barreneche T., Casasoli M., Russell K. et al. . 2004, Comparative mapping between Quercus and Castanea using simple-sequence repeats (SSRs), Theor. Appl. Genet., 108, 558–66. [DOI] [PubMed] [Google Scholar]
- 12.Scotti-Saintagne C., Mariette S., Porth I. et al. . 2004, Genome scanning for interspecific differentiation between two closely related oak species [Quercus robur L. and Q. petraea (Matt.) Liebl.], Genetics, 168, 1615–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bodénès C., Chancerel E., Murat F. et al. . 2012, Comparative mapping in the Fagaceae and beyond using EST-SSRs, BMC Plant Biol., 12, 153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lepoittevin C., Bodénès C., Chancerel E. et al. . 2015, Single-nucleotide polymorphism discovery and validation in high density SNP array for genetic analysis in European white oaks, Mol. Ecol. Res., doi:10.1111/1755-0998.12407. [DOI] [PubMed] [Google Scholar]
- 15.Li H., Kilian A., Zhou M. et al. . 2010, Construction of a high-density composite map and comparative mapping of segregation distortion regions in barley, Mol. Genet. Genomics, 284, 319–31. [DOI] [PubMed] [Google Scholar]
- 16.Ollitraut P., Terol J., Chen C. et al. . 2012, A reference genetic map of C. clementina hort. ex Tan.; citrus evolution inferences from comparative mapping, BMC Genomics, 13, 593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Myburg A.A., Vogl C., Griffin A.R., Sederoff R.R., Whetten R.W.. 2004, Genetics of postzygotic isolation in Eucalyptus: whole-genome analysis of barriers to introgression in a wide interspecific cross of Eucalyptus grandis and E. globulus , Genetics, 166, 1405–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lu H., Romero-Severson J., Bernardo R.. 2002, Chromosomal regions associated with segregation distortion in maize, Theor. Appl. Genet., 105, 622–8. [DOI] [PubMed] [Google Scholar]
- 19.Xu Y., Zhu L., Xiao J., Huang N., McCouch S.R.. 1997, Chromosomal regions associated with segregation distortion of molecular markers in F2, backcross, doubled haploid, and recombinant inbred populations in rice (Oryza sativa L.), Mol. Gen. Genet., 253, 535–45. [DOI] [PubMed] [Google Scholar]
- 20.Fishman L., Willis J.H.. 2001, Evidence for Dobzhansky-Muller incompatibilities contributing to the sterility of hybrids between Mimulus guttatus and M. nasatus, Evolution, 55, 1932–42. [DOI] [PubMed] [Google Scholar]
- 21.Yin T.M., DiFazio S.P., Gunter L.E., Riemenschneider D., Tuskan G.A.. 2004, Large-scale heterospecific segregation distortion in Populus revealed by a dense genetic map, Theor. Appl. Genet., 109, 451–63. [DOI] [PubMed] [Google Scholar]
- 22.Shirasawa K., Isobe S., Hirakawa H. et al. . 2010, SNP discovery and linkage map construction in cultivated tomato, DNA Res., 17, 381–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu X., You J., Guo L. et al. . 2010, Genetic analysis of segregation distortion of SSR markers in F2 population of barley, J. Agric. Sci., 3, 172–7. [Google Scholar]
- 24.Moyle L.C., Olson M.S., Tiffin P.. 2015, Patterns of reproductive isolation in three Angiosperm genera, Evolution, 58, 1195–208. [DOI] [PubMed] [Google Scholar]
- 25.Ouyang Y., Liu Y-G., Zhang Q.. 2010, Hybrid sterility in plant: stories from rice, Curr. Opin. Plant Biol., 13, 186–92. [DOI] [PubMed] [Google Scholar]
- 26.Sweigart A.L., Willis J.H.. 2012, Molecular evolution and genetics of postzygotic reproductive isolation in plants, Biol. Rep., 4, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Klekowski E.J. 1988, Genetic load and its causes in long-lived plants, Trees, 2, 195–203. [Google Scholar]
- 28.Williams C.G., Savolainen O.. 1996, Inbreeding depression in conifers: implications for breeding strategy, For. Sci., 42, 102–17. [Google Scholar]
- 29.Fishman L., Willis J.H.. 2005, A novel meiotic drive locus almost completely distorts segregation in Mimulus (Monkeyflower) hybrids, Genetics, 169, 347–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Steinhoff S. 1993, Results of species hybridization with Quercus robur L. and Quercus petraea (Matt.) Liebl., Ann. Sci. For., 50, 137s–43s. [Google Scholar]
- 31.Ueno S., Le Provost G., Léger V. et al. . 2010, Bioinformatic analysis of ESTs collected by Sanger and pyrosequencing methods for a keustone forest tree species: oak, BMC Genomics, 11, 650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.van Ooijen J.W., In Joinmap® 4, Software for the Calculation of Genetic Maps in Experimental Populations. Edited by Kyazma B.V. Wageningen, Netherlands; 2006. [Google Scholar]
- 33.Voorrips R.E. 2002, MapChart: software for the graphical presentation of linkage maps and QTLs, J. Hered., 93, 77–8. [DOI] [PubMed] [Google Scholar]
- 34.Endelman J., Plomion C.. 2014, LPmerge: an R package for merging genetic maps by linear programming, BioInformatics, 30, 1623–4. [DOI] [PubMed] [Google Scholar]
- 35.Ritter E., Gebhardt C., Salamini F.. 1990, Estimation of recombination frequencies and construction of RFLP linkage maps in plants from crosses between heterozygous parents, Genetics, 125, 645–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ren Y., McGregor C., Zhang Y. et al. . 2014, An integrated genetic map based on four mapping populations and quantitative trait loci associated with economically important traits in watermelon (Citrullus lanatus), BMC Plant Biol., 14, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Alheit K.V., Reif J.C., Maurer H.P. et al. . 2011, Detection of segregation distortion loci in triticale (x Triticosecale Wittmack) based on a high-density DArT marker consensus genetic linkage map, BMC Genomics, 12, 380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bowers J.E., Bachlava E., Brunick R.L., Rieseberg L.H., Knapp S.J., Burke J.M.. 2012, Development of a 10,000 locus genetic map of the sunflower genome based on multiple crosses, G3, 2, 721–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Plomion C., Chancerel E., Endelman J. et al. . 2014, Genome-wide distribution of genetic diversity and linkage disequilibrium in a mass-selected population of maritime pine, BMC Genomics, 15, 171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li Y., Liu S., Qin Z. et al. . 2015, Construction of a high-density, high-resolution genetic map and its integration with BAC-based physical map in channel catfish, DNA Res., 22, 39–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Muñoz-Amatriaín M., Moscou M.J., Bhat P.R. et al. . 2011, An improved consensus linkage map of Barley based on flow-sorted chromosomes and Single Nucleotide Polymorphism markers, Plant Genome, 4, 238–49. [Google Scholar]
- 42.Muñoz-Amatriaian M., Cuesta-Marcos A., Endelman J.B. et al. . 2014, The USDA barley core collection: genetic diversity, population structure, and potential for genome-wide association studies, PLoS ONE, 9, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.de Miguel M., Bartholomé J., Ehrenmann F. et al. . 2015, Evidence of intense chromosomal shuffling during conifer evolution, Genome Biol. Evol., doi:10.1093/gbe/evv185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yu L.X., Barbier H., Rouse M.N. et al. . 2014, A consensus map for Ug99 stem rust resistance loci in wheat, Theor. Appl. Genet., 127, 1561–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lesur I., Le Provost G., Bento P. et al. . 2015, The oak gene expression atlas: insights into Fagaceae genome evolution and the discovery of genes regulated during bud dormancy release, BMC Genomics, 16, 112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Plomion C., Aury J.M., Anselem J. et al. . 2015, Decoding the oak genome: public release of sequence data, assembly, annotation and publication strategies, Mol. Ecol. Res., doi:10.1111/1755-0998.12425. [DOI] [PubMed] [Google Scholar]
- 47.Gaur R., Azam S., Jeena G. et al. . 2012, High-throughput SNP discover y and genotyping for constructing a saturated link age map of Chickpea (Cicer arietinum L.), DNA Res., 19, 357–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ward J., Bhangoo J., Fernández-Fernández F. et al. . 2013, Saturated linkage map construction in Rubus idaeus using genotyping by sequencing and genome-independent imputation, BMC Genomics, 14, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Petroli C., Sansaloni C.P., Carling J. et al. . 2012, Genomic characterization of DArT markers based on high-density linkage analysis and physical mapping to the Eucalyptus genome, PLoS ONE, 7, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kane N.C., King M.G., Barker M.S. et al. . 2009, Comparative genomic and population genetic analysis indicate highly porous genomes and high levels of gene flow between divergent Helianthus species, Evolution, 63, 2061–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jung S., Cestaro A., Troggio M. et al. . 2012, Whole genome comparisons of Fragaria, Prunus and Malus reveal different modes of evolution between Rosaceous subfamilies, BMC Genomics, 13, 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chancerel E., Lamy J.B., Lesur I. et al. . 2013, High-density linkage mapping in a pine tree reveals a genomic region associated with inbreeding depression and provides clues to the extent and distribution of meiotic recombination, BMC Biol., 11, 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Isobe S.N., Hirakawa H., Sato S. et al. . 2013, Construction of an integrated high density Simple Sequence Repeat linkage map in cultivated strawberry (Fragaria 3 ananassa) and its applicability, DNA Res., 20, 79–92, doi:10.1093/dnares/dss035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bartholomé J., Mandrou E., Mabiala A. et al. . 2014, High-resolution genetic linkage maps of Eucalyptus improve Eucalyptus grandis genome assembly, New Phytol., 206, 1283–96. [DOI] [PubMed] [Google Scholar]
- 55.Martínez-García P.J., Parfitt D.E., Ogundiwin E.A. et al. . 2013, High density SNP mapping and QTL analysis for fruit quality characteristics in peach (Prunus persica L.), Tree Genet. Genomes, 9, 19–36. [Google Scholar]
- 56.Yu H.H., Xie W.B., Wand J. et al. . 2011, Gains in QTL detection using an ultra-high density SNPmap based on population sequencing relative to traditional RFLP/SSR markers, PLoS ONE, 6, e17595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fishman L., Stathos A., Beardsley P.M., Williams C.F., Hill J.P.. 2013, Chromosomal rearrangements and the genetics of reproductive barriers in Mimulus (Monkeyflowers), Evolution, 6, 2547–56. [DOI] [PubMed] [Google Scholar]
- 58.Casasoli M., Derory J., Morera-Dutrey C. et al. . 2006, Comparison of quantitative trait loci for adaptive traits between oak and chestnut based on an expressed sequence tag consensus map, Genetics, 172, 533–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Murat F., Van de Peer Y., Salse J.. 2012, Decoding plant and animal genome plasticity from differential paleo-evolutionary patterns and processes, Genome Biol. Evol., 4, 917–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kremer A., Petit R.J.. 1993, Gene diversity in natural populations of oak species, Ann. Sci. For., 50(Suppl. 1), 186s–202s. [Google Scholar]
- 61.Gerber S., Chadoeuf J., Gugerli F. et al. . 2014, High rates of gene flow by pollen and seed in oak populations across Europe, PLoS ONE, 9, e85130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Launey S., Hedgecok D.. 2001, High genetic load in the pacific oyster Crassostrea gigas, Genetics, 159, 255–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kullan A.R.K., van Dyk M.M., Jones N., Kanzler A., Bayley A., Myburg A.A.. 2012, High-density genetic linkage maps with over 2,400 sequence-anchored DArT markers for genetic dissection in an F2 pseudo-backcross of Eucalyptus grandis × E. urophylla, Tree Genet. Genomes, 8, 163–75. [Google Scholar]
- 64.Abadie P., Roussel G., Dencausse B. et al. . 2012, Strength, diversity and plasticity of postmating reproductive barriers between two hybridizing oak species (Quercus robur L. and Quercus petraea (Matt) Liebl.), J. Evol. Biol., 25, 157–73. [DOI] [PubMed] [Google Scholar]
- 65.Lepais O., Petit R.J., Guichoux E. et al. . 2008, Species relative abundance and direction of introgression in oaks, Mol. Ecol., 18, 2228–42. [DOI] [PubMed] [Google Scholar]
- 66.Lepais O., Gerber S.. 2011, Reproductive patterns shape introgression dynamics and species succession within the European white oak species complex, Evolution, 1, 156–70. [DOI] [PubMed] [Google Scholar]
- 67.Brendel O., Le Thiec D., Scotti-Saintagne C., Bodénès C., Kremer A., Guehl J.M.. 2008, Quantitative trait loci controlling water use efficiency and related traits in Quercus robur L., Tree Genet. Genomes, 4, 263–78. [Google Scholar]
- 68.Parelle J., Zapater M., Scotti-Saintagne C. et al. . 2007, Quantitative trait loci of tolerance to waterlogging in a European oak (Quercus robur L.): physiological relevance and temporal effect patterns, Plant, Cell Environment, 30, 422–34. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.