

Abstract
The human gut microbiome has profound influences on the host's health largely through its interference with various intestinal functions. As recent studies have suggested diversity in the human gut microbiome among human populations, it will be interesting to analyse how gut microbiome is correlated with geographical, cultural, and traditional differences. The Japanese people are known to have several characteristic features such as eating a variety of traditional foods and exhibiting a low BMI and long life span. In this study, we analysed gut microbiomes of the Japanese by comparing the metagenomic data obtained from 106 Japanese individuals with those from 11 other nations. We found that the composition of the Japanese gut microbiome showed more abundant in the phylum Actinobacteria, in particular in the genus Bifidobacterium, than other nations. Regarding the microbial functions, those of carbohydrate metabolism were overrepresented with a concurrent decrease in those for replication and repair, and cell motility. The remarkable low prevalence of genes for methanogenesis with a significant depletion of the archaeon Methanobrevibacter smithii and enrichment of acetogenesis genes in the Japanese gut microbiome compared with others suggested a difference in the hydrogen metabolism pathway in the gut between them. It thus seems that the gut microbiome of the Japanese is considerably different from those of other populations, which cannot be simply explained by diet alone. We postulate possible existence of hitherto unknown factors contributing to the population-level diversity in human gut microbiomes.
Keywords: microbiome, gut, metagenome, Japanese
1. Introduction
Various cohort studies of the human gut microbiome based on a metagenomic approach using next-generation sequencing (NGS) have been reported.1–11 These studies included patients with diseases such as obese,1 inflammatory bowel disease,2 type 2 diabetes,3,6 colon cancer,7,9 liver cirrhosis,8 and rheumatoid arthritis,11 as well as numbers of healthy individuals in various countries including the USA, several European countries, and China. In addition, several studies have reported on gut microbiomes of Asian children and natives from rural areas.12–16 These studies suggest that the human gut microbiome is more or less affected by various factors such as diet and host's genetic background,17 and the altered microbiome is associated with diseases.18 Several comparative analyses have also suggested a great diversity in human gut microbiomes at the population level,5,6,12–16,19 and even in those of patients with diseases.6,20 Essentially, human gut microbiomes can be classified into ‘enterotypes’ on the basis of differences in the abundance of a few major species largely linking with dietary habits.21,22
Japanese (JP) have unique dietary culture and habits compared with Western people being reflected in the finding that their gut microbiomes have more genes for aquatic plant-derived polysaccharide-degrading enzymes than those of Americans.23 In addition, Japanese exhibit the highest average life span and very low body mass index (BMI).24 A study on the gut microbiomes of 13 Japanese individuals has been previously published.25 However, the dataset size was too small to allow comparison with other large datasets to evaluate distinct features of the Japanese gut microbiome (JPGM). Therefore, in this study, we collected and analysed the metagenomic data from gut microbiomes of 106 Japanese individuals by sequencing of fecal DNA samples using NGS, and we further explored the unique microbial and functional features of the JPGM using microbiome data from a total of 861 healthy individuals selected from 12 countries including Japan.
2. Materials and methods
2.1. Subjects and fecal sample collection
One hundred and six Japanese volunteers [age: 32 ± 11, BMI: 22 ± 2.7 (mean ± S.D.), and male:female = 64:42] were recruited by Azabu University (Japan) from 2010 to 2013 (Supplementary Table S1). This study was approved by the Human Research Ethics Committee of Azabu University and the Research Ethics Committee of the University of Tokyo, and written consent was obtained from all subjects. No subjects were treated with antibiotics during fecal sample collection. Among them, fecal samples were longitudinally collected twice from 26 individuals every 8 weeks and five times from 9 individuals every 2 weeks, of which 16 individuals were shared with the previous study.26 A total of 168 fecal samples were collected from the 106 individuals. The collected fresh feces were stored under anaerobic conditions in an AneroPack™ (Mitsubishi Gas Chemical Co. Inc., Tokyo, Japan) at 4°C. Within 36 h after sampling, the feces were frozen in 20% glycerol (Wako Pure Chemical Industries, Osaka, Japan)/phosphate buffer saline (PBS) solution (Life Technologies, Tokyo, Japan) by liquid nitrogen and stored at −80°C until ready for use.
2.2. Recovery of bacteria from fecal samples and bacterial DNA isolation
Bacteria and bacterial DNA were prepared as described previously with minor modifications.26,27 In brief, the bacterial DNA was isolated by the enzymatic lysis method using lysozyme (Sigma-Aldrich Co. LCC., Tokyo, Japan) and achromopeptidase (Wako). The DNA samples were purified by treatment with RNase A (Wako), followed by precipitation with 20% PEG solution (PEG6000 in 2.5 M NaCl). The DNA was pelleted by centrifugation, rinsed with 75% ethanol, and dissolved in TE buffer.
2.3. Metagenomic sequencing of fecal DNA
The fecal DNA samples were sequenced using 454 GS FLX Titanium and FLX+ (Roche), Ion PGM and Ion Proton (Life Technologies), and MiSeq (Illumina) sequencing system. The detailed protocols have been described in the Supplementary Text. In brief, after the quality filtering, reads mapped to the human genome (HG19) were removed. Artificially redundant reads from the 454 and Ion PGM/Proton sequencers were removed using a replicate filter.28 From the MiSeq filter-passed reads, reads mapped to the phiX bacteriophage genomes were also removed. Sequencing statistics are summarized in Supplementary Table S1.
2.4. PCR detection of Methanobrevibacter smithii in the Japanese individuals
Methanobrevibacter smithii was detected by PCR using M. smithii 16S rRNA gene-specific primers 5′-ATGCACCTCCTCTCAGCTAGTC-3′ and 5′-AGAGGTACTCCCAGGGTAGAGG-3′. The details have been described in the Supplementary Text.
2.5. Publically available metagenomic sequence data
We collected metagenomic sequence data of individuals from Denmark (DK), Spain (ES), the USA (US), China (CN), Sweden (SE), Russia (RU), Venezuela (VE), Malawi (MW), Austria (AT), France (FR), and Peru (PE). Metagenomic reads from DK2 and ES2 populations were downloaded from http://public.genomics.org.cn. Filter-passed reads from the US4 population were downloaded from the HMP DACC (http://www.hmpdacc.org). Raw reads for DK,29 ES,19 CN,3,8 SE,6 RU,5 AT,9 FR,7 PE,15 US,15 and VE30 were downloaded from public databases and subjected to the quality control prior to use under the same conditions as used for the JP data. The SOLiD reads from RU were subjected to error correction using the SOLiD Accuracy Enhancement Tool (SAET), and the quality control was performed as described previously.5 Raw reads for VE,14 MW,14 and US14 were downloaded from MG-RAST (http://metagenomics.anl.gov) and reads mapped to the human genome were excluded.
2.6. Country-specific metagenomic datasets of healthy individuals
To construct metagenomic datasets from healthy individuals from each country, the data for individuals with BMI ≥30; those designated with the following diseases based on the literature: inflammatory bowel disease, type 2 diabetes, liver cirrhosis, or colorectal cancer; and infants <3 yrs old were excluded from the data collected from a total of 1,734 individuals from the 12 countries. The exclusion of the patient and infant samples was to avoid the incorporation of altered gut microbiomes significantly different from the healthy adult gut microbiome.14,18 Although we could not access the metadata for individuals from US,4 we used all data from individuals with an average BMI of 24 ± 4 (mean ± S.D.) for this cohort. Finally, we selected and used a total of 861 healthy individuals from the 12 countries for the analysis (Supplementary Table S2).
2.7. Construction of microbial reference genomes
To improve the efficiency and accuracy of taxonomic assignment of metagenomic sequences and to reduce excess load in computing, we constructed and used an in-house developed reference genome database from 2,788 complete and 22,317 draft genomes available from GenBank/EBI/DDBJ (as of July 2014), 20 genomes,31 and 2 unpublished genomes. The reference genome database comprises a total of 6,149 genomes representing 2,373 clusters at the species level of Bacteria and Archaea (Supplementary Table S3). Detailed procedures have been described in the Supplementary Text.
2.8. Mapping of metagenomic reads to the reference genomes
One million metagenomic reads per individual were mapped to the reference genomes using Bowtie232 (version 2.2.1) with a 95% identity threshold. The number of multi-hit reads that mapped to several different genomes with identical scores was normalized by the proportion to the number of reads uniquely mapped to the genomes. The relative abundance of each genome was calculated by normalizing the number of reads mapped to the genome by the total number of reads mapped. NCBI taxonomy information was used for taxonomic assignment of phylum, genus, and species for each genome. Genomes that were not assigned to a particular taxonomic rank were assigned to the higher rank classification and designated ‘unclassified higher rank’. Detailed procedures have been described in the Supplementary Text.
2.9. Comparison of microbial compositions
A multi-dimensional scaling (MDS) plot was constructed using the Jensen–Shannon divergence between microbial compositions at the genus level of the 861 individuals. Hierarchical clustering was performed using the Ward method based on the Bray–Curtis distances. A model to predict each country based on microbial composition at the genus level was constructed using the randomForest package in R. The predictive power of the model was evaluated by 10-fold cross-validation with 90% of the training data and 10% of the prediction data. The number of trees was set to 500, and the sample size option was set to the minimum number of individuals among the countries. The receiver operating characteristic curve (ROC) and area under the ROC curves (AUCs) of the predictive model were calculated and plotted with the smooth function using the ROCR and pROC packages. Pearson correlation coefficients (PCCs)6 were used for evaluating similarities between microbial compositions from individuals within and between countries.
To evaluate the effect of methodologies in the metagenomic analysis, three different subsets of fecal samples were subjected to analyses using different protocols in sequencing, DNA preparation, and fecal sample storage steps, respectively. Detailed procedures have been described in the Supplementary Text. The protocols used in the present and other studies are also summarized in Supplementary Table S4.
Permutational Multivariate Analysis of Variance (PERMANOVA) was used to assess the association of age and BMI with the gut microbiome structure using the adonis function in the Vegan package in R. The dietary intake information of the 12 countries was collected from the Food and Agriculture Organization Corporate Statistical (FAOSTAT) database (http://faostat3.fao.org/home/, as of June 2015). Details on the food data have been described in the Supplementary Text.
2.10. Assembly of metagenomic sequences and gene prediction in the JPGM
For each JP individual, the filter-passed reads produced from 454 GS and Ion PGM/Proton and those from MiSeq were assembled using the Newbler assembler (version 2.7), respectively. The contigs generated were further assembled with Minimus233 using the default setting for each individual. Unassembled reads (singletons) from each sequencer in all individuals were combined and reassembled to obtain additional contigs formed between different samples. MetaGeneAnnotator34 was used to predict protein-coding genes (≥100 bp) in the contigs (≥500 bp) and singletons (≥300 bp). Finally, ∼4.9 million (M) non-redundant genes were identified in the JPGM by further clustering the predicted genes using CD-HIT35 with a 95% nucleotide identity and 90% length coverage cut-off.
2.11. Generation of a JPGM and integrated gene catalog merged reference gene set of human gut microbiomes
The integrated gene catalog (IGC),19 which was constructed from metagenomic data of >1,000 gut microbiomes from DK, ES, US, and CN were downloaded from http://meta.genomics.cn/metagene/meta/home. The merged reference gene set was constructed by clustering the JPGM non-redundant genes (4.9 M) and the non-redundant genes (9.9 M) in the IGC using CD-HIT with a 95% nucleotide identity and 90% length coverage cut-off.
2.12. Functional assignment of non-redundant genes in human gut microbiomes
Functional assignment of the non-redundant genes was performed using BLASTP searches (e-value ≤1.0e−5) against the Kyoto Encyclopedia of Genes and Genomes (KEGG) database (release 63) to obtain the KEGG orthologies (KOs). The genes with a best hit to eukaryotic genes were excluded from further analysis. Additionally, the non-redundant genes were searched using BLASTP (e-value ≤1.0e−5) against the eggNOG database36 (version 4.5) to assign them to non-supervised orthologous groups (NOGs). The phylum-level taxonomic assignment of the genes assigned to NOGs was conducted by BLASTN searches to the reference genomes with a ≥65% identity and ≥85% length coverage cut-off.
2.13. Quantification of the annotated genes in human gut microbiomes
One million metagenomic reads per individual were mapped to the JPGM and IGC merged reference gene set using Bowtie2 with a 95% identity cut-off. To adjust the mapping conditions for long reads (e.g. 454 FLX + reads with an average read length of 730 bp) to the short reference genes with an average length of 620 bp, reads >100 bp were split into 100 bp fragments, which were used independently for similarity searches, while fragments <50 bp were discarded. The number of reads that mapped equally to more than one gene was normalized by the proportion of the number of reads uniquely mapped to the genes as was conducted for the mapping analysis to the reference genomes. The quantities of KOs were calculated from the number of reads mapped to them. We first compared the relative abundance of KOs between the 104 JP individuals and the 757 individuals in the other 11 countries using Student's t-test to enumerate the KOs enriched and depleted with statistical significance (adjusted P < 0.01) in the JPGM. Of them, those having the highest and lowest relative abundance among the 12 countries were identified. P-values were corrected for multiple testing using the Benjamini–Hogberg method.
3. Results
3.1. Population-level diversity in the human gut microbiome
We obtained approximately 350 Gb of filter-passed metagenomic reads with an average of 3.4 ± 1.9 Gb (mean ± S.D.) per individual by sequencing fecal DNA samples from the 106 JP individuals using 454 GS, Ion PGM/Proton, or MiSeq sequencers (Supplementary Table S1). To evaluate the microbial composition of the JPGMs, we mapped the metagenomic reads to the reference genomes. The relative abundances of the four dominant phyla were 59.7 ± 14.7% for Firmicutes, 21.8 ± 16.3% for Actinobacteria, 16.7 ± 10.4% for Bacteroidetes, and 1.4 ± 1.6% for Proteobacteria. At the genus level, Bifidobacterium was the most dominant species, accounting for 17.9 ± 15.2%. The results for taxonomic assignment are also presented in Supplementary Fig. S1.
Next, we collected metagenomic data from 11 other countries to characterize the JPGM and to explore population-level variations in human gut microbiomes among the 12 countries. The independent cohort data were combined per country to construct country-specific metagenomic datasets of healthy individuals, from which data for individuals with BMI ≥30, those afflicted with known diseases, or infants <3 yrs were excluded. These country-specific datasets comprising data of a total of 861 healthy individuals including the 104 JP individuals were used for further analysis (Supplementary Table S2).
We obtained the microbial compositions of the 861 individuals from the 12 countries using the same conditions as those used for the JPGM (Supplementary Fig. S2). The MDS plot of the microbial compositions of the 861 individuals showed that they tended to cluster together per country (Fig. 1A). A permutation test confirmed significantly higher similarity of the microbial composition between individuals within a country than between those of different countries (Fig. 1B). To test whether the microbial composition can predict an individual's country of origin, we used random forest analysis6,14 to construct a predictive model for the countries except VE and MW, for which sample numbers were too small to analyse. The result showed that the AUCs ranged from 0.82 to near 1.00 for each country (Fig. 1C), demonstrating the high predictive accuracy of the model. Taken together, these data strongly suggested that the human gut microbiome composition is significantly diverse across the 12 countries.
Figure 1.
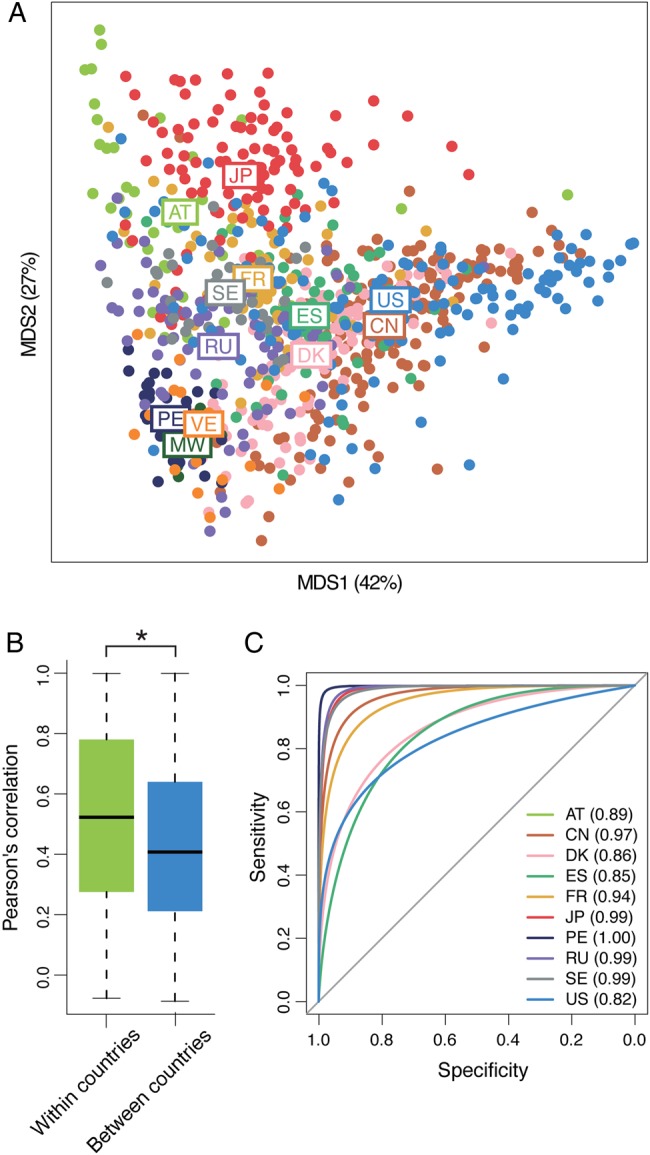
Population-level diversity in human gut microbiomes among the 12 countries. (A) The MDS plot of microbial compositions for all individuals. Each circle represents an individual microbial composition and each colour represents a country of origin. The position based on the average microbial composition for each country is displayed by the abbreviated country name. (B) Comparison of Pearson's correlation coefficients of microbial compositions in individuals within a country and between different countries. Boxes represent the inter-quartile range (IQR), and the lines inside show the median. Whiskers denote the lowest and highest values within 1.5 times the IQR. Asterisk represents P < 0.05. (C) ROC curves and AUC values from the randomForest model. Numbers in parentheses represent the AUC values of the 10 countries.
To examine the effects of different protocols used in the present and other studies on the observed differences in the microbial composition (Supplementary Table S4), we compared and assessed variations in the microbial composition estimated from three different NGS sequencers, four different DNA extraction methods including two enzymatic lysis methods and two commercially available kits based on mechanical disruption of cells, and four different fecal sample storage conditions (Supplementary Table S5). For the fecal sample storage conditions, we focused on assessment of differences in the storage time from defecation until freezing of fecal samples, because it was considered to be varied among the studies. The results revealed that PCCs between the microbial compositions from different protocols were high (from 0.88 to 1.00) in any pair of comparisons between them and significantly higher than those observed for the individuals within and between countries (Supplementary Figs S3 and S4). These data indicated that the methodological differences had no significant effects on the observed variations among the human gut microbiomes.
Hierarchical clustering of the 12 countries based on the average microbial composition at the genus level showed that the JPGM was more similar to microbiomes of AT and SE than that of CN, while those of CN and US were most closely related, but far from the JPGM among the 12 countries (Supplementary Fig. S5). These results indicated that host ethnic and geographical closeness have no large influence on the overall microbial composition of the human gut microbiome. We also assessed the contribution of variations in age and BMI to differences in the microbial abundance by using PERMANOVA. The coefficient of determination for the variation (R2) in age and BMI was 0.16 (P-value = 0.14) and 0.2 (P-value = 0.07), respectively, indicating that both factors had no large influence on the observed results as well.
We further examined the association of diet since it is known to be a major factor influencing microbial composition.22,37 We accessed the FAOSTAT database to collect the dietary intake data (g/capita/day) for 119 food items. Cluster analysis of the 12 countries based on the levels of three main nutrients (carbohydrates, proteins, and lipids) and nine food categories (Supplementary Fig. S6) segregated most of the Western countries from other countries in Asia, South America, and Africa (Supplementary Fig. S7). This indicated that the FAOSTAT data properly represent the current diversity in dietary habits of the 12 countries, allowing for its use in the association analysis with the microbial diversity. Comparison with the dendrogram of the 12 countries based on the microbial composition (Supplementary Fig. S5) revealed several inconsistencies in the relation among the countries. For example, CN had close relations with MW and PE due to the high similarity of dietary component with the high grains/beans and the low animal product level, while it had closer relations with several Western countries such as US, DK, and ES with the high abundance of Bacteroides than MW and PE in the microbial composition. Additionally, SE, FR, and AT were members in the Western group in the dietary component, while those were closer to Japan than other Western countries in the microbial composition. Thus, the population-level diversity in the human gut microbiome may not be relied on diet alone.
3.2. Microbial characterization of the Japanese gut microbiome
We identified 425 genera and 21 phyla of Bacteria and Archaea in the gut microbiomes of the 861 individuals. The genera with an average relative abundance of ≥0.5% in the 12 countries are listed in Supplementary Table S6. When comparing the abundance of the bacterial phyla, the JPGM showed the highest abundance of Actinobacteria. In contrast, the abundance of Bacteroidetes and Proteobacteria in the JPGM was significantly lower than in the microbiomes of various other countries (Fig. 2A). At the genus level, the JPGM was characterized by the highest abundance of Bidfidobacterium, Blautia, Collinsella, Streptococcus, and unclassified Clostridiales, but the lowest abundance of Clostridium, Alistipes, unclassified Firmicutes, Dialister, and Butyrivibrio among the 12 countries (Fig. 2B).
Figure 2.

Taxonomic comparison of gut microbiomes of populations from the 12 countries. Relative abundances of the four dominant phyla (A), the five genera most enriched and depleted in the JPGM (B), and M. smithii (C) in the 12 countries are shown. Vertical axes represent the relative abundance of the species calculated from the number of mapped reads to the reference genomes.
Another characteristic feature of the JPGM was that it has the lowest frequency of M. smithii, a methanogenic archaeon, among the 12 countries (Fig. 2C). Metagenomic mapping analysis showed that this species was detected only in eight (7.7%) JP individuals, while it was detected in a proportion of 39–100% of the individuals in other countries (relative abundance ≥0.0001%, Supplementary Fig. S8a). The lowest prevalence of this archaeon in the JP cohort was also validated by PCR using species-specific 16S primers. The data showed that M. smithii was undetected in 97 (92%) of the 106 JP individuals both in the metagenomic mapping and in the PCR analysis, where 5 were positive in both analyses, 3 were positive only in the mapping analysis, and 1 was positive only in the PCR analysis (Supplementary Fig. S8b).
3.3. Gene numbers in the Japanese gut microbiomes
We performed de novo assembly of filter-passed metagenomic reads from the JP individuals by using the Newbler individually to generate 13 Gb of contigs of ≥500 bp and 0.6 Gb of singletons of ≥300 bp, in which approximately 23 M genes were predicted with MetaGeneAnnotator. By further clustering of the 23 M genes using CD-HIT, we finally obtained 4,854,719 non-redundant genes in the JPGM. Rarefaction analysis of the number of non-redundant genes against the number of individuals sequenced showed that the genes shared by ≥1.9% (2/106) in the JP individuals were almost constant with approximately 100 individuals, while genes unique for an individual (≤1%) increased even beyond 100 individuals (Supplementary Fig. S9a). The number of the genes shared by ≥1.9% (2/106) in the JP individuals represented about 3.8 M, which covered 98.8% of the total reads mapped to the non-redundant genes. These results suggested that the JPGM non-redundant gene set covers most of the genes encoded by the JPGM. In contrast, ≥50% of the JP individuals commonly possessed 0.19 M genes (4.0%), which can be considered to be core genes of the JPGM (Supplementary Fig. S9b).
3.4. Comparison of the JPGM and IGC gene sets
We compared the JPGM gene set with the IGC gene set.19 The clustering of the JPGM (4.9 M) and the IGC genes (9.9 M) generated 11,929,034 non-redundant genes in total, in which 585,856 genes and 202,410 genes were excluded by being merged to longer authentic genes in either of the datasets. As a result, 4,268,863 and 9,676,237 non-redundant genes constituted the JPGM and IGC gene sets, respectively. The data showed that approximately 2.0 M genes were shared by both gene sets, and 2.3 and 7.7 M genes were unique to the JPGM and IGC, respectively (Supplementary Fig. S10a). This limited overlap between the JPGM and IGC gene sets was supported by the mapping analysis of metagenomic reads, in which 45.6% of the JPGM metagenomic reads were mapped to the IGC gene set, while 80.0% were mapped to the JPGM gene set (Supplementary Fig. S10b).
Next, we annotated the functions of the genes with the KEGG database. The analysis identified 5,789 KOs in the JPGM and a total of 6,205 KOs from both gene sets, in which 5,613 KOs (90%) were shared between both datasets, demonstrating a significantly high similarity in functional profiles across the populations despite the small overlap in the gene sequences. It was noted that the IGC-unique 416 KOs included multiple genes related to archaeal methane metabolism, while the JPGM-unique 176 KOs included more genes for spore formation than the IGC-unique KOs (Supplementary Tables S7 and S8).
3.5. Quantitative evaluation of functional profiles of human gut microbiomes
To explore functions that are enriched or depleted in the JPGM compared with microbiomes from the 11 other countries, we mapped the metagenomic reads of the 861 individuals to the JPGM and IGC merged gene set. By comparing the numbers of mapped reads, we identified 563 and 521 KOs having the highest and lowest abundances in the JPGM among the 12 countries with statistical significance (Fig. 3). The overrepresented KOs included functions for carbohydrate metabolism such as glucan 1,3-β-glucosidase (K01210), 6-phospho-β-galactosidase (K01220), and gluconokinase (K00851), and for membrane transport such as the phosphotransferase system of simple sugars including mannose, lactose, and N-acetylgalactosamine (K02796, K02787, and K02746). Thus, metabolic pathways for simple sugars such as mono- and oligosaccharides were significantly enriched in the JPGM compared with the other datasets. On the other hand, the depleted KOs included functions such as cell motility including chemotaxis protein CheX (K03409) and flagellar protein FliO/FilZ (K02418), replication and repair including DNA mismatch repair protein MutL (K03572) and DNA adenine methylase (K06223), suggesting a depletion of functions related to host immunity and DNA damage in the JPGM. Many of the depleted KOs involved in energy metabolism and translation originated from archaea.
Figure 3.

Enriched and depleted functions in the JPGM. Functional categories of the KOs most enriched and depleted in the JPGM compared with those of the other 11 countries are shown. The vertical axis represents the proportion of KOs assigned to the category. Asterisks indicate adjusted P < 0.01 (Fisher's exact test).
In agreement with the lowest prevalence of M. smithii in the JP cohort, many of the KOs involved in methanogenesis were depleted in the JPGM. Of the 25 known KOs involved in methanogenesis, 18 were significantly depleted in the JPGM with the lowest abundance among the 12 countries (Fig. 4 and Supplementary Fig. S11). Conversely, we found a significant enrichment for multiple KOs involved in acetogenesis (the Wood–Ljungdahl pathway) in the JPGM. Of the 17 known KOs involved in acetogenesis, 13 were significantly enriched in the JPGM compared with the other 11 countries, and 5 of them had the highest abundance among the 12 countries (Fig. 4 and Supplementary Fig. S11). These two pathways utilize hydrogen to generate methane and acetate, respectively. Furthermore, the abundance of five genes known to be involved in dissimilatory sulfate reduction (DSR), which is the third pathway for hydrogen metabolism, was similar between the JPGM and other gut microbiomes (Fig. 4). These results indicated that the JPGM had a clear inverse pattern in the abundance of the KOs between methanogenesis and acetogenesis compared with all other microbiomes, suggesting a prominent difference in the pathways for hydrogen utilization in the gut between Japanese and other populations.
Figure 4.

Enriched and depleted genes in the acetogenesis, methanogenesis, and dissimilatory sulfate reduction in the JPGM. Relative abundances of KOs involved in the pathways for hydrogen metabolism in acetogenesis, methanogenesis, and dissimilatory sulfate reduction among the 12 countries are shown. Asterisks indicate adjusted P < 0.01 compared with the abundances of the other 11 countries (Student's t-test).
3.6. Gene families enriched in the Japanese population
We comprehensively surveyed gene families that are frequently present in the JP cohort by using the eggNOG database, which includes more compiled gene families than the KEGG database. The annotation of the merged JPGM and IGC non-redundant genes yielded 51,250 NOGs. In this analysis, we used 10 M metagenomic reads per individual to detect a low content of NOGs, so that 60 individuals having <10 M reads, including all individuals from MW and VE, were excluded from this analysis.
We mapped 10 M reads from 801 individuals to the merged non-redundant genes to detect the NOGs in the individual. For these NOGs, we compared the proportion of individuals possessing them in the JP cohort and with the average proportion of the individuals in the nine other countries (Fig. 5). The results revealed 52 NOGs comprising a total of 1,114 genes that were detected in significantly higher proportions in the JP cohort than in the 9 other datasets using a threshold of a proportion of >70% in the JP cohort, an average proportion of <30% in other countries, and a ratio of JP/others of ≥3 (Supplementary Table S11). Of the 1,114 genes, 63% were taxonomically assigned to the known phyla. Among the 52 NOGs, 30 (58%), 8 (15%), and 5 NOGs (10%) were assigned to only the Actinobacteria, Bacteroidetes, and Firmicutes, respectively, and 9 other NOGs were distributed over more than two phyla (Supplementary Fig. S12a). The high fraction of Actinobacteria may reflect the highest abundance of this phylum in the JP cohort among the 12 countries. Among the eight NOGs assigned to the Bacteroidetes, three NOGs (ENOG4108ZIS, ENOG4108MQB, and ENOG4105WVE), that were detected in approximately 90% of the JP individuals and in about 15% of other populations with the highest ratio of JP/others, were represented by the genes for aquatic plant-derived polysaccharide-degrading enzymes such as β-porphyranase (hydrolase family 16) and β-agarase published previously.23,38 The functional distribution of the 52 NOGs revealed that 35% were of unknown function and no particular function was enriched (Supplementary Fig. S12b).
Figure 5.
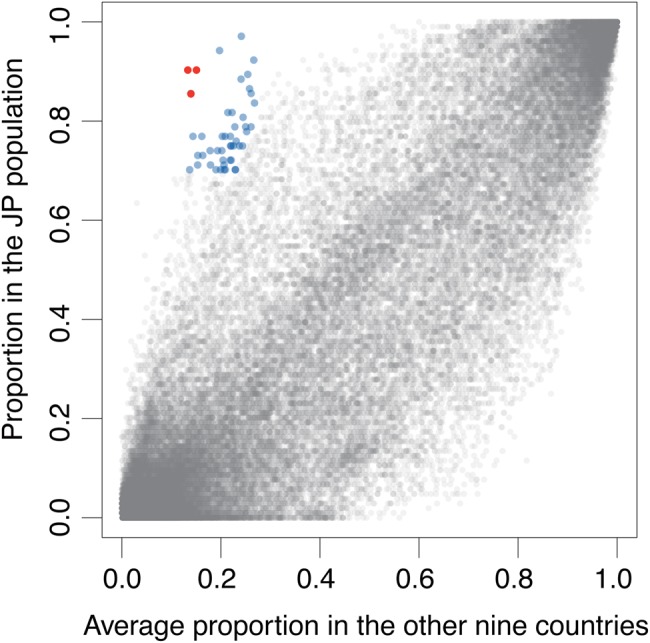
Comparison of the prevalence of NOG gene families between the JP and the other nine populations. The frequency of NOGs in the JP individuals plotted against those in the other nine countries. Each circle represents a NOG. The vertical axis represents the frequency of NOGs detected in the JP individuals. The horizontal axis represents the average frequency of NOGs detected in the individuals of the nine countries. Fifty-two NOGs significantly highly prevalent in the JP cohort compared with the others (the JP >0.7, the others <0.3, and JP/others ≥3) are coloured with blue. Three NOGs (ENOG4108MQB, ENOG4108ZIS, and ENOG4105WVE) were depicted in red.
4. Discussion
In this study, we analysed the metagenomic data from 861 healthy individuals from the 12 countries. The comparative analysis of the high-quality metagenomic datasets revealed a significant population-level diversity in the human gut microbiome across the 12 countries. The accuracy and reliability of this finding are supported by the use of a larger dataset including more populations than those analysed in the previous studies and by the statistical assessment indicating no significant effects of differences in experimental protocols, BMI, and age on the observed results. Thus, we provided the evidence for great variations in structure and function of human gut microbiomes of healthy adults at the population level.
The present study also revealed various features specific to the JPGM. The JPGM showed the highest abundance of the phylum Actinobacteria among the microbiomes of the 12 countries, mainly because of the highest abundance of the genus Bifidobacterium. The high abundance of Bifidobacterium has been also observed in the gut microbiome of Japanese children based on the 16S rRNA gene analysis,12 indicating it is highly prevalent throughout the Japanese population. Bifidobacterium is thought to be a beneficial microbe having more glycoside hydrolases for degrading starch than other intestinal microbes.39,40 Therefore, the high abundance of Bifidobacterium can be considered to be the consequence of the intake of various saccharides in traditional and unique Japanese foods. However, at present, it is unknown exactly which foods or nutrients unique to Japanese contribute to the high abundance of Bifidobacterium.
Additionally, the JPGM was characterized by various unique functional features. For example, the high carbohydrate metabolism capacity in the JPGM may result in the production of high levels of short chain fatty acids and hydrogen as end products, both of which seems to be clinically beneficial.41,42 Concurrently, we found a depletion of deleterious functions such as cell motility, and replication and repair, suggesting a low abundance of the flagellated microbes leading to reduced pro-inflammatory responses by host cells and lowered DNA damage to be repaired in the gut of the Japanese. Together, such a gut ecosystem of benefit functionally might be globally associated with the highest average life span of Japanese in the world.
A remarkable depletion of the archaeon M. smithii was also characteristic of the JPGM, resulting in an overall depletion of genes for methanogenesis. In contrast, genes for acetogenesis, which are exclusively encoded by anaerobic acetogens such as the major species Blautia,43 were enriched in the JPGM as compared with the other gut microbiomes. Both methanogenesis and acetogenesis are considered to be critical pathways for hydrogen consumption in the gut because these pathways are tightly linked with anaerobic fermentation of carbohydrates producing hydrogen.44 Our findings suggest that acetogenesis is the preferable pathway for hydrogen metabolism in the JPGM, while methanogenesis is more actively utilized for hydrogen metabolism in many of the other gut microbiomes. In this context, since the abundance of intestinal M. smithii is positively associated with the level of breath methane,45 our data strongly suggest that M. smithii is the primary factor for ethnic differences in the level of methane in human breath reported previously.46,47
Finally, the similarities and dissimilarities in the microbial composition in the gut microbiome across the 12 countries cannot simply be explained by diet alone, although several papers have suggested its large contribution to the shaping of the gut microbiome.13,14,22 Our findings suggest the existence of factors in addition to, or other than, diet that affect the human gut microbiome, but further studies are required to solve this puzzling difference.
Data availability
All bacterial metagenomic sequences were deposited in DDBJ under accession number PRJDB3601. The information has been summarized in Supplementary Table S12.
Supplementary data
Supplementary data are available at www.dnaresearch.oxfordjournals.org.
Funding
This study was supported in part by a Grant-in-Aid for JSPS Fellows (267812) to S.N., the global COE project of ‘Genome Information Big Bang’ from the Ministry of Education, Culture, Sports, Science, and Technology (MEXT) of Japan to M.H. and K.O., a research project grant from Azabu University to H.M., and a grant from the Core Research for Evolutional Science and Technology (CREST) program of the Japan Science and Technology Agency (JST) to K.O. Funding to pay the Open Access publication charges for this article was provided by an administration grant from the University of Tokyo.
Supplementary Material
Acknowledgements
We thank Drs T. Ito and F. Miura (Kyushu University) for sequencing, Dr M. Umezaki (The University of Tokyo) for support for dietary data analysis, and Dr K. Takanashi, E. Iioka, M. Takagi, C. Shindo, K. Komiya, R. Kurokawa, N. Yamashita, Y. Hattori, E. Omori, H. Kuroyanagi, Y. Takayama, and H. Matsumoto (The University of Tokyo), Y. Noguchi and A. Nakano (Azabu University) for technical support. The super-computing resource was provided by the Human Genome Center (The University of Tokyo).
References
- 1.Turnbaugh P.J., Hamady M., Yatsunenko T. et al. 2009, A core gut microbiome in obese and lean twins, Nature, 457, 480–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Qin J., Li R., Raes J. et al. 2010, A human gut microbial gene catalogue established by metagenomic sequencing, Nature, 464, 59–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Qin J., Li Y., Cai Z. et al. 2012, A metagenome-wide association study of gut microbiota in type 2 diabetes, Nature, 490, 55–60. [DOI] [PubMed] [Google Scholar]
- 4.Human Microbiome Project Consortium 2012, Structure, function and diversity of the healthy human microbiome, Nature, 486, 207–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tyakht A.V., Kostryukova E.S., Popenko A.S. et al. 2013, Human gut microbiota community structures in urban and rural populations in Russia, Nat. Commun., 4, 2469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Karlsson F.H., Tremaroli V., Nookaew I. et al. 2013, Gut metagenome in European women with normal, impaired and diabetic glucose control, Nature, 498, 99–103. [DOI] [PubMed] [Google Scholar]
- 7.Zeller G., Tap J., Voigt A.Y. et al. 2014, Potential of fecal microbiota for early-stage detection of colorectal cancer, Mol. Syst. Biol., 10, 766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Qin N., Yang F., Li A. et al. 2014, Alterations of the human gut microbiome in liver cirrhosis, Nature, 513, 59–64. [DOI] [PubMed] [Google Scholar]
- 9.Feng Q., Liang S., Jia H. et al. 2015, Gut microbiome development along the colorectal adenoma-carcinoma sequence, Nat. Commun., 6, 6528. [DOI] [PubMed] [Google Scholar]
- 10.Backhed F., Roswall J., Peng Y. et al. 2015, Dynamics and stabilization of the human gut microbiome during the first year of life, Cell Host Microbe, 17, 690–703. [DOI] [PubMed] [Google Scholar]
- 11.Zhang X., Zhang D., Jia H. et al. 2015, The oral and gut microbiomes are perturbed in rheumatoid arthritis and partly normalized after treatment, Nat. Med., 21, 895–905. [DOI] [PubMed] [Google Scholar]
- 12.Nakayama J., Watanabe K., Jiang J. et al. 2015, Diversity in gut bacterial community of school-age children in Asia, Sci. Rep., 5, 8397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.De Filippo C., Cavalieri D., Di Paola M. et al. 2010, Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa, Proc. Natl Acad. Sci. USA, 107, 14691–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yatsunenko T., Rey F.E., Manary M.J. et al. 2012, Human gut microbiome viewed across age and geography, Nature, 486, 222–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Obregon-Tito A.J., Tito R.Y., Metcalf J. et al. 2015, Subsistence strategies in traditional societies distinguish gut microbiomes, Nat. Commun., 6, 6505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Martinez I., Stegen J.C., Maldonado-Gomez M.X. et al. 2015, The gut microbiota of rural papua new guineans: composition, diversity patterns, and ecological processes, Cell Rep., 11, 527–38. [DOI] [PubMed] [Google Scholar]
- 17.Goodrich J.K., Waters J.L., Poole A.C. et al. 2014, Human genetics shape the gut microbiome, Cell, 159, 789–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Clemente J.C., Ursell L.K., Parfrey L.W., Knight R.. 2012, The impact of the gut microbiota on human health: an integrative view, Cell, 148, 1258–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li J., Jia H., Cai X. et al. 2014, An integrated catalog of reference genes in the human gut microbiome, Nat. Biotechnol., 32, 834–41. [DOI] [PubMed] [Google Scholar]
- 20.Rehman A., Rauch P., Wang J. et al. 2015, Geographical patterns of the standing and active human gut microbiome in health and IBD, Gut, 65, 238–48. [DOI] [PubMed] [Google Scholar]
- 21.Arumugam M., Raes J., Pelletier E. et al. 2011, Enterotypes of the human gut microbiome, Nature, 473, 174–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu G.D., Chen J., Hoffmann C. et al. 2011, Linking long-term dietary patterns with gut microbial enterotypes, Science, 334, 105–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hehemann J.H., Correc G., Barbeyron T., Helbert W., Czjzek M., Michel G.. 2010, Transfer of carbohydrate-active enzymes from marine bacteria to Japanese gut microbiota, Nature, 464, 908–12. [DOI] [PubMed] [Google Scholar]
- 24.World Health Organization 2015, Global Health Observatory (GHO) data: overweight and obesity. http://www.who.int/gho/ncd/risk_factors/overweight/en/ and Life expectancy http://apps.who.int/gho/data/node.main.688?lang=en/ (January 2016, date last accessed).
- 25.Kurokawa K., Itoh T., Kuwahara T. et al. 2007, Comparative metagenomics revealed commonly enriched gene sets in human gut microbiomes, DNA Res., 14, 169–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim S.W., Suda W., Kim S. et al. 2013, Robustness of gut microbiota of healthy adults in response to probiotic intervention revealed by high-throughput pyrosequencing, DNA Res., 20, 241–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Morita H., Kuwahara T., Oshima K. et al. 2007, An improved DNA isolation method for metagenomic analysis of the microbial flora of the human intestine, Microbes Environ., 22, 214–22. [Google Scholar]
- 28.Gomez-Alvarez V., Teal T.K., Schmidt T.M.. 2009, Systematic artifacts in metagenomes from complex microbial communities, ISME J., 3, 1314–7. [DOI] [PubMed] [Google Scholar]
- 29.Le Chatelier E., Nielsen T., Qin J. et al. 2013, Richness of human gut microbiome correlates with metabolic markers, Nature, 500, 541–6. [DOI] [PubMed] [Google Scholar]
- 30.Clemente J.C., Pehrsson E.C., Blaser M.J. et al. 2015, The microbiome of uncontacted Amerindians, Sci. Adv., 1, e1500183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Atarashi K., Tanoue T., Ando M. et al. 2015, Th17 cell induction by adhesion of microbes to intestinal epithelial cells, Cell, 163, 367–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Langmead B., Salzberg S.L.. 2012, Fast gapped-read alignment with Bowtie 2, Nat. Methods, 9, 357–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Treangen T.J., Sommer D.D., Angly F.E., Koren S., Pop M.. 2011, Next generation sequence assembly with AMOS, Curr. Protoc. Bioinformatics, 30, 11.8.1–11.8.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Noguchi H., Taniguchi T., Itoh T.. 2008, MetaGeneAnnotator: detecting species-specific patterns of ribosomal binding site for precise gene prediction in anonymous prokaryotic and phage genomes, DNA Res., 15, 387–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li W., Godzik A.. 2006, Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences, Bioinformatics, 22, 1658–9. [DOI] [PubMed] [Google Scholar]
- 36.Huerta-Cepas J., Szklarczyk D., Forslund K. et al. 2016, eggNOG 4.5: a hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences, Nucleic Acids Res., 44, D286–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.David L.A., Maurice C.F., Carmody R.N. et al. 2014, Diet rapidly and reproducibly alters the human gut microbiome, Nature, 505, 559–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hehemann J.H., Kelly A.G., Pudlo N.A., Martens E.C., Boraston A.B.. 2012, Bacteria of the human gut microbiome catabolize red seaweed glycans with carbohydrate-active enzyme updates from extrinsic microbes, Proc. Natl Acad. Sci. USA, 109, 19786–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee J.H., O'Sullivan D.J.. 2010, Genomic insights into bifidobacteria, Microbiol. Mol. Biol. Rev., 74, 378–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu S., Ren F., Zhao L. et al. 2015, Starch and starch hydrolysates are favorable carbon sources for bifidobacteria in the human gut, BMC Microbiol., 15, 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tedelind S., Westberg F., Kjerruf M., Vidal A.. 2007, Anti-inflammatory properties of the short-chain fatty acids acetate and propionate: a study with relevance to inflammatory bowel disease, World J. Gastroenterol., 28, 2826–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ohsawa I., Ishikawa M., Takahashi K. et al. 2007, Hydrogen acts as a therapeutic antioxidant by selectively reducing cytotoxic oxygen radicals, Nat. Med., 13, 688–94. [DOI] [PubMed] [Google Scholar]
- 43.Rey F.E., Faith J.J., Bain J. et al. 2010, Dissecting the in vivo metabolic potential of two human gut acetogens, J. Biol. Chem., 285, 22082–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nakamura N., Lin H.C., McSweeney C.S., Mackie R.I., Gaskins H.R.. 2010, Mechanisms of microbial hydrogen disposal in the human colon and implications for health and disease, Annu. Rev. Food Sci. Technol., 1, 363–95. [DOI] [PubMed] [Google Scholar]
- 45.Kim G., Deepinder F., Morales W. et al. 2012, Methanobrevibacter smithii is the predominant methanogen in patients with constipation-predominant IBS and methane on breath, Dig. Dis. Sci., 57, 3213–8. [DOI] [PubMed] [Google Scholar]
- 46.Morii H., Oda K., Suenaga Y., Nakamura T.. 2003, Low methane concentration in the breath of Japanese, J. UOEH, 25, 397–407 (in Japanese). [DOI] [PubMed] [Google Scholar]
- 47.Levitt M.D., Furne J.K., Kuskowski M., Ruddy J.. 2006, Stability of human methanogenic flora over 35 years and a review of insights obtained from breath methane measurements, Clin. Gastroenterol. Hepatol., 4, 123–9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All bacterial metagenomic sequences were deposited in DDBJ under accession number PRJDB3601. The information has been summarized in Supplementary Table S12.