

Abstract
Zoysia is a warm-season turfgrass, which comprises 11 allotetraploid species (2n = 4x = 40), each possessing different morphological and physiological traits. To characterize the genetic systems of Zoysia plants and to analyse their structural and functional differences in individual species and accessions, we sequenced the genomes of Zoysia species using HiSeq and MiSeq platforms. As a reference sequence of Zoysia species, we generated a high-quality draft sequence of the genome of Z. japonica accession ‘Nagirizaki’ (334 Mb) in which 59,271 protein-coding genes were predicted. In parallel, draft genome sequences of Z. matrella ‘Wakaba’ and Z. pacifica ‘Zanpa’ were also generated for comparative analyses. To investigate the genetic diversity among the Zoysia species, genome sequence reads of three additional accessions, Z. japonica ‘Kyoto’, Z. japonica ‘Miyagi’ and Z. matrella ‘Chiba Fair Green’, were accumulated, and aligned against the reference genome of ‘Nagirizaki’ along with those from ‘Wakaba’ and ‘Zanpa’. As a result, we detected 7,424,163 single-nucleotide polymorphisms and 852,488 short indels among these species. The information obtained in this study will be valuable for basic studies on zoysiagrass evolution and genetics as well as for the breeding of zoysiagrasses, and is made available in the ‘Zoysia Genome Database’ at http://zoysia.kazusa.or.jp.
Keywords: Zoysia, turfgrass, genome sequencing, gene prediction, comparative analysis
1. Introduction
Members of the genus Zoysia are some of the most cold- and salt-tolerant plants among warm-season turfgrasses.1,2 Commonly known as zoysiagrasses, Zoysia species offer good turf qualities, including hardiness, salt tolerance, resistance to drought, shade, and heat, and they are preferred for their aesthetic characteristics. Zoysia comprises 11 species, and all of which are allotetraploids (2n = 4x = 40). Originating from East Asia (Japan, Korea, China), these out-crossing and mat-forming perennial grasses have spread throughout the Pacific Rim and have been used for centuries as cover plants for burial mounds according to the customs in the Orient. Five species such as Z. japonica Steudel, Z. matrella (L.) Merrill, Z. pacifica (Goudswaard) M. Hotta & Kuroki [syn. Z. matrella (L.) Merr. var. pacifica Goudswaard], Z. sinica Hance, and Z. macrostachya Franchet & Savatier have been found in Japan, and they exhibit wide morphological and ecological differences.3
Zoysiagrasses have received less attention from the research community than other Poaceae family members. However, several research groups have conducted genetic studies on zoysiagrasses in an attempt to evaluate their genetic variation and clarify their phylogenetic relationships. Studies comparing domestic zoysiagrass accessions and wild species have revealed high rates of introgression and hybridization, particularly among Z. japonica, Z. matrella, and Z. pacifica.4,5 High-density genetic linkage maps have been constructed in many crop species, to promote map-based gene cloning and marker-enhanced breeding. Such maps have been produced for zoysiagrasses based on restriction and amplified fragment length polymorphisms, simple sequence repeats (SSRs), and combinations of these markers, and the maps have been used in quantitative trait loci analyses.4,6,7 Moreover, sequence-tagged high-density genetic maps of Z. japonica were constructed and compared with those of Oryza sativa and Sorghum bicolor, revealing a highly conserved collinearity in the gene order of these genomes.8 In parallel with the construction of genetic maps, transcriptome analyses have been used to identify abiotic stress-related genes9,10 and genes that control tissue-specific anthocyanin pigmentation in Z. japonica.11 A complete sequence of the chloroplast genome of Z. matrella L. Merr has also been published.12
Next-generation sequencing technologies have enabled the acquisition of large sequence datasets from plant nuclear and organelle genomes. These genetic resources offer significant impacts on plant research in all disciplines, including systematics, genetics, co-evolution, conservation genetics, and breeding.13 Whole-genome sequencing along with precise data analysis by means of bioinformatics has allowed the discovery of new genes and regulatory sequences, and can integrate variant types including indels, structural variants and copy number variations.14 Genome and gene sequences have been accumulated for many model and crop plant species. However, much more work is needed to clarify relationships and variations across the Poaceae family, particularly in the subfamily Chloridoideae, which consists of 1,601 species in 131 genera that possess the Kranz or C4 anatomy and chloridoid bicellular microhairs.15 For example, there were only 1,004 sequences from Zoysia spp. found in GenBank at the time of this writing. Here, we report on the generation and comparison of high-quality draft sequences of zoysiagrass genomes derived from native accessions of Z. japonica, Z. matrella and Z. pacifica in Japan. Furthermore, we compared the zoysiagrass genomes with those of related species: O. sativa, S. bicolor, and Brachypodium distachyon.
2. Materials and methods
2.1. Plant materials
Six accessions of three species (Z. japonica, Z. matrella and Z. pacifica) were used in this study. These accessions are maintained at the Kibana Agricultural Science Station, Field Science Center, University of Miyazaki, Japan. An accession of Z. japonica, ‘Nagirizaki’, was collected from Tsushima, Nagasaki. We obtained stolons from ‘Nagirizaki’ plants and cut them into 2–3 cm segments with nodes. The segments were surface sterilized using a standard protocol. The shoot apices were excised from the nodes and cultured on half-strength hormone-free Murashige and Skoog medium16 solidified with 0.7% agar in test tubes for regeneration. The regenerated leaves were used for whole-genome shotgun sequencing. The other accessions were as follows: Z. japonica ‘Miyagi’ from Ishinomaki, Miyagi; Z. japonica ‘Kyoto’ from Kyoto, Kyoto; and Z. matrella ‘Wakaba’, Z. matrella ‘Chiba Fair Green’, and Z. pacifica ‘Zanpa’ from Yomitan-son, Okinawa. Shoots of these accessions were briefly sterilized in 2% (v/v) sodium hypochlorite solution followed by three rinses with sterile water, and transplanted into pots (diameter: 6.0 cm, depth: 5.5 cm) filled with sterilized vermiculite and covered with plastic containers. All accessions were incubated at 31°C under 16 h light (30 µmol m−2 s−1) and 8 h dark condition, and their leaves were used for comparative studies.
2.2. Flow cytometry
Flow cytometry was conducted to estimate the genome sizes of the six Zoysia accessions according to Ishigaki et al.17 Sorghum bicolor ‘Tx623’ was used as the internal standard with a DNA content of 1.67 pg 2C−1.18 The fluorescent intensity of each nuclear suspension was measured using an EPICS XL equipped with a 488 nm argon laser and a long path filter (Beckman Coulter, Inc., CA, USA). The analysis was replicated three times for each sample.
2.3. Shotgun sequencing of zoysiagrass genomes
Whole-genome shotgun sequencing of the Zoysia species was performed using both the HiSeq 2000 and MiSeq platforms (Illumina Inc., CA, USA). Genomic DNA extracted from leaves was used for library construction according to standard protocols. The paired-end (PE) libraries were constructed using the manufacture's protocol (Illumina), and the mate-pair (MP) libraries were constructed using GS Titanium Library Paired End Adaptors (Roche Diagnostics, Basel, Switzerland) as described previously.19 The libraries sequenced and platforms used for each accession are summarized in Supplementary Table S1. For sequencing the genomes of Z. japonica ‘Nagirizaki’, Z. matrella ‘Wakaba’, and Z. pacifica ‘Zanpa’, PE reads with insert sizes of 700 bp were sequenced using the MiSeq platform. For ‘Nagirizaki’, MP reads with insert sizes of 5, 10, and 15 kb were sequenced using the HiSeq 2000 platform. For ‘Wakaba’ and ‘Zanpa’, MP reads with insert sizes of 3 kb were sequenced using HiSeq 2000. For the other three accessions, such as Z. japonica ‘Kyoto’, Z. japonica ‘Miyagi’, and Z. matrella ‘Chiba Fair Green’, PE reads with insert sizes of 300 bp were sequenced using the HiSeq 2000 platform. The k-mer frequency analysis and estimation of genome sizes were performed using SOAPec, ver. 2.0.1,20 using the PE reads of each accession.
2.4. Assembly of genome sequences and RNA-Seq analysis
Genome sequence assembly was performed for Z. japonica ‘Nagirizaki’, Z. matrella ‘Wakaba’, and Z. pacifica ‘Zanpa’ using the MiSeq PE and HiSeq MP reads. The adaptor sequences in the PE and MP reads were trimmed, and polymerase chain reaction (PCR) duplicates in the MP reads were excluded. For ‘Nagirizaki’, the MiSeq and HiSeq 2000 reads were assembled into contigs using Platanus, v1.2.1.21 The scaffolding in Platanus was conducted under strict conditions with options –l 5 and –s 72. For ‘Wakaba’ and ‘Zanpa’, contigs were assembled from the MiSeq PE reads using DISCOVAR de novo ver. 51598 (http://www.broadinstitute.org/software/discovar/blog/) and FERMI, v1.1.22 After the comparison of the status of resulted contigs by two assemblers, the contig sets generated by DISCOVAR de novo were further assembled with the PE and MP reads to generate scaffolds using Opera, v2.0.23 Gaps in the ‘Wakaba’ and ‘Zanpa’ scaffolds were filled by a sequential treatment with the two contig sets (the DISCOVAR de novo-contig set and the FERMI-contig set) using GMcloser.24
For RNA-Seq analysis, PE libraries were prepared using 1 µg of total RNA extracted from young leaves and roots of ‘Nagirizaki’ using the TruSeq Stranded Total RNA with Ribo-Zero Plant kit (Illumina). The constructed libraries were sequenced using the HiSeq 2000 platform. The reads were assembled using Trinity, ver. 2.0,25 with the default parameters except that the min_kmer_cov option was 3, and the resulting assembled contigs with <500 bases were filtered out.
2.5. Validation of assembled sequences
To confirm the genome coverage and the accuracy of the assembled genome sequences, the core eukaryotic genes were mapped using CEGMA, v2.5.26 The quality of the assembled genome sequences was also confirmed using published DNA markers developed by Wang et al.8 The marker sequences of Z. japonica ‘El Toro’ linkage groups were mapped onto the genome sequences of ‘Nagirizaki’ using BLASTN27 with an E-value cut-off of 1E−4 considering the length (95 bp) of the marker sequences.
2.6. Detection of repetitive sequences
We detected known and novel repetitive sequences including transposable elements using the following two steps: First, the repetitive sequences were detected by RepeatScout28 with default parameters. Then, repetitive sequences were detected and masked by RepeatMasker (http://www.repeatmasker.org) against known repetitive sequences in Repbase (http://www.girinst.org/repbase/) and novel repetitive sequences found in the first step. The repetitive sequences were classified according to the results of RepeatMasker. SSR motifs were extracted from the ‘Nagirizaki’, ‘Wakaba’, and ‘Zanpa’ genome sequences and from those of O. sativa (IRGSP-1.0), S. bicolor (Sbicolor_255_v2.0), and B. distachyon (Bdistachyon_283_v2.0) using SciRoKo29 with the MISA mode. The minimum numbers of repeats for SSR motifs were 7, 5, 4, 4 and 4 for di-, tri-, tetra-, penta- and hexa-nucleotides, respectively.
2.7. Gene assignment and annotation
Genes for tRNAs were predicted using tRNAscan-SE, v1.23,30 and rRNA genes were predicted using hmmscan in HMMER, ver. 3.031 with a Rfam 11.0 (http://rfam.xfam.org) database. Genomic protein-coding genes of ‘Nagirizaki’, ‘Wakaba’, and ‘Zanpa’ were predicted using a MAKER pipeline32 as follows: Repetitive sequences were detected and masked using a combination of RepeatModeller (http://www.repeatmasker.org/RepeatModeler.html), RECON,33 RepeatMasker (http://www.repeatmasker.org) and RepeatRunner (https://github.com/Yandell-Lab/RepeatRunner). The training set for ‘Nagirizaki’ was built using BRAKER1 (http://exon.gatech.edu/genemark/braker1.html) with the RNA-Seq data. Prediction of the protein-coding genes was performed using Augustus 3.0.334 with the training set for ‘Nagirizaki’, and snap35 using the hidden Markov model of O. sativa. In addition, the RNA-Seq reads for ‘Nagirizaki’ were mapped onto the masked genome sequences by TopHat,36 and coding regions were detected by Cufflinks37 to generate a gff file indicating transcript units. Also, consensus transcript sequences were constructed by assembly of the RNA-Seq reads with Trinity, and were mapped onto the genome sequences by a BLASTN search and exonerate.38 Finally, the gene models supported by the above lines of evidence were selected using the MAKER pipeline. The predicted genes of ‘Nagirizaki’, ‘Wakaba’, and ‘Zanpa’ were prefixed with ‘Zjn’, ‘Zmw’, and ‘Zpz’, respectively, followed by a scaffold number, sequence version of each scaffold, gene number, and sequence version of each gene, which were connected to each other by dot characters. In addition, information about the gene prediction method was attached with the following signatures: am, assigned by Augustus; sm, assigned by snap; cf, assigned by Cufflinks; es, assigned by exonerate; and cf-es, assigned by Cufflinks and exonerate. The prediction accuracy was attached using the following signatures: mkhc, assigned with high confidence by MAKER (supported by multiple prediction methods); mk, assigned as non-overlapping genes by MAKER; and br, assigned by BRAKER1.
For assumption of the functions of the predicted protein-coding genes, database searches were carried out against the NCBI's non-redundant (nr) protein database (http://www.ncbi.nlm.nih.gov) and the protein sequences of Arabidopsis thaliana (TAIR10) (https://www.arabidopsis.org), O. sativa (RAP-DB) (http://rapdb.dna.affrc.go.jp), S. bicolor, B. distachyon, Setaria italica, Triticum aestivum, Zea mays (Phytozome, v10) (http://phytozome.jgi.doe.gov), and Hordeum vulgare (Ensembl Plants) (http://plants.ensembl.org). The searches were performed using the BLAST program with an E-value cut-off of 1E−10. The translated protein sequences were classified into NCBI's euKaryotic clusters of Orthologous Groups (KOG)39 using BLASTP searches against the KOG database with an E-value cut-off of 1E−10. A KOG id was assigned to each gene based on the top hit of the BLASTP search. Domain searches were performed using InterProScan40 against the InterPro (http://www.ebi.ac.uk/interpro/) database with an E-value cut-off of 1.0. The genes were classified into GO slim categories using the map2slim program41 based on the domain search results. The genes of ‘Nagirizaki’, S. bicolor (Sbicolor_255_v2.0; Phytozome v10), O. sativa (IRGSP-1.0) and B. distachyon (Bdistachyon_283_v2.0; Phytozome v10) were then mapped onto the KEGG (Kyoto Encyclopedia of Genes and Genomes) reference metabolic pathways42 using BLASTP searches against KEGG's KO database with an E-value cut-off of 1E-10, identity ≥25% and length coverage ≥50%. The genes were classified using CD-hit43 with parameters –c = 0.4, -G = 0 and –aS = 0.5.
2.8. Comparison of genes among Z. japonica and three other Poaceae species
To identify common genes among Z. japonica ‘Nagirizaki’ and the three Poaceae species, O. sativa, S. bicolor and B. distachyon, the translated protein sequences were compared by a clustering approach using CD-hit. To compare the gene order, the translated protein sequences on the pseudomolecules of Z. japonica ‘Nagirizaki’ were searched against O. sativa, S. bicolor and B. distachyon using BLAST searches with an E-value cut-off of 1E-100. Synteny plots were made using a perl script and the gnuplot program (http://www.gnuplot.info).
2.9. Polymorphism analysis
The PE reads of each Zoysia accession were aligned onto the genome sequence of ‘Nagirizaki’ using Bowtie 2, ver. 2.2.544 with default parameters, except for the use of an end-to-end mode. SNPs and indel candidates were detected using bam files generated using SAMtools, ver. 0.1.19, and BCFtools, ver. 0.1.19.45 After mapping, the SNP candidates on N or n nucleotides in the reference sequence were excluded. The false-positive SNPs were excluded according to the DP4 field in the VCF format.45
3. Results and discussion
3.1. Sequencing of the Zoysia genomes
3.1.1. Shotgun sequencing and assembly of the Zoysia genomes
A high-quality draft genome sequence of Z. japonica ‘Nagirizaki’ was generated for use, as a reference sequence. PE read data (49.3 Gb) were accumulated using the MiSeq platform. In addition, mate-pair (MP) reads were collected from three MP libraries with insert sizes of 5, 10 and 15 kb using the HiSeq 2000 platform. For Z. matrella ‘Wakaba’ and Z. pacifica ‘Zanpa’, ∼17 Gb of data from PE reads and a single type of MP library (with 3 kb inserts) were accumulated for comparative analyses. The sequence data for each Zoysia accession are summarized in Supplementary Table S1.
The kmer analysis for sequenced reads suggested that all the sequenced genomes had a high proportion of heterozygous alleles (Supplementary Figure S1). We used two computer programs for the primary assemblies: Platanus, which is suitable for assembly of highly heterozygous genomes, and the DISCOVAR de novo assembler, which is capable of generating contigs with high efficiency using PE reads with 250–300 bases of read length. The scaffolds with the highest N50 values were obtained in Platanus for Z. japonica ‘Nagirizaki’ (Supplementary Table S2). After gap-closing by GMcloser, 334 Mb of scaffolds with an N50 length of 2.4 Mb were obtained (Table 1). Ninety percent of the scaffolds (301 Mb) were included in the 167 longest scaffolds. According to the flow cytometry analysis, the genome size of Z. japonica ‘Nagirizaki’ is ∼390 Mb (0.80 pg/2C). This is 15% larger than the estimation by k-mer analysis (340 Mb) (Supplementary Table S3), presumably due to the effect of conserved regions on homeologous chromosome pairs in the tetraploid genome. Thus the genome assembly of Z. japonica ‘Nagirizaki’, designated as ‘Zjn_r1.1’, covered 86% of the whole genome as estimated by flow cytometry.
Table 1.
Assembly statistics of draft genome sequences of the Zoysia species
|
Z. japonica ‘Nagirizaki’ (ZJN_r1.1) |
Z. matrella ‘Wakaba’ (ZMW_r1.0) |
Z. pacifica ‘Zanpa’ (ZPZ_r1.0) |
Z. japonica ‘Nagirizaki’ (ZJN_r1.1_pseudomol) |
|
|---|---|---|---|---|
| Total | ||||
| Number of sequences | 11,786 | 13,609 | 11,428 | 20 |
| Total length (b) | 334,384,427 | 563,438,595 | 397,009,957 | 273,735,632 |
| Ave. length (b) | 28,371 | 41,402 | 34,740 | 13,686,782 |
| Max. length (b) | 8,501,895 | 1,041,506 | 1,506,652 | 21,132,631 |
| Min. length (b) | 500 | 500 | 500 | 7,690,864 |
| N50 length (b) | 2,370,062 | 108,897 | 111,449 | 17,914,192 |
| A | 90,476,466 | 156,060,073 | 110,343,373 | 73,600,141 |
| T | 90,391,770 | 155,985,841 | 110,358,466 | 73,593,144 |
| G | 71,275,668 | 121,996,426 | 85,817,354 | 58,170,503 |
| C | 71,324,485 | 122,016,838 | 85,854,342 | 58,222,241 |
| N | 10,916,038 | 7,379,417 | 4,636,422 | 10,149,603 |
| Total (ATGC) | 323,468,389 | 556,059,178 | 392,373,535 | 263,586,029 |
| G+C% (GC/ATGC) | 44.1 | 43.9 | 43.8 | 44.2 |
| ≥1 kb | ||||
| Number of sequences | 6,554 | 11,227 | 8,730 | – |
| Total length (b) | 330,746,168 | 561,855,676 | 395,175,704 | – |
| Ave. length (b) | 50,465 | 50,045 | 45,266 | – |
| ≥2 kb | ||||
| Number of sequences | 2,069 | 10,304 | 7,340 | – |
| Total length (b) | 324,295,136 | 560,567,885 | 393,244,071 | – |
| Ave. length (b) | 156,740 | 54,403 | 53,575 | – |
| ≥3 kb | ||||
| Number of sequences | 1,149 | 9,902 | 6,930 | – |
| Total length (b) | 322,069,938 | 559,570,268 | 392,259,279 | – |
| Ave. length (b) | 280,305 | 56,511 | 56,603 | – |
| ≥4 kb | ||||
| Number of sequences | 777 | 9,524 | 6,552 | – |
| Total length (b) | 320,784,379 | 558,247,617 | 390,938,668 | – |
| Ave. length (b) | 412,850 | 58,615 | 59,667 | – |
| ≥5 kb | ||||
| Number of sequences | 684 | 9,188 | 6,242 | 20 |
| Total length (b) | 320,368,645 | 556,750,068 | 389,557,068 | 273,735,632 |
| Ave. length (b) | 468,375 | 60,595 | 62,409 | 13,686,782 |
Contigs or scaffolds shorter than 499 bp were excluded.
For Z. matrella ‘Wakaba’ and Z. pacifica ‘Zanpa’, the scaffolds were built by Opera using contigs generated by DISCOVAR de novo because DISCOVAR de novo constructed contigs with higher N50 values than the other assemblers. This may be because only a single MP library was generated for these species. The final assemblies for ‘Wakaba’ and ‘Zanpa’ were generated by gap-closing with GMcloser and resulted in 563 Mb (N50 length, 109 kb) and 397 Mb (N50 length, 111 kb), respectively (Table 1). As with ‘Nagirizaki’, the genome size of ‘Zanpa’ estimated by flow cytometry (370 Mb; 0.76 pg/2C) was larger (by 23%) than the size estimated by k-mer analysis (302 Mb) (Supplementary Table S3). On the other hand, the flow cytometry estimate for ‘Wakaba’ was ∼380 Mb (0.79 pg/2C), which is 11% smaller than that estimated by k-mer analysis (423 Mb) (Supplementary Table S3). This difference was presumably due to the effect of heterozygosity exceeding the effect of tetraploidy. The genome assemblies of ‘Wakaba’ and ‘Zanpa’ were longer than those estimated by flow cytometry, suggesting that they contained redundant scaffolds corresponding to heterozygous alleles. This is likely because the assemblies of these two accessions were obtained with the DISCOVAR de novo and Opera assemblers, which are less able than Platanus to merge homologous sequences corresponding to the same allelic regions. Indeed, the total sizes of the scaffold assemblies of ‘Wakaba’ and ‘Zanpa’ generated by Platanus were 30 and 22% smaller, respectively, than those obtained by DISCOVAR de novo (Supplementary Table S2). Considering the purpose of sequencing for comparative genome analyses, we finally adopted the DISCOVAR de novo assemblies, which presumably contained redundant scaffolds corresponding to heterozygous alleles, for these two accessions. The Z. matrella ‘Wakaba’ and Z. pacifica ‘Zanpa’ genomes were designated as ‘Zmw_r1.0’ and ‘Zpz_r1.0’, respectively.
3.1.2. Authenticity of the assembled sequences
The CEGMA analyses of the assembled genome sequences showed that they completely cover 91.5–94.8% (the highest is for Z. japonica ‘Nagirizaki’), and partially cover over 97%, of the 248 conserved core eukaryotic genes (Supplementary Table S4). These high coverage data strongly indicate that the constructed genome sequences of the three Zoysia species were of high quality and sufficient for subsequent accurate gene modelling and annotation.
The authenticity of the assembled genome sequences was further evaluated by comparing the genome sequence of ‘Nagirizaki’ and the published Z. japonica linkage map of RAD markers.8 We compared 1,230 marker-associated sequences on the Z. japonica ‘El Toro’ linkage map developed by Wang et al.8 with the scaffold sequences of Zjn_r1.1 and found that 1,224 (99.5%) of the sequences were successfully located on Zjn_r1.1. The 135 scaffolds in Zjn_r1.1 were each anchored by three or more markers in the ‘El Toro’ linkage groups and the order of most of the markers was in good agreement with their physical positions on the scaffolds. This result indicates a high degree of authenticity of the Zjn_r1.1 genome assembly.
While comparing the ‘Nagirizaki’ scaffold sequences with the linkage map, we found that 15 scaffolds were anchored to two or three different locations on the linkage map, probably due to mis-scaffolding caused by the presence of repetitive sequences. We separated these scaffolds at the repetitive sequences at the boundaries of the anchored regions, and the latter halves of these scaffolds were designated using numbers starting with ‘2’ or ‘3’ followed by the original four-digit scaffold number (Supplementary Table S5). For example, the scaffold Zjn_sc00037 was anchored by RAD markers at three different locations on the linkage map, and thus it was separated into three scaffolds: Zjn_sc00037, Zjn_sc20037, and Zjn_sc30037. After these revisions, we constructed pseudomolecules of Z. japonica ‘Nagirizaki’ that we designated as Zjn_chr01 – Zjn_chr20 based on the ‘El Toro’ linkage group numbers. These pseudomolecules covered 82% of the Zjn_r1.1 sequence. The pseudomolecules of homeologous chromosome pairs had similar lengths, ranging from the shortest pair: Zjn_chr15 (7.7 Mb) and Zjn_chr16 (8.1 Mb) to the longest pair: Zjn_chr07 (20.3 Mb) and Zjn_chr08 (21.1 Mb). These results confirmed the even distribution of the RAD markers and indicated that the scaffolds in Zjn_r1.1 are sufficient to cover most of the Z. japonica genome (Supplementary Table S6). Therefore, the pseudomolecules of ‘Nagirizaki’ were subjected to subsequent analyses as the reference sequence of Z. japonica, together with the draft genome sequences.
3.2. Characteristic features of the Zoysia genome
3.2.1. Repetitive sequences
Known and novel repetitive sequences, including transposable elements, were identified using RepeatMasker and RepeatScout. A classification of the repetitive elements and their content in each genome is summarized in Supplementary Table S7. The genome sequences of ‘Nagirizaki’ (Zjn_r1.1), ‘Wakaba’ (Zmw_r1.0), and ‘Zanpa’ (Zpz_r1.0) had total repeat contents of 40.9, 45.9 and 44.7%, respectively. The total repeat content in the pseudomolecules of the Z. japonica ‘Nagirizaki’ genome (35.6%) was lower than those of the other draft sequences because the unanchored scaffolds, which were likely to contain more repetitive sequences, were not included in the pseudomolecules. Based on the methods for detection of repetitive sequences used in this study, the contents of repetitive sequences in the Zoysia species were similar to that of B. distachyon (37.7% of total genome size, 272 Mb in Bdistachyon_283_v2.0), and smaller than those of O. sativa (48.7% of 373 Mb in IRGSP-1.0) and S. bicolor (64.8% of 727 Mb in Sbicolor_255_v2.0).
The SSRs in the draft genome sequences were detected using SciRoKo software, and their contents in each draft genome are summarized in Supplementary Table S8. The total numbers of the SSR motifs in the Zoysia species were 24,383 in Z. japonica ‘Nagirizaki’ (7.07 SSRs per 100 kb), 41,883 in Z. matrella ‘Wakaba’ (7.20 SSRs per 100 kb), and 29,271 in Z. pacifica ‘Zanpa’ (7.15 SSRs per 100 kb). These frequencies are similar to the frequency for B. distachyon: 7.80 SSRs per 100 kb (total number: 21,818). The frequencies of di-nucleotides in the three Zoysia species were nearly the same and were close to that of S. bicolor. On the other hand, the frequencies of tri-nucleotides in the three Zoysia species were fewer than those in O. sativa, S. bicolor and B. distachyon.
3.2.2. RNA-coding genes
Genes for tRNAs were predicted using tRNAscan-SE and rRNA genes were predicted using HMMER3 searches against the Rfam database. Totals of 514, 827 and 597 tRNA genes (Supplementary Table S9), and 59, 47 and 52 rRNA genes were assigned to the draft genome sequences of ‘Nagirizaki’, ‘Wakaba’ and ‘Zanpa’, respectively. The numbers of predicted non-coding RNA genes were roughly proportional to the lengths of the assembled genome sequences of the three species. Although the genome size of Z. japonica ‘Nagirizaki’ (390 Mb) is similar to that of O. sativa (373 Mb), the number of predicted tRNA genes in ‘Nagirizaki’ (514) is ∼73% of that in O. sativa (705).
3.3. Characteristic features of the protein-coding genes
3.3.1. Prediction of protein-coding genes and annotation
The protein-coding genes were predicted for the three Zoysia species using multiple computer programs as described in the Materials and methods section. The number of coding sequences predicted in ‘Nagirizaki’ (Zjn_r1.1) was 59,271 with an N50 length of 1,119 bp (Table 2). Of these, 49,103 sequences were assigned to the pseudomolecules of ‘Nagirizaki’. When the predicted genes on the homeologous chromosome pairs were compared in detail, more than half of the genes seemed to be lost from one of the pairs (Supplementary Table S10). This explained why the number of predicted genes in tetraploid Z. japonica is less than twice those in the diploid Poaceae species O. sativa (35,626 genes) and S. bicolor (33,032 genes). On the other hand, the total numbers of protein-coding genes predicted on the draft genome sequences of ‘Wakaba’ and ‘Zanpa’ were 95,079 and 65,252, with N50 lengths of 1,065 and 1,062 bp, respectively (Table 2). The higher numbers of genes predicted for these two accessions presumably reflects the presence of redundant scaffolds corresponding to heterozygous alleles in Zmw_r1.0 and Zpz_r1.0.
Table 2.
Statistics of genes predicted in the draft genome sequences of Zoysia species
|
Z. japonica ‘Nagirizaki’ |
Z. matrella ‘Wakaba’ |
Z. pacifica ‘Zanpa’ |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Total | mkhc | mk | br | Total | mkhc | mk | Total | mkhc | mk | |
| Total | ||||||||||
| Number of sequences | 59,271 | 16,205 | 37,605 | 5,461 | 95,079 | 24,398 | 70,681 | 65,252 | 16,455 | 48,797 |
| Total length (b) | 49,483,110 | 16,398,294 | 27,579,822 | 5,504,994 | 76,439,178 | 23,712,921 | 52,726,257 | 52,368,507 | 16,358,223 | 36,010,284 |
| Ave. length (b) | 835 | 1,012 | 733 | 1,008 | 804 | 972 | 746 | 803 | 994 | 738 |
| Max. length (b) | 11,838 | 11,838 | 6,666 | 5,439 | 13,647 | 13,647 | 6,240 | 14,421 | 14,421 | 6,735 |
| Min. length (b) | 108 | 150 | 150 | 108 | 150 | 153 | 150 | 150 | 150 | 150 |
| N50 length (b) | 1,119 | 1,254 | 987 | 1,275 | 1,065 | 1,179 | 999 | 1,062 | 1,206 | 978 |
| G+C% | 59.3 | 53.2 | 63.2 | 58.3 | 59.6 | 52.6 | 62.7 | 59.6 | 52.7 | 62.8 |
| ≥100 b | ||||||||||
| Number of sequences | 59,271 | 16,205 | 37,605 | 5,461 | 95,079 | 24,398 | 70,681 | 65,252 | 16,455 | 48,797 |
| Total length (b) | 49,483,110 | 16,398,294 | 27,579,822 | 5,504,994 | 76,439,178 | 23,712,921 | 52,726,257 | 52,368,507 | 16,358,223 | 36,010,284 |
| Ave. length (b) | 835 | 1,012 | 733 | 1,008 | 804 | 972 | 746 | 803 | 994 | 738 |
| ≥500 b | ||||||||||
| Number of sequences | 36,762 | 12,446 | 20,166 | 4,150 | 58,044 | 18,851 | 39,193 | 39,673 | 12,864 | 26,809 |
| Total length (b) | 41,915,830 | 15,083,250 | 21,825,114 | 5,007,466 | 63,962,067 | 21,739,386 | 42,222,681 | 43,778,979 | 15,084,588 | 28,694,391 |
| Ave. length (b) | 1,140 | 1,212 | 1,082 | 1,207 | 1,102 | 1,153 | 1,077 | 1,103 | 1,173 | 1,070 |
| ≥1 kb | ||||||||||
| Number of sequences | 17,091 | 6,436 | 8,605 | 2,050 | 25,866 | 9,153 | 16,713 | 17,496 | 6,342 | 11,154 |
| Total length (b) | 27,773,167 | 10,647,366 | 13,635,474 | 3,490,327 | 40,863,282 | 14,576,319 | 26,286,963 | 27,907,539 | 10,272,498 | 17,635,041 |
| Ave. length (b) | 1,625 | 1,654 | 1,585 | 1,703 | 1,580 | 1,593 | 1,573 | 1,595 | 1,620 | 1,581 |
| ≥5 kb | ||||||||||
| Number of sequences | 56 | 41 | 10 | 5 | 53 | 41 | 12 | 42 | 35 | 7 |
| Total length (b) | 342,564 | 262,044 | 54,009 | 26,511 | 350,736 | 283,644 | 67,092 | 277,953 | 239,226 | 38,727 |
| Ave. length (b) | 6,117 | 6,391 | 5,401 | 5,302 | 6,618 | 6,918 | 5,591 | 6,618 | 6,835 | 5,532 |
The predicted protein-coding genes of Z. japonica ‘Nagirizaki’, O. sativa, S. bicolor, and B. distachyon were assigned to the functional categories of NCBI's euKaryotic clusters of Orthologous Groups (KOG). The percentages of proteins in each KOG category are shown in Supplementary Figure S2. The four species showed similar percentages of proteins in each functional category. The predicted genes were then classified into GO slim categories based on the results of the domain searches using InterProScan against the InterPro database. The percentages of genes in the root categories Biological Process (BP), Cellular Components (CC) and Molecular Function (MF) were also similar among the four related species (Supplementary Figure S3).
The metabolic pathways of Z. japonica ‘Nagirizaki’, O. sativa, S. bicolor, and B. distachyon were then compared by mapping the genes of each genome onto the reference map of pathways in the ‘Metabolism’ group of the KEGG database. As a result, most of the enzymes found in Z. japonica ‘Nagirizaki’ were conserved in other three Poaceae species (Supplementary Table S11). However, there were eight enzymes found only in Z. japonica ‘Nagirizaki’, which were classified into seven pathways including ‘Sesquiterpenoid and triterpenoid biosynthesis’, ‘Benzoxazinoid biosynthesis’, and ‘Benzoate degradation’ (Supplementary Table S11). The entire results from the mapping of the metabolic pathways are available on the ‘Zoysia Genome Database’ at http://zoysia.kazusa.or.jp.
3.3.2. Gene characteristics of Zoysia species
Plant adaptation to high saline environments is mediated by complex interactions among salt-stress responsive genes and transcription factors (TFs).46 The NAC family is one of the largest groups of plant-specific TFs involved in plant development and abiotic stress. A member of the NAC family, ANAC102, was reported to play a role in salinity tolerance in Arabidopsis.47 To elucidate the high salt tolerance mechanisms in Zoysia species, we searched for ANAC102-like genes in the three Zoysia genome sequences. Two ANAC102-like genes were identified in each Zoysia genome (Supplementary Figure S4). Among the first group (Zjn_sc00013.1.g08180.1, Zmw_sc04858.1.g00100.1, and Zpz_sc00127.1.g00380.1) we found high degrees of similarity in amino acid sequences and small number of mutations, and each gene was equally represented in the three species. The second group (Zjn_sc00014.1.g08280.1, Zmw_sc02298.1.g00200.1, Zmw_sc02531.1.g00150.1 and Zpz_sc01716.1.g00180.1) was similar to the first group in amino acid sequences but had differences in the mutation sites. However, Z. japonica and Z. pacifica each had a distinctive sequence feature in the corresponding 5′ upstream region, and both features were found in Z. matrella (Supplementary Figure S5). Based on the positions of these ANAC102-like genes on the ‘Nagirizaki’ pseudomolecules, two copies were retained on both homeologous chromosomes (Zjn_chr01 and Zjn_chr02). These differences of ANAC102-like genes may provide some contribution to salt tolerance in the Zoysia species.
The B-box type zinc-finger family of TFs are also key factors in regulatory networks such as those related to salt stress.48 Specifically, the Salt Tolerance/B-Box Zinc-Finger Protein 24 (STO/BBX24) of A. thaliana was found to increase salt tolerance in yeast (Saccharomyces cerevisiae),49 and the overexpression of STO/BBX24 enhances the root growth of Arabidopsis seedlings in a high salt medium.50 To investigate the contribution of STO/BBX24 to salt tolerance in Zoysia species, we compared the structures of the STO/BBX24 genes in the three Zoysia species. We found two structurally distinguishable sequences in corresponding regions of the Z. japonica and Z. pacifica genomes, while both alleles were found in Z. matrella (Supplementary Figure S6). Moreover, mutations were found in the 5′ upstream region of each STO/BBX24 gene (Supplementary Figure S7). Zoysia matrella tends to possess higher level of salt tolerance than Z. japonica and Z. pacifica.2,51 The polymorphisms that we found in the ANAC102 and STO/BBX24 genes may imply that these genes contribute to the variation in salt tolerance levels among these three Zoysia species.
Another zinc-binding B-box domain protein that is involved in plant responses to salinity and dehydration is a leucine zipper (bZIP) TF.52,53 Sporobolus virginicus is another halophyte turfgrass in the tribe Zoysiae, along with the Zoysia species. A comprehensive analysis of transcripts involved in salt tolerance in, S. virginicus revealed five bZIP genes with root-specific salt-responsive expression.54 Only two of these bZIP genes (PK07387.1 and evm_27.model.AmTr_v1.0_scaffold00081.79 in PlantTFDB3.055) were conserved in the Zoysia genomes. This suggests that Zoysia species may have different mechanisms for salt tolerance than those in S. virginicus.
Anthocyanins are a class of phenolic compounds responsible for the red to purple pigmentation in plants. They are involved in plant defenses and stress avoidance due to their antioxidant properties.56 Ahn et al.11 reported that zoysiagrasses exhibit red to purple pigments in their spike and stolon tissues. A transcriptome analysis of Z. japonica accessions with either green or purple tissues was used to identify two genes related to anthocyanin biosynthesis: anthocyanidin synthase (ANS) and dihydroflavonol 4-reductase (DFR). In another study, a Ginko biloba ANS gene showed synergistic patterns of expression in anthocyanin accumulation and abiotic stress responses.57 Therefore, ANS genes are implicated in responses to adverse environment stresses including high levels of salinity.57 A single ANS1 orthologue was identified in each of Z. japonica and Z. pacifica, and these genes were designated as ZjANS1 and ZpANS1 (Supplementary Figure S8). A comparison of the ZjANS1 and ZpANS1 sequences revealed a number of SNPs and indels in the 5′ upstream regions and 27 SNPs in the coding regions, including an SNP in the termination codon of ZjANS1 that extends the coding region for an additional 27 bp. These polymorphisms may reflect the diversity of anthocyanin pigmentation within and between species of the genus Zoysia, as described in previous reports.11,58 As is the case for the ANAC102 and STO/BBX24, the ANS1 gene was heterozygous in Z. matrella, and both ZjANS1 and ZpANS1 were identified in the ‘Wakaba’ genome assembly (Supplementary Figure S9). Zoysia matrella shows wide variations in morphological traits, most of which are intermediate between those of Z. japonica and Z. pacifica. Tanaka et al.5 conducted an SSR marker analysis and suggested that Z. matrella might represent an interspecific hybrid between Z. japonica and Z. pacifica. The combinations of the ANAC102, STO/BBX24, and ANS1 alleles in the three Zoysia genomes provide additional supportive evidence for this hypothesis.
3.4. Comparative genome analysis
3.4.1. Comparison among Z. japonica and other Zoysia species
In order to investigate the phylogenetic relationships among the Zoysia species, we collected genome sequence reads from three additional accessions, Z. japonica ‘Kyoto’, Z. japonica ‘Miyagi’, and Z. matrella ‘Chiba Fair Green’. The reads from these three accessions and those from Z. matrella ‘Wakaba’ and Z. pacifica ‘Zanpa’ were aligned against the reference genome of Z. japonica ‘Nagirizaki’. We identified a total of 7,424,163 SNPs including 3,660,698 SNPs in two or more accessions (common SNPs) (Table 3). Additionally, we found a total of 852,488 short indels of 1–60 bp; of which, 319,551 were common to two or more accessions (Supplementary Table S12). These total numbers of SNPs and indels are well correlated with the previously reported phylogenetic relationships among Zoysia species based on DNA marker analyses.4,59,60 In Z. pacifica ‘Zanpa’, the majority of SNPs and indels were homozygous in each allele, whereas Z. matrella ‘Wakaba’ and ‘Chiba Fair Green’ had higher numbers of heterozygous SNPs and indels (Table 3).
Table 3.
Summary of SNPs between Z. japonica ‘Nagirizaki’ and other Zoysia species
| All SNPs | Common SNP sites |
Specific SNP sites |
|||||
|---|---|---|---|---|---|---|---|
| Total number | ALT/homo | Hetero | Total number | ALT/homo | Hetero | ||
| Total of 5 accessions | 7,424,163 | 3,660,698 | – | – | 3,763,465 | – | – |
| Z. japonica | |||||||
| Kyoto | 1,011,037 | 749,624 | 434,991 | 314,633 | 261,413 | 90,960 | 170,453 |
| Miyagi | 1,055,665 | 789,694 | 627,166 | 162,528 | 265,971 | 177,898 | 88,073 |
| Z. matrella | |||||||
| Wakaba | 3,929,947 | 2,896,705 | 964,659 | 1,932,046 | 1,033,242 | 384,846 | 648,396 |
| Chiba Fair Green | 3,182,483 | 2,511,362 | 813,474 | 1,697,888 | 671,121 | 210,065 | 461,056 |
| Z. pacifica | |||||||
| Zanpa | 4,228,938 | 2,697,220 | 2,315,529 | 381,691 | 1,531,718 | 958,605 | 573,113 |
To investigate the homozygous and heterozygous features of the Zoysia genomes, the genotype of each SNP locus was classified into one of the three types: homozygous with the reference allele (R), homozygous with an alternative allele (A), or heterozygous (H). The zoysiagrass accessions were compared in the following order: Z. japonica ‘Kyoto’, Z. japonica ‘Miyagi’, Z. matrella ‘Wakaba’, Z. matrella ‘Chiba Fair Green’, and Z. pacifica ‘Zanpa’. The most common genotype pattern was R-R-H-H-A, with 670,724 SNP sites (18.3% of total), followed by R-R-H-R-A (13.4%), R-R-A-R-A (5.8%), and R-R-R-H-A (5.8%) (Supplementary Table S13). A similar tendency was observed in the pattern of common indels (Supplementary Table S14). The results indicate that most of the polymorphisms among the three Zoysia species are between Z. japonica and Z. pacifica, and these sites are often heterozygous in Z. matrella. These data, along with the heterozygosity of the ANAC102, STO/BBX24, and ANS1 genes in Z. matrella, and the results from our previous study using SSR markers5 all support the high probability that Z. matrella is an interspecific hybrid between Z. japonica and Z. pacifica. This possibility would further support a hypothesis that Z. matrella is a homoploid hybrid species since the nature of Z. matrella reflects the criteria for homoploid hybrid speciation. These criteria include: similar chromosome numbers with parental species, ability to backcross with parental species, intermediate phenotypic traits, and ecological divergence from parents.61 The information obtained in this study will provide new clues about the origin of Zoysia. In addition, our results highlight the importance of hybridization and polyploidization in grass diversification, which have implications for conservation.
3.4.2. Comparisons with other Poaceae species
Synteny among the Poaceae genomes was investigated by comparing the positions of genes on the Z. japonica ‘Nagirizaki’ pseudomolecules with their positions in the genome sequences of O. sativa, S. bicolor, and B. distachyon (see Materials and methods section for details). As shown in a Venn diagram in Supplementary Figure S10, 8,596 of 53,625 genes (16.0%) were conserved in these four Poaceae species, while 17,288 genes (32.2%) occurred in clusters only found in Z. japonica. Based on the homologous genes thus identified and their positions on the pseudomolecules, we generated dot-plot diagrams between Z. japonica and the three other Poaceae species. The results revealed extensive collinearities in all cases, and remarkable collinearities between the entire genomes of O. sativa and S. bicolor (Fig. 1 and Supplementary Figs S11 and S12). The comparison between Z. japonica and S. bicolor clearly revealed the following rearrangements: the middle section of S. bicolor chromosome 1 corresponded with the middle sections of Z. japonica chromosomes 7 and 8, and the middle section of S. bicolor chromosome 2 corresponded with the middle sections of Z. japonica chromosomes 19 and 20 (Fig. 1). The rearranged regions of chromosomes 7 and 8 and chromosomes 19 and 20 in Z. japonica corresponded to chromosomes 10 and 9 in O. sativa, respectively (Supplementary Figure S11). These rearrangements between the Z. japonica and S. bicolor genomes support the possibility that ‘invading’ chromosomes, the paleo-ancestors of chromosomes 10 and 9, integrated into different sets of chromosomes in Z. japonica and S. bicolor, as discussed in a previous study using RAD markers of Z. japonica.8
Figure 1.
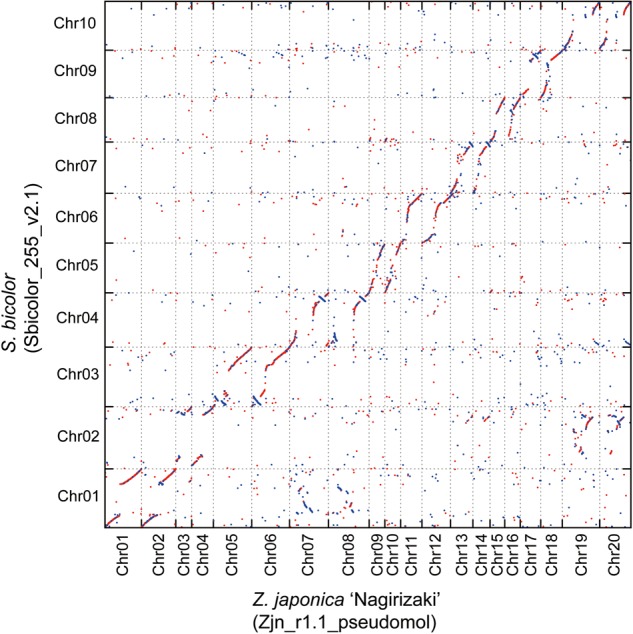
Comparison between the Z. japonica ‘Nagirizaki’ and S. bicolor genomes. The translated protein sequences on the pseudomolecules of Z. japonica ‘Nagirizaki’ and S. bicolor were compared by BLAST searches with an E-value cut-off of 1E−100, and the top hits were plotted. A hit on the same strand between both species is coloured in red, while hits on reverse strands are coloured in blue.
3.5. Databases and data retrieval
The draft genome sequences, gene and protein sequences, and annotation files (gff files) of Z. japonica ‘Nagirizaki’ (Zjn_r1.1), Z. matrella ‘Wakaba’ (Zmw_r1.0) and Z. pacifica ‘Zanpa’ (Zpz_r1.0) can be retrieved from the Zoysia Genome Database (http://zoysia.kazusa.or.jp). The pseudomolecule sequences of Z. japonica ‘Nagirizaki’ are also provided. In this database, the BLAST searches against genome sequences and genes are available by inputting query sequences in the BLAST search page. Users can then browse the BLAST search results against databases with annotations, such as NCBI's NR database and TAIR10 pep.
The accession number in NCBI's BioProject database for the raw sequence data obtained in this study is PRJDB4210. The scaffolds of ‘Nagirizaki’ (Zjn_r1.1), ‘Wakaba’ (Zmw_r1.0), and ‘Zanpa’ (Zpz_r1.0) are available from DDBJ/EMBL/NCBI under the accession numbers BCLF01000001–BCLF01011786 (11,786 entries), BCLG01000001–BCLG01013609 (13,609 entries), and BCLH01000001–BCLH01011428 (11,428 entries), respectively. The llumina PE and MP reads from MiSeq and HiSeq 2000 are available from DDBJ's DRA database under the accession number DRA004073. BioSample accession numbers of the species are as follows: SAMD00041943 (Z. japonica ‘Nagirizaki’), SAMD00041944 (Z. matrella ‘Wakaba’), SAMD00041945 (Z. pacifica ‘Zanpa’), SAMD00041942 (Z. japonica ‘Kyoto’), SAMD00041941 (Z. japonica ‘Miyagi’), and SAMD00041940 (Z. matrella ‘Chiba Fair Green’).
Supplementary data
Supplementary data are available at www.dnaresearch.oxfordjournals.org.
Funding
This work was supported by the Kazusa DNA Research Institute Foundation. Funding to pay the Open Access publication charges for this article was provided by the Kazusa DNA Research Institute Foundation.
Supplementary Material
Acknowledgements
We thank T. Fujishiro, C. Jige, K. Kawashima, Y. Kishida, M. Kohara and N. Nakazaki for excellent technical assistance. Thanks are also due to the Chiba Prefectural Agriculture and Forestry Research Center and Mr. Ippei Katayama in Kyoto Prefectural Katsura Senior High School for kindly providing the plant materials.
References
- 1.Huang B., DaCosta M., Jiang Y.. 2014, Research advances in mechanisms of turfgrass tolerance to abiotic stresses: from physiology to molecular biology, Crit. Rev. Plant Sci., 33, 141–89. [Google Scholar]
- 2.Marcum K.B. 1999, Salinity tolerance mechanisms of grasses in the subfamily Chloridoideae, Crop Sci., 39, 1153–60. [Google Scholar]
- 3.Kitamura F. 1989, The climate of Japan and its surrounding areas and the distribution and classification of zoysiagrasses, Int. Turfgrass Soc. Res. J., 6, 17–21. [Google Scholar]
- 4.Kimball J.A., Zuleta C.M., Kenworthy K.E., Lehman V.G., Harris-Shultz K.R., Milla-Lewis S.R.. 2013, Genetic relationships in Zoysia species and the identification of putative interspecific hybrids using simple sequence repeat markers and inflorescence traits, Crop Sci., 53, 285–95. [Google Scholar]
- 5.Tanaka H., Tokunaga R., Muguerza M. et al. 2016, Genetic structure and speciation of zoysiagrass ecotypes collected in Japan, Crop Sci., 56, 818–26. [Google Scholar]
- 6.Cai H., Li M., Wang X., Yuyama N., Hirata M.. 2014, In: Cai H., Yamada T., Kole C. (eds.) Genetics, Genomics and Breeding of Forage Crops, CRC Press, Florida, pp. 168–86. [Google Scholar]
- 7.Guo H., Ding W., Chen J. et al. 2014, Genetic linkage map construction and QTL mapping of salt tolerance traits in zoysiagrass (Zoysia japonica), PLoS One, 9, e107249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang F., Singh R., Genovesi A.D. et al. 2015, Sequence-tagged high-density genetic maps of Zoysia japonica provide insights into genome evolution in Chloridoideae, Plant J., 82, 744–57. [DOI] [PubMed] [Google Scholar]
- 9.Wei S., Du Z., Gao F. et al. 2015, Global transcriptome profiles of ‘Meyer’ zoysiagrass in response to cold stress, PLoS One, 10, e0131153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xie Q., Niu J., Xu X. et al. 2015, De novo assembly of the Japanese lawngrass (Zoysia japonica Steud.) root transcriptome and identification of candidate unigenes related to early responses under salt stress, Front. Plant Sci., 6, 610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ahn J.H., Kim J.S., Kim S. et al. 2015, De novo transcriptome analysis to identify anthocyanin biosynthesis genes responsible for tissue-specific pigmentation in zoysiagrass (Zoysia japonica Steud.), PLoS One, 10, e0124497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tanaka H., Hirakawa H., Muguerza M. et al. 2016, The complete chloroplast genome sequence of Zoysia matrella, Crop Sci., doi:10.2135/cropsci2015.08.0517. [Google Scholar]
- 13.Godden G.T., Jordon-Thaden I.E., Chamala S. et al. 2012, Making next-generation sequencing work for you: Approaches and practical considerations for marker development and phylogenetics, Plant Ecol. Diversity, 5, 427–50. [Google Scholar]
- 14.Sims D., Sudbery I., Ilott N.E., Heger A., Ponting C.P.. 2014, Sequencing depth and coverage: key considerations in genomic analyses, Nat. Rev. Genet., 15, 121–32. [DOI] [PubMed] [Google Scholar]
- 15.Paterson A.H., Freeling M., Sasaki T.. 2005, Grains of knowledge: Genomics of model cereals, Genome Res., 15, 1643–50. [DOI] [PubMed] [Google Scholar]
- 16.Murashige T., Skoog F.. 1986, A revised medium for rapid growth and bio assays with tobacco tissue cultures, Physiol. Plant, 15, 473–97. [Google Scholar]
- 17.Ishigaki G., Gondo T., Ebina M., Suenaga K., Akashi R.. 2010, Estimations of genome size in Brachiaria species, Grassl. Sci., 56, 240–2. [Google Scholar]
- 18.Price H.J., Dillon S.L., Hodnett G., Rooney W.L., Ross L., Johnston J.S.. 2005, Genome evolution in the genus Sorghum (Poaceae), Ann. Bot., 95, 219–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yagi M., Kosugi S., Hirakawa H. et al. 2014, Sequence analysis of the genome of carnation (Dianthus caryophyllus L.), DNA Res., 21, 231–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang X., Chockalingam S.P., Aluru S.. 2013, Survey of error-correction methods for next-generation sequencing, Brief Bioinform., 14, 56–66. [DOI] [PubMed] [Google Scholar]
- 21.Kajitani R., Toshimoto K., Noguchi H. et al. 2014, Efficient de novo assembly of highly heterozygous genomes from whole-genome shotgun short reads, Genome Res., 24, 1384–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li H. 2012, Exploring single-sample SNP and INDEL calling with whole-genome de novo assembly, Bioinformatics, 28, 1838–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gao S., Sung W.K., Nagarajan N.. 2011, Opera: reconstructing optimal genomic scaffolds with high-throughput paired-end sequences, J. Comput. Biol., 18, 1681–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kosugi S., Hirakawa H., Tabata S.. 2015, GMcloser: closing gaps in assemblies accurately with a likelihood-based selection of contig or long-read alignments, Bioinformatics, 31, 3733–41. [DOI] [PubMed] [Google Scholar]
- 25.Grabherr M.G., Haas B.J., Yassour M. et al. 2011, Full-length transcriptome assembly from RNA-Seq data without a reference genome, Nat. Biotechnol., 29, 644–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Parra G., Bradnam K., Korf I.. 2007, CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes, Bioinformatics, 23, 1061–7. [DOI] [PubMed] [Google Scholar]
- 27.Altschul S.F., Madden T.L., Schaffer A.A. et al. 1997, Gapped BLAST and PSI-BLAST: a new generation of protein database search programs, Nucleic Acids Res., 25, 3389–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Price A.L., Jones N.C., Pevzner P.A.. 2005, De novo identification of repeat families in large genomes, Bioinformatics, 21(Suppl 1), i351–8. [DOI] [PubMed] [Google Scholar]
- 29.Kofler R., Schlotterer C., Lelley T.. 2007, SciRoKo: a new tool for whole genome microsatellite search and investigation, Bioinformatics, 23, 1683–5. [DOI] [PubMed] [Google Scholar]
- 30.Lowe T.M., Eddy S.R.. 1997, tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence, Nucleic Acids Res., 25, 955–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Eddy S.R. 2009, A new generation of homology search tools based on probabilistic inference, Genome Inform., 23, 205–11. [PubMed] [Google Scholar]
- 32.Holt C., Yandell M.. 2011, MAKER2: an annotation pipeline and genome-database management tool for second-generation genome projects, BMC Bioinformatics, 12, 491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bao Z., Eddy S.R.. 2002, Automated de novo identification of repeat sequence families in sequenced genomes, Genome Res., 12, 1269–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stanke M., Waack S.. 2003, Gene prediction with a hidden Markov model and a new intron submodel, Bioinformatics, 19(Suppl 2), ii215–25. [DOI] [PubMed] [Google Scholar]
- 35.Korf I. 2004, Gene finding in novel genomes, BMC Bioinformatics, 5, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Trapnell C., Pachter L., Salzberg S.L.. 2009, TopHat: discovering splice junctions with RNA-Seq, Bioinformatics, 25, 1105–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Trapnell C., Williams B.A., Pertea G. et al. 2010, Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation, Nat. Biotechnol., 28, 511–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Slater G.S., Birney E.. 2005, Automated generation of heuristics for biological sequence comparison, BMC Bioinformatics, 6, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tatusov R.L., Fedorova N.D., Jackson J.D. et al. 2003, The COG database: an updated version includes eukaryotes, BMC Bioinformatics, 4, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Quevillon E., Silventoinen V., Pillai S. et al. 2005, InterProScan: protein domains identifier, Nucleic Acids Res., 33, W116–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.The Gene Ontology Consortium. 2000, Gene ontology: tool for the unification of biology, Nat. Genet., 25, 25–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ogata H., Goto S., Sato K., Fujibuchi W., Bono H., Kanehisa M.. 1999, KEGG: Kyoto encyclopedia of genes and genomes, Nucleic Acids Res., 27, 29–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fu L., Niu B., Zhu Z., Wu S., Li W.. 2012, CD-HIT: accelerated for clustering the next-generation sequencing data, Bioinformatics, 28, 3150–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Langmead B., Trapnell C., Pop M., Salzberg S.L.. 2009, Ultrafast and memory-efficient alignment of short DNA sequences to the human genome, Genome Biol., 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li H., Handsaker B., Wysoker A. et al. 2009, The Sequence Alignment/Map format and SAMtools, Bioinformatics, 25, 2078–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gupta B., Huang B.. 2014, Mechanism of salinity tolerance in plants: physiological, biochemical, and molecular characterization, Int. J. Genomics, 2014, 701596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Christianson J.A., Wilson I.W., Llewellyn D.J., Dennis E.S.. 2009, The low-oxygen induced NAC domain transcription factor ANAC102 affects viability of Arabidopsis thaliana seeds following low-oxygen treatment, Plant Physiol., 149, 1724–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gangappa S.N., Botto J.F.. 2014, The BBX family of plant transcription factors, Trends Plant Sci., 19, 460–70. [DOI] [PubMed] [Google Scholar]
- 49.Lippuner V., Cyert M.S., Gasser C.S.. 1996, Two classes of plant cDNA clones differentially complement yeast calcineurin mutants and increase salt tolerance of wild-type yeast, J. Biol. Chem., 271, 12859–66. [DOI] [PubMed] [Google Scholar]
- 50.Nagaoka S., Takano T.. 2003, Salt tolerance-related protein STO binds to a Myb transcription factor homologue and confers salt tolerance in Arabidopsis, J. Exp. Bot., 54, 2231–7. [DOI] [PubMed] [Google Scholar]
- 51.Qian Y.L., Engelke M.C., Foster M.J.V.. 2000, Salinity effects on zoysiagrass cultivars and experimental lines, Crop Sci., 40, 488–92. [Google Scholar]
- 52.Jakoby M., Weisshaar B., Dröge-Laser W. et al. 2002, bZIP transcription factors in Arabidopsis, Trends Plant Sci., 7, 106–11. [DOI] [PubMed] [Google Scholar]
- 53.Liu X., Chu Z.. 2015, Genome-wide evolutionary characterization and analysis of bZIP transcription factors and their expression profiles in response to multiple abiotic stresses in Brachypodium distachyon, BMC Genomics, 16, 227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yamamoto N., Takano T., Tanaka K. et al. 2015, Comprehensive analysis of transcriptome response to salinity stress in the halophytic turf grass Sporobolus virginicus, Front. Plant Sci., 6, 241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jin J., Zhang H., Kong L., Gao G., Luo J.. 2014, PlantTFDB 3.0: a portal for the functional and evolutionary study of plant transcription factors, Nucleic Acids Res., 42, D1182–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Manetas Y. 2006, Why some leaves are anthocyanic and why most anthocyanic leaves are red?, Flora, 201, 163–77. [Google Scholar]
- 57.Xu F., Cheng H., Cai R. et al. 2008, Molecular cloning and function analysis of an anthocyanidin synthase gene from Ginkgo biloba, and its expression in abiotic stress responses, Mol. Cells, 26, 536–47. [PubMed] [Google Scholar]
- 58.Choi J.S. 2010, Morphological characteristics of medium-leaf type zoysiagrasses (Zoysia spp.) and their classification using RAPDs, Kor. Turfgrass Sci., 24, 88–96. [Google Scholar]
- 59.Yaneshita M., Nagasawa R., Engelke M.C., Sasakuma T.. 1997, Genetic variation and interspecific hybridization among natural populations of zoysiagrasses detected by RFLP analyses of chloroplast and nuclear DNA, Genes Genet. Syst., 72, 173–9. [DOI] [PubMed] [Google Scholar]
- 60.Tsuruta S., Hashiguchi M., Ebina M. et al. 2005, Development and characterization of simple sequence repeat markers in Zoysia japonica Steud, Grassl. Sci., 51, 249–57. [Google Scholar]
- 61.Rieseberg L.H., Willis J.H.. 2007, Plant speciation, Science, 317, 910–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.