

Abstract
Two series of analogues of the tetrahydroprotoberberine (THPB) alkaloid (±)-stepholidine that a) contain various alkoxy substituents at the C10 position and, b) were de-rigidified with respect to (±)-stepholidine, were synthesized and evaluated for affinity at dopamine and σ receptors in order to evaluate effects on D3 and σ2 receptor affinity and selectivity. Small n-alkoxy groups are best tolerated by D3 and σ2 receptors. Among all compounds tested, C10 methoxy and ethoxy analogues (10 and 11 respectively) displayed the highest affinity for σ2 receptors as well as σ2 versus σ1 selectivity and also showed the highest D3 receptor affinity. De-rigidification of stepholidine resulted in decreased affinity at all receptors evaluated; thus the tetracyclic THPB framework is advantageous for affinity at dopamine and σ receptors. Docking of the C10 analogues at the D3 receptor, suggest that an ionic interaction between the protonated nitrogen atom and Asp110, a H-bond interaction between the C2 phenol and Ser192, a H-bond interaction between the C10 phenol and Cys181 as well as hydrophobic interactions of the aryl rings to Phe106 and Phe345, are critical for high affinity of the compounds.

1. Introduction
The dopamine D3 receptor has received considerable attention, especially over the past two decades, mainly due to its prominent role in mediating craving for addictive substances.1-3 In that regard, selective D3 antagonists have been sought after as potential therapeutics for cocaine addiction and the treatment of cocaine relapse.4 Additionally, a growing body of evidence in the literature suggests that D3 antagonism may be a valuable antipsychotic therapeutic strategy.5-7 Several selective D3 antagonists have been identified that bear the key pharmacophoric elements of a phenylpiperazine motif connected to an aryl amide unit via a 4 carbon spacer.8,9,1 Other D3-preferring scaffolds are also known, although the phenypiperazine template has been most prolific in terms of the number of compounds reported. Representative examples of selective D3 antagonists are depicted in Figure 1.
Figure 1.

Structures of representative selective D3 antagonists
Despite advances in understanding the structure-activity relationships (SARs) of such D3 scaffolds, in most cases, clinical translation of the resulting optimized compounds has been limited, primarily due to pharmacokinetic issues.3 There is thus a constant search for new structural templates endowed with selective D3 antagonist activity as such compounds could serve as useful leads for development as in vivo biological tools and therapeutic leads, especially pertinent to the realms of neuropsychiatric disorders and drug abuse.
There are two types of σ receptors - σ1 and σ2, as borne out by differential binding of selective radioligands as well as other biochemical and pharmacological studies.10-12 σ receptors are distributed in the central nervous system (CNS) and in peripheral tissue.13,14 Within the CNS, σ1 receptors are concentrated in the hippocampus and other limbic regions which control cognition and emotion.15,16 Thus these receptors have been implicated in a number of neuropsychiatric disorders and have been targets for the development of treatment interventions for such diseases.17-20 σ2 receptors are found predominantly in regions responsible for motor functions in the CNS and are also highly expressed in the lung, liver and kidneys.15,16 The σ1 receptor has been cloned but the σ2 receptor has not, and there is some debate about the identity of the σ2 receptor.21 Recent studies suggest that the σ2 receptor is Progesterone Receptor Membrane Component 1 (PGRMC1).22,23
σ2 receptors are overexpressed in tumor cells and are upregulated in rapidly proliferating cells and this receptor has attracted interest as a biomarker for cancer proliferation.24,25 Some σ2 ligands (particularly σ2 receptor agonists) display cytotoxic activity.26 Thus, σ2 ligands have been of interest for the development of "stand alone" antitumor drugs, targeted drug delivery agents and Positron Emission Tomography (PET) tools for imaging the proliferative status of various cancers.27-30 σ2 receptor ligands have been predominantly from four structural classes: 6,7-dimethoxytetrahydroisoquinolines, indoles, tropanes and cyclohexylpiperazines (Figure 2). Many of these compounds lack sufficient selectivity or are otherwise hampered by unfavorable pharmacokinetic drawbacks that limit their usefulness as drugs, anticancer drug delivery vehicles or tools. New, potent and selective σ2 receptor ligands are sought after to fully exploit the potential of the σ2 receptor as a target for cancer diagnosis and therapy.
Figure 2.

Structural classes of σ2ligands and representative examples
The tetrahydroprotoberberine (THPB) alkaloid (−)-stepholidine (1, Figure 3) shows affinity for dopamine D1 and D2 receptors and has been the focus of a number of structure-activity relationship (SAR) studies in this regard.31,32 The antipsychotic and anti-drug abuse potential of (−)-stepholidine has been attributed to its unusual D1 agonist/D2 antagonist profile. (−)-Stepholidine also shows good affinity for σ2 (Ki = 54 nM) and D3 receptors (Ki = 30 nM), as revealed by data from the Psychoactive Drug Screening Program (PDSP) database.33 Given the comparatively high σ2 and D3 affinity of 1, we considered that the THPB template may be exploitable towards the development of novel potent and selective σ2 receptor or D3 receptor ligands. However, no SAR studies have been performed on THPBs in relation to their σ2 or D3 receptor affinity. Furthermore, computational evaluations on THPBs particularly at the D3 receptor (for which a crystal structure is available)34 are lacking in the literature. Such studies could propel our understanding of the potential utility of this scaffold towards obtaining novel, selective σ2 or D3 receptor ligands. Our explorations in this regard are described herein.
Figure 3.

Structure of (−)-stepholidine (1)
2. Results and discussion
Stepholidine was used as the lead molecule for our SAR evaluations. To initiate this study, we decided to prepare a series of racemic C10 stepholidine analogues in order to probe the tolerance of this position for larger substituent groups at both receptors. THPBs are structurally similar to the 6,7-dimethoxytetrahydroisoquinoline class of σ2 ligands in that both possess a tetrahydroisoquinoline motif. Tetrahydroisoquinoline sub-structures are also found in a number of D3 antagonist ligands (eg. SB277011A, Figure 1).1 However, known 6,7-dimethoxytetrahydroisoquioline σ2 ligands, like the known D3-preferring tetrahydroisoquinolines are flexible, extended molecules whereas the THPB scaffold is obviously a more rigid structure. To elucidate the importance of structural rigidity of the THPB core on σ2 and D3 receptor affinity, less rigid stepholidine analogues were prepared and evaluated. Given that THPBs have been known to show affinity for D1 and D2 receptors, the analogues were also assessed at D1 and D2 receptors to further gauge selectivity. Computational studies were also performed to help to rationalize observed affinities.
2.1 Chemistry
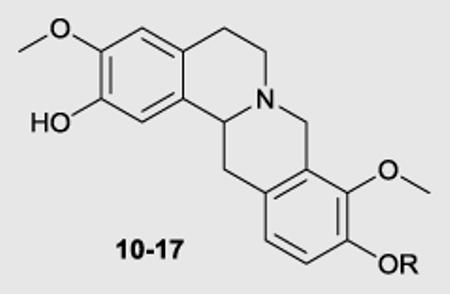
We engaged a route that we have recently reported for the synthesis of the THPB 10 for the synthesis of the C10 alkoxy derivatives.35 This route is depicted in Scheme 1. Commercially available phenol 2 was reacted with ethoxymethyl chloride. The protected aryl bromide (3) thus formed was then treated with LDA (generated from reaction of n-BuLi with diisopropylamine) to afford diester 4. Selective hydrolysis of the aliphatic ester in 4 gave acid 5. Compound 5 was coupled to amine 6 and the resulting amide (7) was subjected to cyclization under Bischler-Napieralski conditions. Cyclization was accompanied by removal of the ethoxymethyl protecting group. The imine which formed was not isolated, but was treated thereafter with sodium borohydride, whereupon cyclization to afford the 8-oxoprotoberberine core ensued (compound 8). Reduction of the 8-oxo group of 8 with lithium aluminum hydride gave the THPB 9 containing the unprotected C10 phenol group. Williamson ether alkylation of phenol 9 with various alkyl halides afforded the C9 alkoxy/C2 benzyl protected derivatives. Finally, the benzyl group of these intermediates was removed by treatment with HCl in refluxing methanol to obtain the racemic C10 analogues 10-17. Compound (±)-1 was obtained by deprotection of the benzyl group of phenol 9 in similar fashion as for other C10 analogues.
Scheme 1.

Synthesis of C10 analogues. Reagents and conditions: (a) DIPEA, (chloromethoxy)ethane, DCM, 98%; (b) diisopropylamine, n-BuLi, DMM, THF, −78 °C, 54%; (c) K2CO3, 1:1 H2O/MeOH, reflux, 95%; (d) CDI, THF, 78%; (e) i) POCl3, CH3CN, reflux; ii) NaBH4, MeOH, 79% over 2 steps; (f) LAH, THF, reflux, 84%; (g) i) appropriate alkyl halide, DMF, CH3CN, rt to reflux; ii) conc. HCl, MeOH, reflux; 64-77% over 2 steps; (h) conc. HCl, MeOH, reflux, 81 %
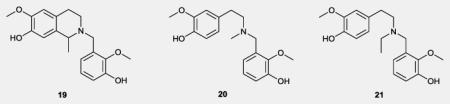
The route for preparation of the flexible analogues is shown in Scheme 2. Here, primary amine 6 was converted to the secondary amine 18 via reductive amination with 3-benzyloxy-2-methoxybezaldehyde. Pictet-Spengler cyclization followed by acidic debenzylation were implemented to convert 18 to the tetrahydroisoquinoline analogue 19. A sequence of reductive amination and benzyl group hydrolysis reactions gave tertiary amines 20 and 21 from the secondary amine 18.
Scheme 2.

Synthesis of flexible analogues. Reagents and conditions: a) Na(OAc)3BH, DCM, rt, appropriate aldehyde; b) i) CF3COOH, 1,1-diethoxyethane, DCM, rt; ii) Conc. HCl, MeOH, reflux, 60% over two steps; c) Conc. HCl, MeOH, reflux.
2.2 Biological evaluations
2.2.1 SAR on C10 analogues
Compounds (±)-1, 10-17 and 19-21 were assayed for binding affinity to human dopamine D1, D2 and D3 receptors and σ1 and σ2 receptors by the Psychoactive Drug Screening Program (PDSP). Details of the screening protocol may be found online at the PDSP website (https://pdspdb.unc.edu/pdspWeb/). In brief, the compounds were initially assayed in quadruplicate at a 10 μM concentration at D1, D2, D3, σ1 and σ2 receptors. Compounds that showed an inhibition of binding of >50% were progressed to secondary assays to measure Ki. The Ki determinations were performed via 12-point concentration-response curves in triplicate (unless noted otherwise). This data is presented in Tables 1 (for C10 analogues) and 2 (for de-rigidified analogues).
Table 1.
Affinity data (Ki in nM) for C10 analogues at dopamine and σ receptors
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
|
| ||||||||||
| Ki (nM)a | Selectivity | |||||||||
|
|
||||||||||
| Compound | R | D1b | D2c | D3d | σ1e | σ2f | D2/D1 | D3/D1 | D2/D3 | σ1/σ2 |
| (±)−1 | H | 5.6g | 115.5g | 101g | 269 | 9.0 | 20.6 | 18.0 | 1.1 | 30 |
| 10 | Me | 15 | 102 | 37 | 106 | 1.0 | 6.8 | 2.5 | 2.8 | 106 |
| 11 | Et | 12.4 | 159 | 44 | 124 | 1.3 | 12.8 | 3.5 | 3.6 | 95 |
| 12 | n-Pr | 7.8 | 99 | 53 | 57 | 6.7 | 12.7 | 6.8 | 1.9 | 8.5 |
| 13 | n-Bu | 11 | 121 | 65 | 13 | 49 | 11 | 5.9 | 1.9 | 0.27 |
| 14 | n-Pent | 19 | 283 | 101 | 119 | 97 | 14.9 | 5.3 | 2.8 | 1.22 |
| 15 | n-Hex | 40 | NAh | 220 | 37 | 107 | NDi | 5.5 | NDi | 0.35 |
| 16 | Hydroxypropyl | 6.7 | 211 | 110 | 53 | 3.8 | 31.5 | 16.4 | 1.9 | 13.9 |
| 17 | Cyclopropylmethyl | 12 | 54 | 54 | 12 | 2.4 | 4.5 | 4.5 | 1 | 5 |
Experiments carried out in triplicate - SEM values are within 13% of reported Ki;
[3H]SCH2390 used as radioligand/(+)−butaclamol used as reference compound - Ki = 2.3 nM;
[3H]N-methylspiperone used as radioligand/haloperidol used as reference compound - Ki = 9.6 nM;
[3H]N-methylspiperone used as radioligand/chlorpromazine used as reference compound - Ki = 11.0 nM;
[3H](+)−pentazocine used as radioligand/haloperidol used as reference compound - Ki = 1.1 nM;
[3H]DTG used as radioligand/haloperidol used as reference compound - Ki = 10.0 nM;
experiments carried out in duplicate;
NA- not active (<50% inhibition in primary assay therefore no secondary assay was performed);
ND - not determined
(±)-1 displayed approximately 20-fold stronger affinity for the D1 receptor as compared to the D2 and D3 receptors (Ki of 5.6, 115.5 and 101 nM at D1, D2 and D3 respectively). For C10 alkoxy derivatives alkylation tends to reduce D1 affinity, with the analogues showing a 1.2 to 7-fold decrease in affinity as compared to (±)-1. The C10 phenol plays role but is not absolutely required for D1 affinity, given the relatively small drop in affinity seen for some analogues (for example 12 and 16). This result is in accordance with observations from previous SAR work on the THPB framework by others.32,31 Methyl and ethyl substituents resulted in an approximately 2-fold decrease in affinity (compounds 10 and 11 respectively). In the n-propyl (12) to n-hexyl (15) homologous series, a progressive decrease in affinity was observed. The D1 receptor affinity seen for the hydroxypropyl analogue 16 was comparable to that observed for (±)-1 and the n-butyl analogue 13 (6.7, 5.6 and 11 nM respectively). This suggests that the hydroxyl group in 16 plays a relatively minor role in binding to the D1 receptor. The presence of the hydroxypropyl substituent in 16 restores the D1 affinity of stepholidine, improves the selectivity versus the D2 receptor (20.6 versus 31.5 for D2/D1 selectivity for stepholidine and 16 respectively) and retains the selectivity versus the D3 receptor (18 versus 16-fold for D3/D1 selectivity). Among the alkylated analogues, a 3 carbon chain length seems to be optimal for D1 affinity (consider 12 and 16). The cyclopropylmethyl analogue (17) had similar affinity to the n-butyl analogue 13 and ethyl analogue 11. Thus some degree of branching on the alkyl chain may be tolerated for D1 affinity.
On a whole, the D2 affinity of the analogues was lower than their D1 affinities. Thus the compounds retained selective affinity for the D1 receptor, as was observed in the lead molecule (±)-1. However, most compounds had lower than the 20-fold D1 selectivity seen for (±)-1 (except for compounds 15 and 16). Compounds in the homologous series from a methyl group to an n-butyl chain (10-13), had D2 affinities that are similar to (±)-1, ranging from 99 to 159 nM. However, compounds with the larger n-pentyl and n-hexyl chains had reduced affinities; in fact, the n-hexyl analogue (15) did not show any appreciable affinity for the D2 receptor (<50% inhibition) in the primary assay. The cyclopropylmethyl analogue (17) was the only compound in this series that displayed a significantly higher affinity for the D2 receptor than (±)-1.
There was a clear trend in the methyl to n-hexyl series at the D3 receptor, where an increase in the alkyl chain length was associated with a progressive decrease in affinity for the receptor. The hydroxypropyl analogue had similar affinity for the D3 receptor as compared to stepholidine. In the case of the cyclopropylmethyl analogue, D3 receptor affinity was improved two-fold as compared to stepholidine (54 nM vs 101 nM).
At the σ1 receptor, all analogues tested showed increased affinity as compared to (±)-1 (ranging from approximately 2-fold to 22-fold increase in σ1affinity). Therefore, the C10 phenol functionality is not required for σ1 receptor affinity. No clear SAR trend could be discerned in the homologous series 10-15. The n-butyl analogue 13 had the highest affinity of the compounds in this series [Ki = 13 nM for 13 vs 269 nM for (±)-1]. The hydroxypropyl analogue 16 had lower affinity than 13 (53 vs 13 nM) which suggests that the hydroxyl group in 16 does not have any significant binding interactions with the σ1 receptor (akin to observations at the D1 receptor). The cyclopropylmethyl and n-butyl analogues had similar σ1 affinities-approximately 12 nM (a situation that mirrors results for 17 at the D1 receptor). It thus appears that the cyclopropylmethyl group may function as a less hydrophobic bioisostere of the n-butyl group in this series at σ1 and D1 receptors.
A clear trend was seen at the σ2 receptor for analogues 10-15, wherein there was a progressive decrease in σ2 receptor affinity as the alkyl chain was extended. Compounds 10 and 11 had the highest affinity of all compounds assayed (1 nM and 1.3 nM respectively), representing an approximately 7 - to 9-fold higher σ2 receptor affinity than (±)-1. With respect to binding at σ receptors, both 10 and 11 exhibited higher σ2 receptor selectivity than compound (±)-1 [approximately 100-fold for 10 and 11 versus 30-fold for compound (±)-1]. All other analogues had decreased selective affinity for the σ2 receptor as compared to the σ1 receptor. In fact, this σ receptor selectivity was reversed for compounds 13 and 15 which both showed approximately 3-fold higher selectivity for σ1 versus σ2 receptors. Analogues 10-12 had higher affinity than (±)-1, indicating that small C10 alkoxy groups are better tolerated than the C10 phenolic group at the σ2 receptor. Compounds 16 and 17 displayed very high σ2 receptor affinity, comparable to 10 and 11; thus there is some tolerance for small polar or hydrophobic groups at C10 for σ2 affinity.
2.2.2 SAR on flexible analogues
All de-rigidified analogues showed significantly lower affinity than (±)-1 for dopamine D1, D2 and D3 receptors. Thus, the intact THPB core seems to be very important for affinity to dopamine receptors. Compounds 20 and 21 which lacked a tetrahydroisoquinoline motif had lower affinities for the D1, D2 and D3 receptor than compound 19 which possesses such a motif. Therefore it would appear that the presence of an intact tetrahydroisoquinoline moiety contributes significantly to dopamine receptor affinity.
De-rigidification did not significantly improve σ1 receptor affinity. Only a 2-fold increase in σ1 affinity was seen for compounds 19 and 20 as compared to (±)-1; a decrease in σ1 affinity was observed with compound 21. All flexible analogues showed a deterioration in σ2 receptor affinity as compared to the lead (227 - 788 nM vs 9 nM). However, in general the de-rigidified analogues showed higher selectivity for σ receptors as compared to dopamine receptors suggesting that molecular flexibility is better tolerated at σ receptors within this series.
2.2.3 Cytotoxicity evaluation
As alluded to before, some compounds have shown cytotoxic actions that are attributable to their interaction with σ receptors. We were curious to determine the extent to which our newly identified high affinity σ2 receptor ligands displayed cytotoxicity. Therefore, isocorypalmine (10), the most potent and selective σ2 ligand identified in this study, was selected for cytotoxicity evaluation in an Alamar Blue assay.36 Compound 10 lacked significant cytotoxic activity in this assay, with an IC50 of 178 μM. Since σ2 receptor agonists are generally cytotoxic,37-39 this result tends to imply that compound 10 is a σ2 receptor antagonist (although this needs to be ratified with further assays).
2.2.4 Receptor docking studies at the D3 receptor
The phenolic group at C10 of stepholidine does not seem to play a major role in its binding to dopamine D1 and D2 receptors (given the similarity in D1 and D2 affinities of the analogues as compared to stepholidine). In the case of the D3 receptor, a number of analogues had moderately improved affinity as compared to stepholidine. Therefore it seems that the C10 phenolic group is not an absolute necessity for affinity of stepholidine to the D3 receptor.
Based on the SAR trend observed (increasing alkyl chain length decreases D3 affinity), it is apparent that the C10 alkoxy substituents of the analogues might project into a shallow hydrophobic pocket in the receptor. To get further clarity into this possibility, and to aid in rationalizing the D3 affinity trend observed, docking evaluations were performed on analogues (±)-1, 10-17 and 19-21 at the D3 receptor.
Investigation of the docked ligand poses and recognition of key protein-ligand interactions provides insights into the SAR outcomes of the series of C10 analogues and the de-rigidified compounds with respect to D3. In this study, ligand docking was performed retrospectively in order to elucidate and rationalize the measured D3 affinity data. Overall, the calculated binding energies for stepholidine and the C10 analogues are in reasonable agreement with activities derived from the experimental data as shown in Table 3. Compounds (±)-1, 10-17 and 19-21 were docked into a pre-prepared 3-dimensional structure of the D3 receptor derived from the crystal structure with PDB code 3PBL.34 A representation of the docked poses for the ligands (±)-1, 10-15 and 17 within the D3 receptor binding pocket is depicted in Figure 4. Each ligand generated multiple binding modes and the structures shown display the top ranked poses according to Glidescore (+ligand strain). These poses maintain key receptor-ligand interactions, including the critical protonated tertiary N – Asp110 salt bridge motif, H-bonds to Ser192 and Cys181 and hydrophobic interactions to Phe106 and Phe345 (Figure 4a). The analogues are positioned such that the aryl substituents project into hydrophobic cavities in the receptor (depicted in Figure 4b for compound 10). The estimated binding energies according to Glidescore are very similar for this series of C10 analogue ligands, covering a narrow range from −7.9 to −7.3 kcal/mol. Overall, the relatively small decrease in the predicted binding energy as the length of the alkyl chain increases for compounds 10-15 qualitatively matches the decrease in the measured affinity for these ligands.
Table 3.
Predicted binding affinity from Glidescore and ligand strain energy for C10 and flexible analogues at the D3 receptor prepared from the crystal structure with PDB code 3PBL
| Compound | R | Glidescore | Ki at D3 (nM) |
|---|---|---|---|
| (±)−1 | H | −7.9 | 101 |
| 10 | Me | −7.5 | 37 |
| 11 | Et | −7.4 | 44 |
| 12 | n-Pr | −7.5 | 53 |
| 13 | n-Bu | −7.4 | 65 |
| 14 | n-Pent | −7.5 | 101 |
| 15 | n-Hex | −7.3 | 220 |
| 16 | Hydroxypropyl | −8.1 | 110 |
| 17 | Cyclopropylmethyl | −7.3 | 54 |
| 19 | −6.2 | 1367 | |
| 20 | −6.3 | 2680 | |
| 21 | −6.4 | 1811 |
Figure 4.

a) Docked poses of the lead molecule (±)-1 and the C10 analogues 10-15 and 17. Key hydrogen bonding interactions are given by the yellow dashed lines and hydrophobic interactions by the turquoise dashed lines. b) compound 10 docked in binding pocket
Quantitatively, the binding energy differences are very small compared to the wider, yet still relatively low, variation in measured affinities. The Glidescore value for stepholidine (−7.9 kcal/mol) was found to be lower than that for the other C10 analogues, including the cyclopropylmethyl analogue (−7.3 kcal/mol), in contrast to the experimental measurements. However, in a similar manner to the measured affinities, the hydroxypropyl analogue, with docked pose illustrated in Figure 5, had a predicted binding energy that is close to that of stepholidine (−8.1 kcal/mol vs −7.9 kcal/mol, respectively). Examination of the D3 receptor binding site shows that the alkyl, cyclopropylmethyl and hydroxypropyl groups of the C10 analogues fit into the extracellular hydrophobic region of the binding pocket, consisting of the extracellular loops and the junction of several helices, without incurring any significant clashes with the protein structure. Hence, the small difference in predicted binding energies for these systems is unsurprising. The hydroxyl group in compound 16 also forms an additional H-bond to Glu90.
Figure 5.

Docked pose of the hydroxypropyl analogue, compound 16.
In agreement with the experimental affinity measurements at the D3 receptor, the de-rigidified, flexible compounds 19-21 are predicted to have significantly lower binding energies than the above more rigid C10 analogues according to the in silico docking outputs. The Glidescore values for the three compounds are very similar, ranging only from −6.4 kcal/mol to −6.2 kcal/mol. Analysis of the generated poses and the key protein-ligand interactions (depicted in Figures 6 and 7) suggests that the lowered affinity and predicted binding energy for these de-rigidified analogues is a consequence of (i) extra ligand flexibility, which leads to a small amount of strain for the ligand to fit in the site compared to the nearest minimized ligand conformation, and (ii) a lower lipophilic interaction score with the binding site residues due to lack of the tetrahydroisoquinoline motif (20, 21) and the THBP core (19). Each of these effects is relatively small (< 1 kcal/mol), however, the overall consequence is to reduce the binding affinity of the flexible analogues.
Figure 6.

Docked pose of the flexible analogue, compound 19.
Figure 7.

Docked poses of the flexible analogues, compounds 20 and 21.
Others have investigated docking of stepholidine at the D3 receptor computationally.40 As compared to prior work, we found similarities in the key residues that are important for affinity of stepholidine (and the C10 analogues), particularly Asp110, Ser192 and Phe345. Apart from the key protonated nitrogen-Asp110 salt bridge interaction, in both studies Ser192 interacts with an aromatic hydroxyl group via a hydrogen bond and Phe345 interacts with an aromatic ring via aromatic stacking (Fig 4a). The main difference is that the orientation of stepholidine in our Glide docking study is reversed compared to the AutoDock derived pose in the previous work. Thus in our case Ser192 is H-bonded to the phenolic hydroxyl in ring A and Phe345 interacts with ring A, whereas in prior work Ser192 and Phe345 have interactions with the ring D hydroxyl and ring D respectively. Given the close to symmetric structure of the molecule and its high rigidity, this reversal has little impact on the interactions with the receptor pocket, except for the formation of an additional H-bonding interaction between the phenolic group in ring D of stepholidine and the backbone O of Cys181 in our docked pose.
In order to assess the binding stability of flexible compounds 19-21 the ligands were simulated in complex with D3 (crystal structure 3pbl).34 Ligand parameters were generated in Generalized Amber Force Field (GAFF),41 protein parameters were assigned from AMBERFF14SB,42 and the system was solvated in a 10 Å buffer of Tip4pew (~12000 water molecules). Ligands were allowed to move freely, however, protein heavy atoms were restrained with 2.5 kcal/Å2 harmonic restraints to maintain model accuracy (in the absence of lipids). The starting configuration utilized for these simulations was the top ranked docked poses discussed above. These configurations were minimized and heated to 300 K, then equilibrated for 20 ns in NPT, and finally run in a 100 ns production NVT molecular dynamics simulation sampling every 1 ps.
The results of these simulations show that the docking poses are stable within this D3 configuration. Two metrics were utilized to assess ligand stability throughout the production simulations: ligand RMSD and the distance between the ligand’s center of mass and Asp110 (critical for binding). Ligand RMSD was computed for each frame of the production trajectories compared to the ligand configuration in the original docked pose. This analysis shows that while ligand positions fluctuated over the course of the simulation, however, this did not differ greatly from their starting poses (Figure 8). The distance between ligand center of mass and Asp110 was utilized to determine whether the ligand maintained this key contact over the course of the simulation. Though the distance does change by a small amount in compounds 19 and 21, all three ligands maintain a bound state during the simulation (Figure 9). These data suggest that compounds 19-21 maintain a configuration similar to the predicted docked pose during a molecular dynamics simulation and that these predicted poses are stable with respect to the crystal D3 configuration. This supports the assertion that the difference in binding affinity is not likely due to destabilized binding, but rather the added ligand flexibility and decreased lipophilic interactivity.
Figure 8.

RMSD of flexible compounds 19 (left), 20 (middle) and 21 (right) between all atom coordinates at each frame, compared to the coordinates of the docked pose (starting configuration). All three compounds display limited motion within the binding site, fluctuating about their original docked pose.
Figure 9.

Distance between flexible compounds 19 (left), 20 (middle), and 21 (right) center of mass and the key aspartate Asp110 over the course of a 100 ns simulation. All three ligands maintain this contact throughout the sampled time.
3. Conclusions
The purpose of this study was to shed light on how substituent groups at the C10 position of THPBs as well as molecular rigidity impact σ2 receptor and D3 receptor affinity and selectivity (as compared to σ1, D1 and D2 receptors). Among the 5 receptors evaluated, the C10 THPB analogues seem to exhibit a general preference for the D1 receptor. The lowest D1 affinity seen for any analogue was 40 nM (for compound 15). Thus the C10 position seems to be fairly tolerant of alkoxy substituent groups with respect to D1 affinity. Of all C10 analogues tested, the lowest affinities were seen for D2 and σ1 receptors. Among the 3 dopamine receptors, affinity was generally highest for D1 receptor and lowest for the D2 receptor. Overall, the general order of affinities for C10 analogues at the 5 receptors evaluated is D1>σ-2>D3>D2≈σ1. However, individual compounds may exhibit high σ2 receptor affinity and selectivity. This is the situation with compounds 10 and 11. Compound 10 is 15-fold more selective for the σ2 receptor as compared to the D1 receptor, thirty-seven-fold more selective as compared to the D3 receptor and approximately 100-fold more selective as compared to the σ1 receptor. A similar selectivity profile was obtained for compound 11. The data suggests that relatively small n-alkoxy groups can be accommodated at C10 for high σ2 affinity and σ2 versus σ1 selectivity. It appears that high σ2 affinity may be obtained (perhaps to the detriment of selectivity) with small branched alkoxy groups (as is seen in for compound 17). However the generality of this premise will be need to be tested via the evaluation of a larger set of analogues. Compound 10 is the racemic form of the known natural product isocorypalmine which has been reported to possess anti-cocaine effects in animals.43 It is not clear at this time to what extent σ2 receptor affinity may play in this activity of naturally-occurring isocorypalmine. Rigidity of the THPB core seems to be important to impart high σ2 receptor affinity but de-ridification of the THPB core negatively impacts dopamine receptor affinity to a greater extent than σ receptor affinity.
The ligand binding energies from the D3 receptor docking studies show some similarities and minor discrepancies compared to the experimentally derived affinities. This is not surprising as empirical scoring functions, such as Glidescore used in this study, cannot rank order correctly and quantitatively discriminate all protein-ligand complexes based on the predicted scores. Such scoring functions work best in terms of distinguishing ligands as active or inactive, in contrast to quantitative rank ordering. In the current work, the relatively small series of compounds analyzed in the biological assays and in silico simulations, the relatively small range of affinities observed and the formation of multiple ligand binding modes in the D3 receptor binding pocket represent additional causes for the absence of a clear correlation between the docking outcomes and the experimentally derived affinities. Further exploration of a significantly larger and structurally more diverse ligand library would be required to provide deeper understanding and quantitative evaluation of the effectiveness of our in silico modeling process. Nevertheless, despite these issues, the predictive model successfully differentiated the flexible analogues from the more rigid systems and allowed for the identification of important protein-ligand interactions within the D3 receptor binding site that should potentially be conserved in the search for more active and selective compounds. The lack of crystal structures for σ receptors precluded similar studies at these receptors; this would have been similarly insightful, had it been possible.
This study is the first to investigate structural requirements of stepholidine analogues for affinity to σ receptors and the dopamine D3 receptor, via the utility of a combination of SAR and computational techniques. Our work has provided important revelations into the future design of THPB-based D3 and σ2 ligands and has uncovered compounds with higher D3 and σ2 receptor affinity than the lead, such as compound 10 and the new THPBs 11, 12 and 17. These compounds may be useful as leads for further SAR explorations to optimize σ2 and D3 affinity and selectivity. In closing, we posit that the THPB scaffold represents a good starting point from which to mine ligands with interesting mono- and multi-receptor profiles, particularly at dopamine D3 and σ2 receptors.
4. Experimental
4. 1 General methods
All moisture-sensitive and oxygen-sensitive reactions were carried out in flame-dried glassware under a nitrogen atmosphere. Solvents and all other reagents were purchased at the highest commercial quality from Aldrich and Fisher Scientific USA and used without further purification. Anhydrous sodium sulfate was used as drying agent for work-up of reactions. HRESIMS spectra were obtained using an Agilent 6520 QTOF instrument. 1H NMR and 13C NMR spectra were recorded using Bruker DPX-500 spectrometer (operating at 500 MHz for 1H; 125 MHz, for 13C) or Bruker Avance III spectrometer (operating at 400 MHz for 1H; 100 MHz, for 13C) using CDCl3 as solvent, unless stated otherwise. Tetramethylsilane (δ 0.00 ppm) served as an internal standard in 1H NMR and CDCl3 (δ 77.0 ppm) in 13C NMR unless stated otherwise. Chemical shift (δ 0.00 ppm) values are reported in parts per million and coupling constants in Hertz (Hz). Splitting patterns are described as singlet (s), doublet (d), triplet (t), and multiplet (m). Reactions were monitored by TLC with Whatman Flexible TLC silica gel G/UV 254 precoated plates (0.25 mm). TLC plates were visualized by UV (254 nm) and by staining in an iodine chamber. Flash column chromatography was performed with silica gel 60 (EMD Chemicals, 230-400 mesh, 0.063 μm particle size).
4.2 Chemistry
General procedure for synthesis of compounds 10-17 (as described for compound 10): 3,9,10-Trimethoxy-5,8,13,13a-tetrahydro-6H-isoquinolino[3,2-a]isoquinolin-2-ol (10)
Compound 9 (0.1 g, 0.24 mmol) was dissolved in DMF. Potassium carbonate (0.07 g, 0.48 mmol) and methyl iodide (0.02 mL, 0.36 mmol) were added and the reaction mixture was stirred overnight at rt. The crude reaction solution was filtered and the filtrate was evaporated to dryness. The residue was refluxed in a mixture of MeOH (5 mL) and concentrated HCl (3 mL) for 3 h. The MeOH was evaporated and the crude mixture was basified with ammonia solution, and extracted with DCM (3 × 15 mL). The combined organic layer was dried over anhydrous Na2SO4 and concentrated to give the crude product, which was purified by flash column chromatography on silica gel (2% MeOH/DCM) to give compound 10 (0.052 g, 64%) as an off-white solid; mp 208-210 °C; 1H NMR (CDCl3, 400 MHz) δ 6.87-6.77 (m, 3H), 6.60 (s, 1H), 5.56 (s, 1H), 4.24 (d, J = 15.7 Hz, 1H), 3.87 (s, 3H), 3.85 (s, 6H), 3.54-3.50 (m, 2H), 3.26-3.11 (m, 3H), 2.81 (dd, J = 16, 11.5 Hz, 1H), 2.68-2.61 (m, 2H); 13C NMR (CDCl3, 100 MHz) δ 150.2, 145.1, 143.9, 143.9, 130.6, 128.7, 127.9, 126.1, 124.0, 111.3, 111.0, 110.6, 60.2, 59.3, 59.3, 55.9, 54.0, 51.7, 36.3, 29.2; HRMS (ESI) m/z calcd. for C20H23NO4 [M+H]+, 342.1700, found 342.1704.
10-Ethoxy-3,9-dimethoxy-5,8,13,13a-tetrahydro-6H-isoquinolino[3,2-a]isoquinolin-2-ol (11)
Yield 67%; off-white powder, mp 186-188 °C ; 1H NMR (CDCl3, 400 MHz) δ 6.85-6.77 (m, 3H), 6.60 (s, 1H), 4.24 (d, J = 15.8 Hz, 1H), 4.06 (q, J = 7.0 Hz, 2H), 3.88 (s, 3H), 3.87 (s, 3H), 3.52 (d, J = 15.3 Hz, 2H), 3.27-3.10 (m, 3H), 2.84-2.61 (m, 3H), 1.44 (t, J = 7.0 Hz, 3H); 13C NMR (CDCl3, 100 MHz) δ 147.5, 143.5 143.2, 142.0, 128.7, 126.7, 125.9, 124.2, 122.0, 110.5, 109.5, 108.7, 62.5, 58.2, 57.4, 54.0, 52.2, 49.8, 34.4, 27.3, 13.1; HRMS (ESI) m/z calcd. for C21H25NO4 [M+H]+, 356.1856, found 356.1861.
3,9-Dimethoxy-10-propoxy-5,8,13,13a-tetrahydro-6H-isoquinolino[3,2-a]isoquinolin-2-ol (12)
Yield 68%; brown solid, mp 87-90 °C; 1H NMR (CDCl3, 500 MHz) δ 6.84-6.78 (m, 3H), 6.60 (s, 1H), 4.23 (d, J = 15.8 Hz, 1H), 3.94 (dt, J = 6.7, 1.8 Hz, 2H), 3.87 (s, 6H), 3.51 (d, J = 15.3 Hz, 2H), 3.25-3.10 (m, 3H), 2.80 (dd, J = 15.6, 11.4 Hz, 1H), 2.68-2.60 (m, 2H), 1.84 (sext, J = 7.1 Hz, 2H), 1.06 (t, J = 7.4 Hz, 3H); 13C NMR (CDCl3, 125 MHz) δ 149.6, 145.3, 145.1, 144.0, 130.6, 128.6, 127.8, 126.0, 123.8 112.3, 111.3, 110.6, 70.4, 60.2, 59.2, 55.9, 54.1, 51.7, 36.3, 29.2, 22.8, 10.7; HRMS (ESI) m/z calcd. for C22H27NO4 [M+H]+, 370.2013, found 370.2020.
10-Butoxy-3,9-dimethoxy-5,8,13,13a-tetrahydro-6H-isoquinolino[3,2-a]isoquinolin-2-ol (13)
Yield 73%; orange oil; 1H NMR (CDCl3, 400 MHz) δ 6.84-6.77 (m, 3H), 6.60 (s, 1H), 4.24 (d, J = 15.7 Hz, 1H) 3.98 (dt, J =1.0, 6.45 Hz, 2H), 3.86 (s, 6H), 3.52 (d J = 15.0 Hz, 2H), 3.25-3.10 (m, 3H), 2.80 (dd, J = 15.2, 11.3 Hz, 1H), 2.67-2.60 (m, 2H), 1.80 (quint, J = 6.9 Hz, 2H), 1.52 (sex, J = 7.6 Hz, 2H), 0.98 (t, J = 7.4 Hz, 3H); 13C NMR (CDCl3, 100 MHz) δ 149.6, 145.3, 145.1, 143.9, 130.5, 128.5, 127.7, 126.0, 123.8, 112.2, 111.3, 110.6, 68.5, 60.1, 59.2, 55.9, 54.0, 51.6, 36.2, 31.5, 29.2, 19.4, 13.9; HRMS (ESI) m/z calcd. for C23H29NO4 [M+H]+, 384.2169, found 384.2173.
3,9-Dimethoxy-10-(pentyloxy)-5,8,13,13a-tetrahydro-6H-isoquinolino[3,2-a]isoquinolin-2-ol (14)
Yield 65%; brown oil; 1H NMR (CDCl3, 400 MHz ) δ 6.84-6.76 (m, 3H), 6.60 (s, 1H), 4.23 (d, J = 15.8 Hz, 1H), 3.97 (dt, J =1.5, 6.55 Hz, 2H), 3.87 (s, 6H), 3.51 (d, J = 15.2 Hz, 2H), 3.26-3.10 (m, 3H), 2.80 (dd, J = 15.7, 11.5 Hz, 1H), 2.68-2.60 (m, 2H), 1.82 (quint, J = 6.7 Hz, 2H), 1.49-1.36 (m, 4H), 0.93 (t, J = 7.2 Hz, 3H); 13C NMR (CDCl3, 100 MHz) δ 149.6, 145.3, 145.0, 143.9, 130.6, 128.6, 127.7, 126.1, 123.8, 112.3, 111.3, 110.6, 68.9, 60.2, 59.2, 55.9, 54.6, 51.6, 36.3, 29.2, 29.1, 28.3, 22.5, 14.0; HRMS (ESI) m/z calcd. for C24H31NO4 [M+H]+, 398.2326, found 398.2326.
10-(Hexyloxy)-3,9-dimethoxy-5,8,13,13a-tetrahydro-6H-isoquinolino[3,2-a]isoquinolin-2-ol (15)
Yield 70%; dark brown oil; 1H NMR (CDCl3, 500MHz ) δ 6.84-6.76 (m, 3H), 6.60 (s, 1H), 4.23 (d, J = 15.7 Hz, 1H), 3.97 (t, J = 6.6 Hz , 2H), 3.86 (s, 6H), 3.53-3.50 (m, 2H), 3.26-3.10 (m, 3H), 2.80 (dd, J = 15.8, 11.6 Hz, 1H), 2.68-2.60 (m, 2H), 1.81 (quin, J = 6.7 Hz, 2H), 1.48 (quint, J= 7.2, 2H), 1.34 (sex, J = 3.8 Hz, 4H), 0.91 (t, J= 7.0 Hz, 3H); 13C NMR (CDCl3, 125 MHz) δ 149.6, 145.3, 145.0, 143.9, 130.6, 128.6, 127.7, 126.1, 123.8, 112.3, 111.4, 110.6, 68.9, 60.2, 59.2, 55.9, 54.1, 51.7, 36.3, 31.6, 29.4, 29.2, 25.8, 22.6, 14.0; HRMS (ESI) m/z calcd. for C25H33NO4 [M+H]+, 412.2482, found 412.2429.
10-(3-Hydroxypropoxy)-3,9-dimethoxy-5,8,13,13a-tetrahydro-6H-isoquinolino[3,2-a]isoquinolin-2-ol (16)
Yield 76%; reddish-brown solid, mp 73-78 °C; 1H NMR (CDCl3, 500MHz ) 6.87-6.80 (m, 3H), 6.60 (s, 1H), 4.22 (d, J = 15.7 Hz, 1H), 4.16 (td, J = 6.1, 1.5 Hz, 2H), 3.91-3.84 (m, 8H), 3.52 (d, J = 15.4 Hz, 2H), 3.27-3.10 (m, 3H), 2.80 (dd, J = 15.7, 11.6 Hz, 1H), 2.68-2.61 (m, 2H), 2.07 (quint, J = 5.8 Hz, 2H); 13C NMR (CDCl3, 125 MHz) δ 149.4, 145.4, 145.1, 143.9, 130.5, 128.8, 128.4, 126.1, 124.1, 112.5, 111.3, 110.6, 67.5, 61.0, 60.3, 59.2, 55.9, 54.0, 51.6, 36.3, 32.1, 29.2; HRMS (ESI) m/z calcd. for C22H27NO5 [M+H]+, 386.1884, found 386.1897.
10-(Cyclopropylmethoxy)-3,9-dimethoxy-5,8,13,13a-tetrahydro-6H-isoquinolino[3,2-a]isoquinolin-2-ol (17)
Yield 81%; yellow oil; 1H NMR (CDCl3, 500 MHz ) δ 6.83-6.76 (m,3H), 6.60 (s, 1H), 4.23 (d, J = 15.7 Hz, 1H), 3.90-3.83 (m, 8H), 3.51 (d, J = 15.1 Hz, 2H), 3.26-3.11 (m, 3H), 2.80 (dd, J = 15.0, 12.0 Hz, 1H), 2.68-2.62 (m, 2H), 1.31-1.27 (m, 1H), 0.61 (q, J = 5.7 Hz, 2H), 0.36 (q, J = 4.85 Hz, 2H); 13C NMR (CDCl3, 125 MHz) δ 149.5, 145.7, 145.0, 143.9, 130.6, 128.6, 128.1, 126.1, 123.8, 113.1, 111.3, 110.6, 73.9, 60.2, 59.2, 55.9, 54.1, 51.6, 36.3, 29.2, 10.5, 3.2, 3.2; HRMS (ESI) m/z calcd. for C23H27NO4 [M+H]+, 382.2013, found 382.2019.
N-(3-(Benzyloxy)-2-methoxybenzyl)-2-(4-(benzyloxy)-3-methoxyphenyl)ethan-1-amine (18)
Amine 6 (0.25g, 0.97 mmol) and 3-(benzyloxy)-2-methoxybenzaldehyde (0.24 g ,0.97 mmol) were dissolved in DCM and the solution stirred at 0 °C for 10 min. Sodium triacetoxyborohydride (0.31 g, 1.5 mmol) was added to the reaction mixture at 0 °C and allowed to stir for 3 h at rt. The reaction mixture was washed with saturated NaHCO3 and the organic layer was dried over sodium sulfate and concentrated to give the compound 18 (0.4 g, 83%) as a brown oil; 1H NMR (CDCl3, 500MHz ) δ 7.40-7.27 (m, 10H), 6.98-6.95 (m, 1H), 6.89-6.6.85 (m, 2H), 6.79-6.74 (m, 2H), 6.66 (dd, J = 1.6, 8.1 Hz, 1H), 5.12 (s, 2H), 5.10 (s, 2H), 3.85-3.79 (m, 8H), 2.86 (t, J = 7.3, 2H), 2.77 (t, J = 7.3 Hz, 2H); 13C NMR (CDCl3, 125 MHz) δ 151.6, 149.5, 147.7, 146.5, 137.3, 137.0, 133.5, 133.0, 128.6, 128.5, 128.0, 127.7, 127.3, 127.2, 123.8, 122.2, 122.1, 121.3, 120.6, 114.7, 114.1, 113.3, 112.4, 111.9, 71.1, 70.1, 60.7, 55.9, 50.3, 48.8, 35.7 ; HRMS (ESI) m/z calcd. for C31H33NO4 [M+H]+, 484.2488, found 484.2475.
2-(3-Hydroxy-2-methoxybenzyl)-6-methoxy-1-methyl-1,2,3,4-tetrahydroisoquinolin-7-ol (19)
Compound 18 (0.3 g, 0.6 mmol) was dissolved in DCM. 1,1 diethoxyethane (0.13 mL, 0.9 mmol) and TFA (0.14 mL, 1.8 mmol) were added to the reaction mixture and allowed to stir o.n. The reaction mixture was washed with saturated NaHCO3 and organic layer was dried over sodium sulfate and evaporated to dryness. The residues were refluxed in a mixture of MeOH (5 mL) and concentrated HCl (3 mL) for 3 h. The methanol was evaporated and the crude mixture was basified with ammonia solution, and extracted with dichloromethane (3 × 10 mL). The combined organic layer was dried over sodium sulfate and concentrated to give the crude product, which was purified by flash column chromatography on silica gel (3% MeOH/DCM) to give the compound 19 (0.12 g, 60%) as a yellow powder, mp 56-58 °C; 1H NMR (CDCl3, 500 MHz) δ 7.00-6.98 (m, 2H), 6.88 (dd, J =6.85, 2.8 Hz, 1H), 6.64 (s, 1H), 6.55 (s, 1H), 3.85-3.79 (m, 8H), 3.67 (d, J = 13.6 Hz, 1H), 3.07-3.02 (m, 1H), 2.83-2.77 (m, 1H), 2.68 (dt, J = 12.5, 4.5 Hz, 1H), 2.58 (dt, J = 13, 4.5 Hz, 1H), 1.37 (d, J = 6.6 Hz, 3H); 13C NMR (CDCl3, 125 MHz) δ 149.0, 145.9, 144.9, 143.6, 133.0, 132.7, 125.6, 124.6, 122.1, 114.3, 113.0, 110.6, 61.8, 56.3, 55.9, 52.2, 44.0, 27.0, 20.0; HRMS (ESI) m/z calcd. for C19H23NO4 [M+H]+, 330.1700, found 330.1707.
General procedures for synthesis of compounds 20 and 21 (as described for compound 20): 3-(((4-Hydroxy-3-methoxyphenethyl)(methyl)amino)methyl)-2-methoxyphenol (20)
Amine 18 (0.2g, 0.41 mmol) and formaldehyde (0.06 mL, 1.64 mmol) were dissolved in DCM and allowed to stir the solution at 0 °C for 10 minutes. Sodium triacetoxy borohydride (0.13g, 0.62 mmol) was added in to the reaction mixture at 0 °C and allowed it to stir for 3 h at room temperature. Reaction mixture was washed with saturated NaHCO3 and organic layer was dried over sodium sulfate and concentrated. The residues were refluxed in to the mixture of methanol (5 mL) and concentrated HCl (3 mL) for 3 h. Methanol was evaporated and the crude mixture was basified with ammonia solution, and extracted with dichloromethane (10 mL × 3). The combined organic layers were dried over sodium sulfate and concentrated to give the crude product, which was purified by flash column chromatography on silica gel (3% methanol/dichloromethane) to give the compound 20 (0.1g, 74%) as a yellow oil; 1H NMR (CDCl3, 500 MHz) δ 7.0-6.89 (m, 3H), 6.82 (d, J = 8.1 Hz, 1H), 6.68-6.66 (m, 2H), 3.86 (s, 3H), 3.80 (s, 3H), 3.64 (s, 2H), 2.79 (s, 2H), 2.70 (s, 2H), 2.33 (s, 3H); 13C NMR (CDCl3, 125 MHz) δ 149.0, 146.3, 145.9, 143.8, 124.7, 122.5, 121.2, 121.2, 114.2, 114.1, 114.0, 111.2, 61.7, 55.9, 55.7, 52.2, 42.0, 33.3; HRMS (ESI) m/z calcd. for C18H23NO4 [M+H]+, 318.1700, found 318.1693
3-((Ethyl(4-hydroxy-3-methoxyphenethyl)amino)methyl)-2-methoxyphenol (21)
Yield 68%, brown oil; 1H NMR (CDCl3, 500MHz ) δ 6.98-6.93 (m, 2H), 6.87 (dd, J = 2.0, 7.5 Hz, 1H), 6.80 (d, J = 8.4 Hz, 1H), 6.65-6.64 (m, 2H), 3.85 (s, 3H), 3.80 (s, 3H), 3.67 (s, 2H), 2.71 (s, 4H), 2.62 (q, J = 6.8 Hz, 2H), 1.1 (t, J = 7.0 Hz, 3H); 13C NMR (CDCl3, 125 MHz) δ 149.0, 146.3, 145.7, 143.7, 124.6, 122.3, 121.3, 121.3, 114.3, 114.1, 114.1, 111.2, 61.7, 55.8, 55.3, 51.9, 47.5, 33.0, 11.8; HRMS (ESI) m/z calcd. for C19H25NO4 [M+H]+, 332.1856, found 332.1851.
Supplementary Material
Table 2.
Kidata for flexible analogues
|
|||||
|---|---|---|---|---|---|
|
| |||||
| Ki (nMa) | |||||
|
| |||||
| Compound | D1b | D2c | D3d | σ1e | σ2f |
| 19 | 833 | 1095 | 1367 | 130 | 301 |
| 20 | 3111 | >10,000 | 2680 | 122 | 227 |
| 21 | >10000 | >10000 | 1811 | 350 | 788 |
Experiments carried out in triplicate - SEM values are within 13% of reported Ki;
[3H]SCH2390 used as radioligand/(+)−butaclamol used as reference compound - Ki = 2.3 nM;
[3H]N-methylspiperone used as radioligand/ haloperidol used as reference compound - Ki = 9.6 nM;
[3H]N-methylspiperone used as radioligand/chlorpromazine used as reference compound - Ki = 11.0 nM;
[3H](+)−pentazocine used as radioligand/ haloperidol used as reference compound - Ki = 1.1 nM;
[3H]DTG used as radioligand/ haloperidol used as reference compound - Ki = 10.0 nM
5. Acknowledgements
This publication was made possible by Grant Numbers 1SC1GM092282 (WWH), G12MD007599 (WWH), and 5SC3GM095417 (TK) from the National Institutes of Health. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH or its divisions. Ki determinations, and receptor binding profiles were generously provided by the National Institute of Mental Health's Psychoactive Drug Screening Program, Contract # HHSN-271-2008-00025-C (NIMH PDSP). The NIMH PDSP is directed by Bryan L. Roth MD, PhD at the University of North Carolina at Chapel Hill and Project Officer Jamie Driscol at NIMH, Bethesda MD, USA. For experimental details please refer to the PDSP website http://pdsp.med.unc.edu/ and click on "Binding Assay" or "Functional Assay" on the menu bar.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Micheli F, Heidbreder C. Expert Opin. Ther. Pat. 2013;23:363. doi: 10.1517/13543776.2013.757593. [DOI] [PubMed] [Google Scholar]
- 2.Heidbreder C. CNS Neurol. Disord. Drug Targets. 2008;7:410. doi: 10.2174/187152708786927822. [DOI] [PubMed] [Google Scholar]
- 3.Keck TM, John WS, Czoty PW, Nader MA, Newman AH. J Med Chem. 2015;58:5361. doi: 10.1021/jm501512b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Heidbreder Naunyn Schmiedebergs Arch. Pharmacol. 2013;386:167. doi: 10.1007/s00210-012-0803-6. [DOI] [PubMed] [Google Scholar]
- 5.Luedtke RR, Rangel-Barajas C, Malik M, Reichert DE, Mach RH. Curr. Pharm. Des. 2015;21:3700. doi: 10.2174/1381612821666150724100830. [DOI] [PubMed] [Google Scholar]
- 6.Pich EM, Collo Eur Neuropsychopharmacol. 2015;25:1437. doi: 10.1016/j.euroneuro.2015.07.012. [DOI] [PubMed] [Google Scholar]
- 7.Gross G, Wicke K, Drescher KU. Naunyn Schmiedebergs Arch. Pharmacol. 2013;386:155. doi: 10.1007/s00210-012-0806-3. [DOI] [PubMed] [Google Scholar]
- 8.Hackling AE, Stark H. Chembiochem. 2002;3:946. doi: 10.1002/1439-7633(20021004)3:10<946::AID-CBIC946>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 9.Newman AH, Grundt P, Nader MA. J. Med. Chem. 2005;48:3663. doi: 10.1021/jm040190e. [DOI] [PubMed] [Google Scholar]
- 10.Bouchard P, Quirion R. Neuroscience. 1997;76:467. doi: 10.1016/s0306-4522(96)00221-7. [DOI] [PubMed] [Google Scholar]
- 11.Quirion R, Bowen WD, Itzhak Y, Junien JL, Musacchio JM, Rothman RB, Su TP, Tam SW, Taylor DP. Trends Pharmacol. Sci. 1992;13:85. doi: 10.1016/0165-6147(92)90030-a. [DOI] [PubMed] [Google Scholar]
- 12.McCann DJ, Weissman AD, Su TP. Synapse. 1994;17:182. doi: 10.1002/syn.890170307. [DOI] [PubMed] [Google Scholar]
- 13.Hellewell SB, Bruce A, Feinstein G, Orringer J, Williams W, Bowen WD. Eur. J. Pharmacol. 1994;268:9. doi: 10.1016/0922-4106(94)90115-5. [DOI] [PubMed] [Google Scholar]
- 14.McCann DJ, Su TP. Eur. J. Pharmacol. 1990;188:211. doi: 10.1016/0922-4106(90)90004-h. [DOI] [PubMed] [Google Scholar]
- 15.Guitart X, Codony X, Monroy X. Psychopharmacology (Berl) 2004;174:301. doi: 10.1007/s00213-004-1920-9. [DOI] [PubMed] [Google Scholar]
- 16.Walker JM, Bowen WD, Walker FO, Matsumoto RR, De Costa B, Rice KC. Pharmacol. Rev. 1990;42:355. [PubMed] [Google Scholar]
- 17.Matsumoto RR, Nguyen L, Kaushal N, Robson MJ. Adv. Pharmacol. 2014;69:323. doi: 10.1016/B978-0-12-420118-7.00009-3. [DOI] [PubMed] [Google Scholar]
- 18.Robson MJ, Noorbakhsh B, Seminerio MJ, Matsumoto RR. Curr. Pharm. Des. 2012;18:902. doi: 10.2174/138161212799436601. [DOI] [PubMed] [Google Scholar]
- 19.Fishback JA, Robson MJ, Xu YT, Matsumoto RR. Pharmacol. Ther. 2010;127:271. doi: 10.1016/j.pharmthera.2010.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Matsumoto RR. Expert Rev. Clin. Pharmacol. 2009;2:351. doi: 10.1586/ecp.09.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Abate C, Niso M, Infantino V, Menga A, Berardi F. Eur. J. Pharmacol. 2015;758:16. doi: 10.1016/j.ejphar.2015.03.067. [DOI] [PubMed] [Google Scholar]
- 22.Zeng C, Garg N, Mach RH. Mol. Imaging Biol. 2015 doi: 10.1007/s11307-015-0891-z. [DOI] [PubMed] [Google Scholar]
- 23.Xu J, Zeng C, Chu W, Pan F, Rothfuss JM, Zhang F, Tu Z, Zhou D, Zeng D, Vangveravong S, Johnston F, Spitzer D, Chang KC, Hotchkiss RS, Hawkins WG, Wheeler KT, Mach RH. Nat. Commun. 2011;2:380. doi: 10.1038/ncomms1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mach RH, Smith CR, al-Nabulsi I, Whirrett BR, Childers SR, Wheeler KT. Cancer Res. 1997;57:156. [PubMed] [Google Scholar]
- 25.Wheeler KT, Wang LM, Wallen CA, Childers SR, Cline JM, Keng PC, Mach RH. Br. J. Cancer. 2000;82:1223. doi: 10.1054/bjoc.1999.1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zeng C, Rothfuss J, Zhang J, Chu W, Vangveravong S, Tu Z, Pan F, Chang KC, Hotchkiss R, Mach RH. Br. J. Cancer. 2012;106:693. doi: 10.1038/bjc.2011.602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.van Waarde A, Rybczynska AA, Ramakrishnan NK, Ishiwata K, Elsinga PH, Dierckx RA. Biochim. Biophys. Acta. 2015;1848:2703. doi: 10.1016/j.bbamem.2014.08.022. [DOI] [PubMed] [Google Scholar]
- 28.Mach RH, Zeng C, Hawkins WG. J. Med. Chem. 2013;56:7137. doi: 10.1021/jm301545c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hornick JR, Spitzer D, Goedegebuure P, Mach RH, Hawkins WG. Surgery. 2012;152:S152. doi: 10.1016/j.surg.2012.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Makvandi M, Tilahun ED, Lieberman BP, Anderson RC, Zeng C, Xu K, Hou C, McDonald ES, Pryma DA, Mach RH. Biochem. Biophys. Res. Commun. 2015;467:1070. doi: 10.1016/j.bbrc.2015.09.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sun H, Zhu L, Yang H, Qian W, Guo L, Zhou S, Gao B, Li Z, Zhou Y, Jiang H, Chen K, Zhen X, Liu H. Bioorg. Med. Chem. 2013;21:856. doi: 10.1016/j.bmc.2012.12.016. [DOI] [PubMed] [Google Scholar]
- 32.Qian W, Lu W, Sun H, Li Z, Zhu L, Zhao R, Zhang L, Zhou S, Zhou Y, Jiang H, Zhen X, Liu H. Bioorg. Med. Chem. 2012;20:4862. doi: 10.1016/j.bmc.2012.05.057. [DOI] [PubMed] [Google Scholar]
- 33.Roth BL, Lopez E, Beischel S, Westkaemper RB, Evans JM. Pharmacol. Ther. 2004;102:99. doi: 10.1016/j.pharmthera.2004.03.004. [DOI] [PubMed] [Google Scholar]
- 34.Chien EY, Liu W, Zhao Q, Katritch V, Han GW, Hanson MA, Shi L, Newman AH, Javitch JA, Cherezov V, Stevens RC. Science. 2010;330:1091. doi: 10.1126/science.1197410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gadhiya S, Ponnala S, Harding WW. Tetrahedron. 2015;71:1227. doi: 10.1016/j.tet.2015.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nakayama GR, Caton MC, Nova MP, Parandoosh Z. J. Immunol. Methods. 1997;204:205. doi: 10.1016/s0022-1759(97)00043-4. [DOI] [PubMed] [Google Scholar]
- 37.Behensky AA, Yasny IE, Shuster AM, Seredenin SB, Petrov AV, Cuevas J. J. Pharmacol. Exp. Ther. 2013;347:458. doi: 10.1124/jpet.113.208348. [DOI] [PubMed] [Google Scholar]
- 38.Rybczynska AA, Dierckx RA, Ishiwata K, Elsinga PH, van Waarde A. J. Nucl. Med. 2008;49:2049. doi: 10.2967/jnumed.108.053876. [DOI] [PubMed] [Google Scholar]
- 39.Korpis K, Weber F, Wunsch B, Bednarski PJ. Pharmazie. 2014;69:917. [PubMed] [Google Scholar]
- 40.Li B, Li W, Du P, Yu KQ, Fu W. J. Phys. Chem. B. 2012;116:8121. doi: 10.1021/jp3049235. [DOI] [PubMed] [Google Scholar]
- 41.Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA. J. Comput. Chem. 2004;25:1157. doi: 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- 42.Maier JA, Martinez C, Kasavajhala K, Wickstrom L, Hauser KE, Simmerling C. J. Chem. Theory Comput. 2015;11:3696. doi: 10.1021/acs.jctc.5b00255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xu W, Wang Y, Ma Z, Chiu YT, Huang P, Rasakham K, Unterwald E, Lee DY, Liu-Chen LY. Drug Alcohol Depend. 2013;133:693. doi: 10.1016/j.drugalcdep.2013.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.